
INTRODUCTION
Cancer has been a major health problem worldwide, with an estimate of more than 4,500 new cases each day in 2014 [1]. Numerous factors, including environment [2], lifestyle [3] and genetic defects [4, 5], contribute to tumorigenesis and development of cancer. The development of high-throughput profiling technology has enabled characterization of cancer cells from perspective of genome, epigenome and transcriptome [6, 7]. Several studies have succeeded in identifying tumor biomarkers for cancer detection, diagnosis or prognosis determination for specific types of cancer, such as estrogen receptor and progesterone receptor in breast cancer [8] and prostate-specific antigen in prostate cancer [9]. Although high heterogeneity was observed between the transcriptomic landscape of distinct types of cancer [10], cancer cells share characteristics such as dys-regulated cell growth and potential to invade compared to normal cells [11]. Consequently, biomarkers might be either specific to a particular type of cancer or general to multiple types of cancer.
The majority of previous efforts have focused on protein coding genes (PCGs). However, since lncRNAs have been implicated to play important roles in multiple biological processes such as cell cycle [12], cell apoptosis [13] as well as signal pathway [14, 15], lncRNAs might function as tumor suppressors or oncogenic drivers [16]. Dys-regulated expression of some lncRNAs, such as HOTAIR, PCAT1 and SNHG1, has been considered as indicator of several human cancers [17–19]. However, the molecular mechanism of lncRNA functions in cancer biology is still poorly understood.
In this study, microarray test was applied to obtain expression profiles of both protein coding genes and lncRNAs in tumor and paired adjacent non-tumor tissues from 76 patients (20 with gastric cancer, 20 with colon cancer, 16 with liver cancer and 20 with lung cancer). The microarray platform is custom designed covering both kind of transcripts of 21,789 mRNAs and 39,311 lncRNAs. A collection of 157 lncRNAs as well as 316 PCGs with dys-regulated expression in tumor tissues compared with adjacent non-tumor tissues in all of four types of cancer was observed. The possible functions of the identified 157 common DE-lncRNAs were further inferred based on their correlation with PCGs from both perspective of expression and genomic coordinates. Besides, PCGs and lncRNAs whose expression showed tissue specifity in any type of cancer were also identified. Part of the results were validated by quantitative PCR (qPCR) in external patient samples. In summary, this study has discovered cancer- and tissue- associated PCGs and lncRNAs through integrative analysis of expression profile revealed by custom designed microarray, thus providing a systematic summary of expression pattern and biological relevance of lncRNAs in cancer.
RESULTS
Identification of PCGs and lncRNAs as candidate common biomarkers for cancer
The global expression profile of both PCGs and lncRNAs in four types of cancer tissues (gastric, colon, liver, and lung) and adjacent non-cancerous tissues were examined by a custom microarray platform (see Materials and Methods for details). Hundreds of PCGs and lncRNAs show differential expression in each type of cancer (Figure 1A).
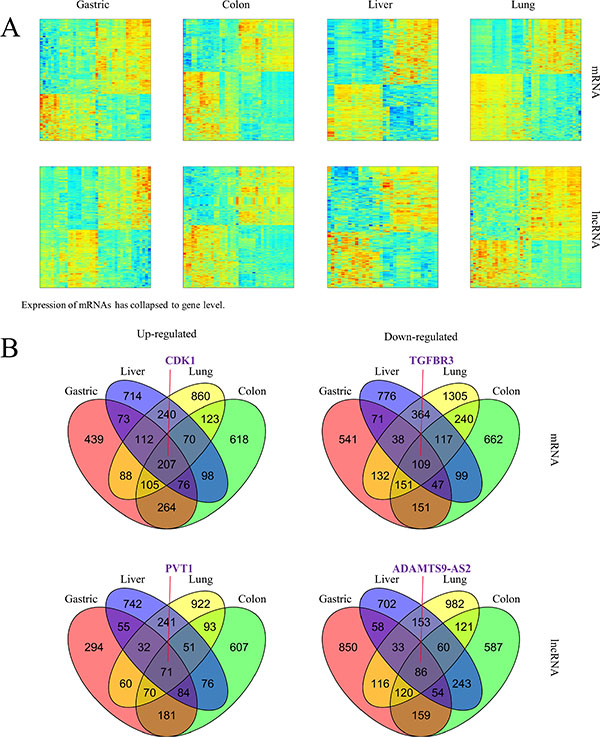
Figure 1: Altered expression of mRNAs and lncRNAs across cancer types. (A) Hierarchically clustered heatmaps of mRNAs and lncRNAs that are differentially expressed (student t-test corrected p-value < 0.05 and fold change > 1.5) in each type of cancer tissues compared to adjacent non-cancerous tissues. (B) Venn diagrams showing up-regulated and down-regulated mRNAs and lncRNAs whose dys-regulated expression pattern was shared by four types of cancer. Literature curated cancer biomarkers were indicated as examples.
We first examined the expression patterns of several well-known cancer-related lncRNAs revealed by our microarray experiments [20]. Notably, 8 of the 12 lncRNAs examined showed differential expression between tumors and non-tumor tissues in at least one type of cancer (Supplementary Figure S1). However, they could hardly serve as biomarkers common to multiple types of cancer other than a few examples. Besides, several of them might produce confusion. For example, as shown in our data, HOTTIP was indicated to have potential oncogenic function in liver cancer and gastric cancer as it showed up-regulated expression in tumors compared to adjacent non-tumor tissues while its association with colon cancer and lung cancer was not found. UCA1 is a lncRNA reported to promote cell proliferation in both breast cancer [21] and bladder cancer [22]. Of the cancer types used in our microarray experiments, UCA1 showed its oncogenic potential in gastric, colon and lung but had expression characteristic of tumor suppressor in liver. Although such lncRNAs might explain complex and heterogeneous nature of different cancer types [23], it would be more desirable to discover PCGs or lncRNAs as indicators in various types of cancer. As a consequence, further efforts were taken to identify PCGs and lncRNAs with up-regulated or down-regulated expression in all of four cancer types (Student t-test, false discovery rate (FDR) < 0.05, fold change (FC) > 1.5), resulting in 207 PCGs and 71 lncRNAs as potential oncogenes and 109 PCGs and 86 lncRNAs as potential tumor suppressors common to these four cancer types (Figure 1B, Supplementary File 1). These include some known cancer biomarkers such as PCGs CDK1 [24, 25] and TGFBR3 [26], as well lncRNAs PVT1 [27, 28] and ADAMTS9-AS2 [29]. In order to validate the alterations of PCG and lncRNA expression obtained from microarray data, we validated a subset of them across a panel of external samples by qRT-PCR (Figure 2A; the validation in gastric cancer was absent due to lack of additional samples). The qRT-PCR result showed high consistency with microarray data (Figure 2B).
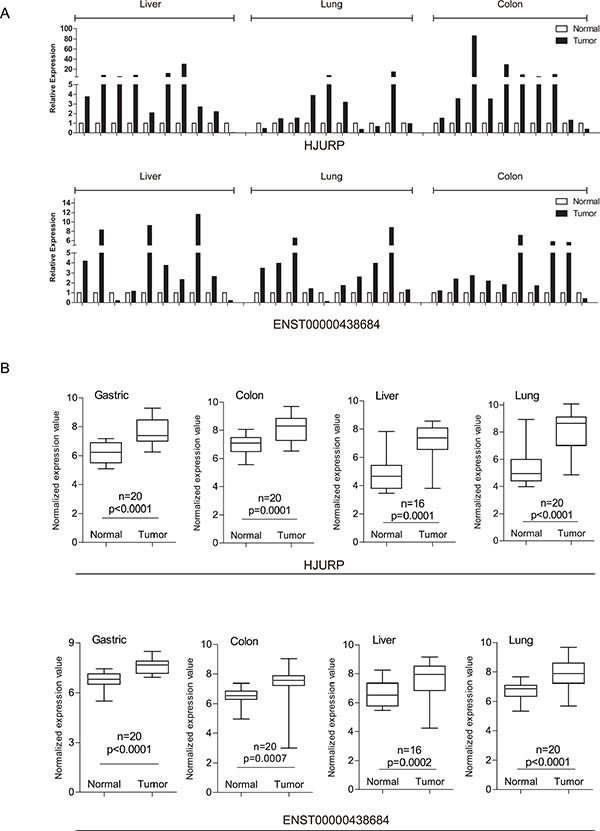
Figure 2: qRT-PCR validation of lncRNA and mRNA expression in samples from external patients. (A) Expression of common DE-mRNAs/lncRNAs (upper panel representing a mRNA and lower panel representing a lncRNA). (B) Boxplots represent common DE-mRNAs/lncRNAs’ expression based on the microarray data. (upper panel representing a mRNA and lower panel representing a lncRNA).
Next, gene ontology enrichment analysis was performed in the common DE-PCGs (Figure 3A). Up-regulated PCGs were enriched in cell cycle related biological processes while down-regulated PCGs were associated with cell adhesion, consistent with the common characteristics of cancer which refer to promoted cell proliferation and activated cell migration. Similarly, it could be inferred that common DE-lncRNAs might also play important roles in regulating essential biological processes and dys-regulated expression of them would lead to abnormality.
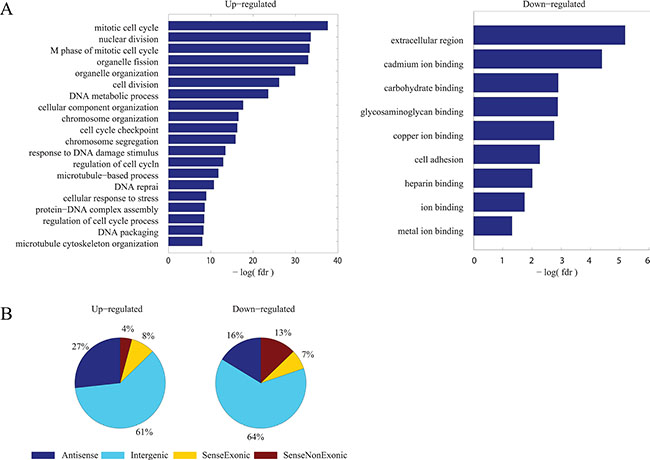
Figure 3: Signatures of common dys-regulated mRNAs and lncRNAs. (A) Functional enrichment result by DAVID for common up-regulated and down-regulated PCGs respectively. The resulted GO terms of biological processes with a FDR < 0.1 were considered statistically significant and depicted. (B) Distribution of common up-regulated and down-regulated lncRNAs according to their genomic context association with PCGs.
Classifying lncRNAs into different subgroups according to their genomic context association with PCGs results in the largest subgroup to be intergenic lncRNAs [30, 31]. Similar proportion of different subgroups was observed in these 157 common DE-lncRNAs (Figure 3B).
Functional prediction of common DE-lncRNAs
Gene Set Enrichment Analysis (GSEA) [32, 33] was performed in order to gain insights into the biological significance of the identified DE-lncRNAs which might serve as biomarkers common to four types of cancer. Pearson correlation coefficients between expression profiles of mRNAs and common DE-lncRNAs across all tissues were calculated based on which common DE-lncRNAs associated gene sets were identified. Unsupervised hierarchical clustering of enrichment score of KEGG pathways clearly separated common up-regulated lncRNAs from common down-regulated lncRNAs (Figure 4). Particularly, the majority of common up-regulated lncRNAs were significantly associated with cell cycle, similar to the function enrichment result of common up-regulated PCGs. Besides, some of the common up-regulated lncRNAs were functionally related to spliceosome. The signaling pathways including WNT pathways and MAPK pathways, which these common DE-lncRNAs might be involved in, were also indicated. Since our knowledge of lncRNAs had been far less than PCGs, the expression profile association between lncRNAs and PCGs would be beneficial clues to understand functions of lncRNAs.
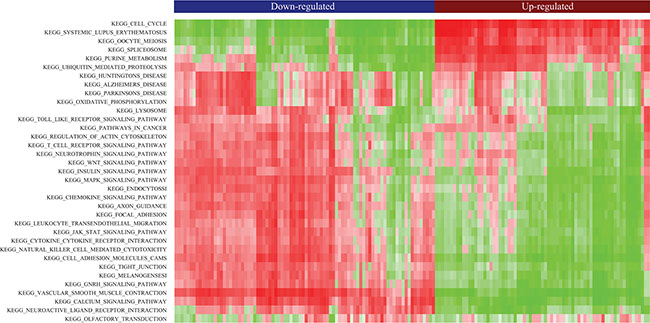
Figure 4: Heatmap of clustered pathway enrichment scores for common DE-lncRNAs. Red (green) denotes positive (negative) nominal enrichment scores in gene set enrichment analysis (GSEA) for KEGG pathways.
Another aspect of association between lncRNAs and PCGs is about genomic coordinates. Several lncRNAs have been known to function by cis-acting mechanism [34–36]. For example, Kcnq1ot1 has a negative control of its neighboring PCGs [37, 38]. In order to figure out the possibility that the common DE-lncRNAs function in cis, the expression correlation between the common DE-lncRNAs and their genomic neighbor genes (the nearest PCGs for intergenic lncRNAs, the PCGs on the opposite strand of antisense lncRNAs, the host PCGs of intronic lncRNAs and overlapping PCG for sense-exonic lncRNAs) was investigated. Compared with the expression correlation with neighbor PCGs of genome-wide lncRNAs covered by the microarray platform, there is a significant positive correlation for that of common DE-lncRNAs (Supplementary Figure S2). Examples representing common DE-lncRNAs which might have a role of regulating gene expression in cis were also shown (Figure 5).
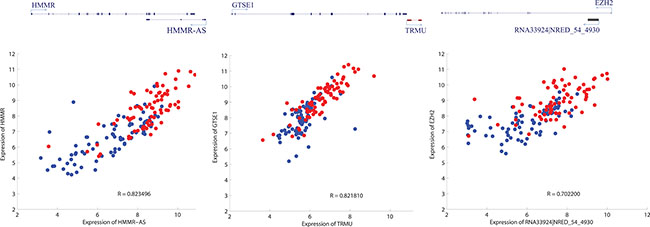
Figure 5: Three examples depicting potential cis regulation of common DE-lncRNAs on mRNAs. Upper panel is simplified schematic diagram representing the relative genomic location of lncRNAs and their neighboring PCGs (left: antisense; middle: intergenic; right: intronic); lower panel is scatter plot characterizing the expression profile correlation of the pair of genomic interacting lncRNA and mRNA (red and blue representing cancer tissues and adjacent non-cancerous tissues respectively).
Some lncRNAs are reported to play important roles in tumorigenesis by acting as competing endogenous RNAs (ceRNAs) [39–42]. In order to explore the possibility that lncRNAs might regulate the expression level of neighboring PCGs via ceRNA pathway, a tri-color network was constructed to elucidate interactions among common DE-lncRNAs, neighboring PCGs and miRNAs (Figure 6A). Interactions between common DE-lncRNAs and neighboring PCGs involved both their genomic position interaction and expression profile correlation (correlation coefficient > 0.45 and p-value < 0.01) while miRNA targets prediction result by miRanda [43, 44] linked miRNAs and DE-lncRNAs or neighboring PCGs. Minimum tri-color submotifs were identified. For example, CCT5 and its antisense lncRNA (RNA58651 nominated in-house) had positively correlated expression profile and both of them were predicted to be targeted by miR-1183 (Figure 6B). Another example is RNA34433, a novel intergenic lncRNA located downstream of NTRK3. RNA34433 showed down-regulated expression in tumor tissues compared to adjacent non-tumor tissues in four types of cancer while NTRK3 had been reported to be a potential tumor suppressor [45]. Both RNA34433 and NTRK3 were predicted to be targeted by has-miR-297 (Figure 6C), whose potential role in cancer genesis should be further investigated. MiR-34a, a literature-curated tumor suppressor [46–48], had a higher degree than other miRNAs in the network. Moreover, a significant enrichment of experimentally determined miR-34a targets was obtained in neighboring PCGs of common up-regulated lncRNAs (Supplementary Figure S3) through Gene ontology enrichment analysis [49].
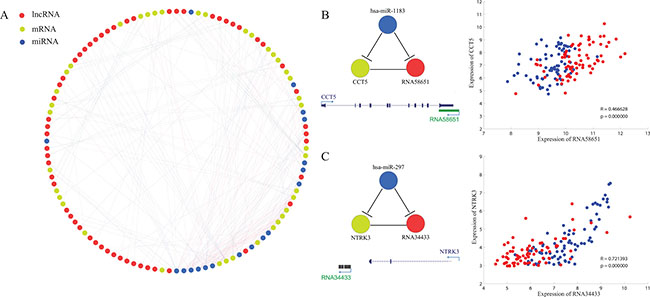
Figure 6: Prediction of lncRNAs as ceRNAs of their cis mRNAs. (A) Tri-color network consisting of lncRNAs (red), mRNAs (yellow) and miRNAs (blue). (B and C) Two examples showing the minimal ceRNA motif (In the scatter plot, red and blue representing cancer tissues and adjacent non-cancerous tissues respectively, same with that of Figure 5) (B) an antisense lncRNA with oncogenic expression profile. (C) an intergenic lncRNA with tumor suppressor-like expression profile.
Identification of tissue-specific PCGs and lncRNAs
Unsupervised hierarchical clustering of all tissues using common DE-lncRNAs apparently separated cancer tissues from adjacent non-cancerous tissues, but hardly distinguish different tissues (Supplementary Figure S4). Yet, higher degree of similarity between gastric and colon, as well as that between liver and lung, was observed in both cancer tissues and adjacent non-cancerous tissues, suggesting the indicative potential of lncRNAs for the origins and functions of different tissues. Consequently, a self-organizing map (SOM) based approach was applied in order to identify lncRNAs and mRNAs which might explain the specific characteristics of each type of gastric, colon, liver and lung tissues. lncRNAs and PCGs were first classified into hexagonal units, each of which represented a set of lncRNAs and PCGs whose expression profile are most similar to each other (Supplementary Figure S5). Then the color of each unit was assigned according to the overall expression level of lncRNAs and PCGs, brighter color representing higher expression while darker representing lower. Units with higher overall expression level in one type of tissues than others were determined to comprise lncRNAs and PCGs with tissue specificity which was subjected to later functional enrichment analysis (Figure 7A). Take gastric as an example, three units numbered 102, 103 and 104 respectively had obviously brighter color representing higher overall expression level in gastric tissues than other three types of tissues. An enrichment of digestion function was observed for the 41 PCGs, suggesting that the 35 lncRNAs in the same unit might also participate in the gastric-specific physiological process. Similarly, lncRNAs and PCGs with expression specificity in colon, liver and lung were identified respectively (see full list of tissue-specific lncRNAs and mRNAs in Supplementary File 2). qRT-PCR validation was performed on random selected tissue-specific lncRNAs and was in good agreement with microarray result (Figure 7B and Supplementary Figure S6).
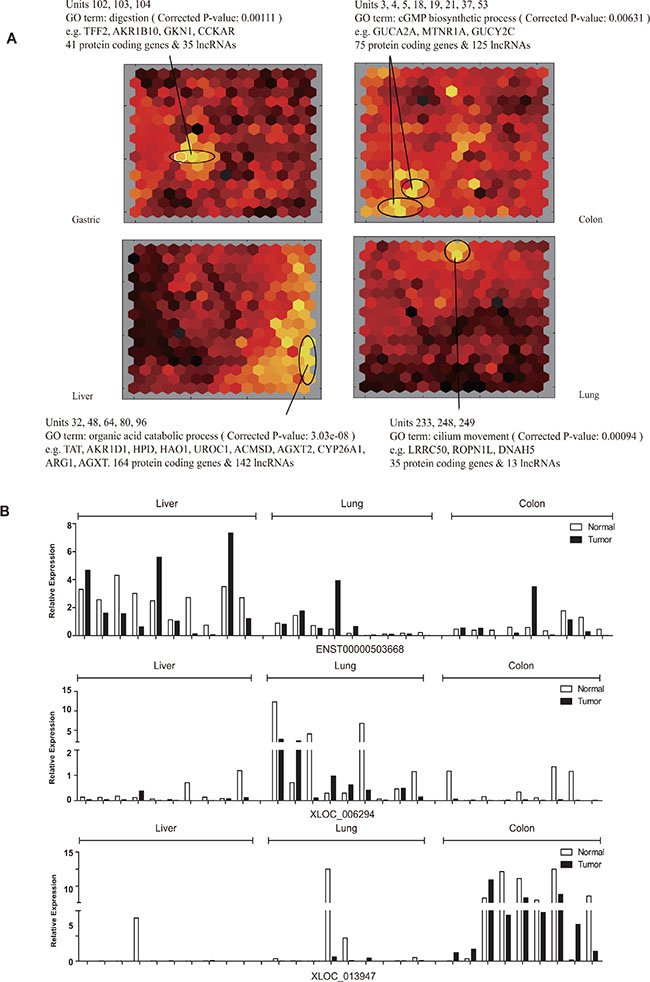
Figure 7: Transcriptome comparison by the self-organizing map (SOM). (A) Schematic illustration of expression profiles of four types of tissues depicted by SOM. Each hexagonal grid is a cluster of lncRNAs and PCGs (see also Figure S5). Grids showing significant expression specificity in each type of tissues were circled. Significantly enriched Gene Ontology (GO) terms for PCGs of circled clusters were indicated. (B) Expression of tissue-specific lncRNAs (upper, middle and lower panel representing an example of lncRNA specifically expressed in liver cancer, lung cancer and colon cancer, respectively).
DISCUSSION
Previous efforts to study the pathogenesis of diseases have been focused on PCGs. However, the accumulating discoveries of lncRNA functions in various biological processes have revealed the potential of lncRNAs acting as cancer biomarkers. The majority of biomarker lncRNAs reported by now are derived from researches based on particular type of cancer, such as SChLAP1 to be identified as a prostate cancer–associated lncRNA [50]. Considering that different types of cancer share common characteristics, we launched this study in order to identify lncRNAs with the potential to serve as common biomarkers for multiple types of cancer. By incorporating gastric, colon, liver and lung cancer tissues accompanied with paired control non-cancerous tissues into gene expression detection by a custom designed microarray platform covering 39,311 lncRNA transcripts, a total of 157 lncRNAs were identified as potential common biomarkers with expression pattern of either oncogenes or tumor suppressors. On the other hand, we also generated a list of lncRNAs and PCGs whose expression might explain the specific origins or functions of different tissues.
Despite the potential role of the identified common DE-lncRNAs as biomarkers general to different types of cancer, how the dys-regulation of their expression would prompt tumorigenesis remains a challenging problem. In this study, the association between lncRNAs and PCGs was established from the perspective of both expression correlation and genomic interaction in order to infer the possible functions of the common DE-lncRNAs. However, the mechanisms by which the lncRNAs function should still be further explored by taking advantage of experimental approaches. Both microarray and RNA-seq are high-throughput technologies for reliable assessment of transcript abundance [51]. Microarray was chosen in this study for its low cost as well as flexibility compared with RNA-seq. Meanwhile, an integrative application of high-throughput sequencing data beyond level of gene expression, such as PARS-Seq [52] which enables genome-scale reconstruction of RNA secondary structure and CLIP-Seq [53] which allows detection of massive interacting RNAs for a specific protein, will contribute to better understanding of the functions and regulatory mechanisms of lncRNAs.
CONCLUSIONS
In conclusion, we identified lncRNAs whose dys-regulated expression was shared among four types of cancer as well as lncRNAs whose expression was specifically active in specific type of cancer, suggesting potential contribution of lncRNAs to tumorigenesis and histogenesis of different tissues. In addition, due to the lack of sufficient knowledge about functions and mechanisms of the majority of lncRNAs, we inferred the possible functions of common DE-lncRNAs by establishing their association with PCGs, thus providing clues for further mechanism exploration by experimental approaches.
MATERIALS AND METHODS
Tissue samples
Paired cancer and adjacent non-cancerous tissues from 20 patients with gastric cancer, 20 patients with colon cancer, 16 patients with liver cancer and 20 patients with lung cancer were collected with informed consent from Chinese PLA General Hospital and Peking University Cancer Hospital & Institute. 30 paired samples of patients with liver, lung and colon cancer for external validation were gathered from First People’s Hospital of Foshan. All samples were collected by surgical operation and quickly stored in −80°C.
RNA extraction and reverse transcription
Frozen tissue was cut into 2−4mm3 for homogenization. Total RNA was isolated with TRIzol reagent (Invitrogen, 15596–018) according to the manufacturer’s instruction. Genomic DNA was removed using recombinant DNase I system (Ambion AM2235, Ambion), The RNA quantity was measured by NanoDrop spectrophotometer (Thermo Scientific, USA) and the integrity was assessed using agarose gel electrophoresis, the 28S/18S ratio was about equal to 2.0. All steps were performed under RNase-free conditions.
5 ug total RNA of each sample was reverse transcribed into cDNA with the SuperScript III First-strand synthesis system (Invitrogen, 18080–051) using random hexamers following the manufacture’s protocol.
The custom designed microarray platform
The custom designed microarray platform was manufactured by Agilent, consisting of probes for 21,789 PCGs and 39,311 lncRNA transcripts. LncRNA transcripts were collected from a number of different sources including NONCODE [31], H-InvDB [54], UCSC, Ensembl [55], LincRNA Catalog [56] and so on (see Supplementary File 4 for details about lncRNA collection). At least one probe was designed for each lncRNA transcript. Of all of the probes designed for lncRNA transcripts, 28,937 are specific for lncRNA transcripts and do not overlap with protein coding loci. These lncRNA transcripts with unique probes were subjected to further analysis.
Bioinformatics analysis of microarray data
The expression of lncRNAs and PCGs in obtained samples was examined using the microarray platform described above. Feature Extraction (Agilent Technologies, CA) software was used to extract all features of the data obtained from the scanned images. The lncRNA + mRNA array data were subjected for background subtraction and quality control by the GeneSpring software (Agilent). Quantile normalization was carried out on the whole set of probes for PCGs and unique probes for lncRNA transcripts for each type of tissues. Expression values were log2-scale transformed and then probes for mRNAs were collapsed down to gene level.
Hierarchical clustering was performed using cluster 3.0 [57] with complete linkage and centred Pearson correlation. The normalized and log2-scaled expression values were centred on the median before performing unsupervised hierarchical clustering. PCGs and lncRNAs were determined to be differentially expressed with two-tailed Student’s t-test p-value < 0.05 (after FDR correction) and fold change greater than 1.5 between tumor samples and adjacent control samples.
Construction of tri-color network
The construction of tri-color network consists of three steps: (i) prediction of miRNA targets of mRNAs and lncRNAs by miRanda; (ii) for each of the common DE-lncRNAs, calculate its expression profile correlation with its genomic nearest neighboring PCG; (iii) visualization of network by Cytoscape [58]. In the network, different types of RNAs were discriminated from each other by different colors (red, yellow, blue represents lncRNA, mRNA and miRNA respectively). Different color of edges represents different types of interactions. miRNA targeting of lncRNAs or mRNAs were represented by red lines while the genomic interaction and expression correlation between lncRNAs and mRNAs were represented by blue and light green respectively.
Quantitative real-time PCR
Microarray data were validated by quantitative real-time PCR (qRT-PCR). The primers for validating selected genes were designed by software primer premier 5.0 and the IDT web server (http://sg.idtdna.com/Primerquest/Home/Index). The specificity of all the primers was confirmed by UCSC BLAT tool (http://genome.ucsc.edu/cgi-bin/hgBlat). All the primers were tested in Trans-Start top Green qPCR Supermix reaction (TransGen Biotech, AQ131–03) following the manufacture’s protocol. The optimal primers were selected for quantitative validation.
The qRT-PCR assays were carried out on the Rotor-Gene Q real-time PCR cycler (Qiagen, 9001630) according to manufacturer’s instruction. For each gene, qRT-PCR reactions were performed in technical triplicate, with 18S rRNA as internal control gene for normalization. The relative expression was calculated with the 2−ΔΔCT method. The primer sequences were listed in Supplementary File 3.
Functional enrichment analysis
Functional enrichment analysis of DE-PCGs were performed using the DAVID Bioinformatics Tool [59]. Gene set enrichment analysis was performed for common DE-lncRNAs as previously described [32]. For each lncRNA, the Pearson correlation coefficients with all PCGs based on their expression in all samples were calculated as the weight subjected to GSEA. Then the degree to which a specific pathway related genes were overrepresented was calculated as the enrichment score representing the correlation between the lncRNA and the pathways. A positive/negative enrichment score with higher value means higher positive/negative correlation between the lncRNA and the pathway. The GSEA software and the gene set database were downloaded from http://www.broadinstitute.org/gsea/.
Accession numbers
The NCBI GEO accession number for the microarray data reported in this paper is GSE70880.
ACKNOWLEDGMENTS
We thank Prof. Hengmi Cui, Prof. Zhi Lu, and Dr. Guifeng Wei for helpful discussion on this project. We thank Prof. Yan Li from Institute of Biophysics, CAS, and Prof. Yuejian Wang, Jing Wu from First People’s Hospital of Foshan for providing different set of tissue samples for validation. We also thank all other members from Chen Lab for discussions about the experiments.
GRANT SUPPORT
This work was supported by grants from Chinese Academy of Science Strategic Project of Leading Science and Technology (XDA01020402) and the National High Technology Research and Development Program (“863” Program) of China (2012AA020402 and 2012AA02A202).
CONFLICTS OF INTEREST
The authors declare that there is no conflicts of interest that could be perceived as prejudicing the impartiality of the research reported.
REFERENCES
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA: a cancer journal for clinicians. 2014; 64:9–29.
2. Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K. Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000; 343:78–85.
3. Irigaray P, Newby JA, Clapp R, Hardell L, Howard V, Montagnier L, Epstein S, Belpomme D. Lifestyle-related factors and environmental agents causing cancer: an overview. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2007; 61:640–658.
4. Loeb KR, Loeb LA. Significance of multiple mutations in cancer. Carcinogenesis. 2000; 21:379–385.
5. Hahn WC, Weinberg RA. Modelling the molecular circuitry of cancer. Nat Rev Cancer. 2002; 2:331–341.
6. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MD, Miller CA, Welch JS, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013; 502:333–339.
7. Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013; 45:1127–1133.
8. Weigel MT, Dowsett M. Current and emerging biomarkers in breast cancer: prognosis and prediction. Endocr Relat Cancer. 2010; 17:R245–262.
9. Crawford ED, Ventii K, Shore ND. New biomarkers in prostate cancer. Oncology (Williston Park). 2014; 28:135–142.
10. Wyatt AW, Mo F, Wang K, McConeghy B, Brahmbhatt S, Jong L, Mitchell DM, Johnston RL, Haegert A, Li E, Liew J, Yeung J, Shrestha R, et al. Heterogeneity in the inter-tumor transcriptome of high risk prostate cancer. Genome Biol. 2014; 15:426.
11. Rutkowski MJ, Sughrue ME, Kane AJ, Mills SA, Parsa AT. Cancer and the complement cascade. Mol Cancer Res. 2010; 8:1453–1465.
12. Tripathi V, Shen Z, Chakraborty A, Giri S, Freier SM, Wu X, Zhang Y, Gorospe M, Prasanth SG, Lal A, Prasanth KV. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet. 2013; 9:e1003368.
13. Holdt LM, Hoffmann S, Sass K, Langenberger D, Scholz M, Krohn K, Finstermeier K, Stahringer A, Wilfert W, Beutner F, Gielen S, Schuler G, Gabel G, et al. Alu elements in ANRIL non-coding RNA at chromosome 9p21 modulate atherogenic cell functions through trans-regulation of gene networks. PLoS Genet. 2013; 9:e1003588.
14. Arun G, Akhade VS, Donakonda S, Rao MR. mrhl RNA, a long noncoding RNA, negatively regulates Wnt signaling through its protein partner Ddx5/p68 in mouse spermatogonial cells. Mol Cell Biol. 2012; 32:3140–3152.
15. Li R, Zhang L, Jia L, Duan Y, Li Y, Bao L, Sha N. Long non-coding RNA BANCR promotes proliferation in malignant melanoma by regulating MAPK pathway activation. PLoS One. 2014; 9:e100893.
16. Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer discovery. 2011; 1:391–407.
17. Kogo R, Shimamura T, Mimori K, Kawahara K, Imoto S, Sudo T, Tanaka F, Shibata K, Suzuki A, Komune S, Miyano S, Mori M. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011; 71:6320–6326.
18. Prensner JR, Iyer MK, Balbin OA, Dhanasekaran SM, Cao Q, Brenner JC, Laxman B, Asangani IA, Grasso CS, Kominsky HD, Cao X, Jing X, Wang X, et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol. 2011; 29:742–749.
19. Cao WJ, Wu HL, He BS, Zhang YS, Zhang ZY. Analysis of long non-coding RNA expression profiles in gastric cancer. World J Gastroenterol. 2013; 19:3658–3664.
20. Du Z, Fei T, Verhaak RG, Su Z, Zhang Y, Brown M, Chen Y, Liu XS. Integrative genomic analyses reveal clinically relevant long noncoding RNAs in human cancer. Nat Struct Mol Biol. 2013; 20:908–913.
21. Huang J, Zhou N, Watabe K, Lu Z, Wu F, Xu M, Mo YY. Long non-coding RNA UCA1 promotes breast tumor growth by suppression of p27 (Kip1). Cell Death Dis. 2014; 5:e1008.
22. Wang Y, Chen W, Yang C, Wu W, Wu S, Qin X, Li X. Long non-coding RNA UCA1a(CUDR) promotes proliferation and tumorigenesis of bladder cancer. Int J Oncol. 2012; 41:276–284.
23. Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, Topisirovic I. Targeting the translation machinery in cancer. Nature reviews Drug discovery. 2015.
24. Kang J, Sergio CM, Sutherland RL, Musgrove EA. Targeting cyclin-dependent kinase 1 (CDK1) but not CDK4/6 or CDK2 is selectively lethal to MYC-dependent human breast cancer cells. BMC Cancer. 2014; 14:32.
25. Liu P, Kao TP, Huang H. CDK1 promotes cell proliferation and survival via phosphorylation and inhibition of FOXO1 transcription factor. Oncogene. 2008; 27:4733–4744.
26. Sharifi N, Hurt EM, Kawasaki BT, Farrar WL. TGFBR3 loss and consequences in prostate cancer. Prostate. 2007; 67:301–311.
27. Guan Y, Kuo WL, Stilwell JL, Takano H, Lapuk AV, Fridlyand J, Mao JH, Yu M, Miller MA, Santos JL, Kalloger SE, Carlson JW, Ginzinger DG, et al. Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer. Clin Cancer Res. 2007; 13:5745–5755.
28. Tseng YY, Moriarity BS, Gong W, Akiyama R, Tiwari A, Kawakami H, Ronning P, Reuland B, Guenther K, Beadnell TC, Essig J, Otto GM, O'Sullivan MG, et al. PVT1 dependence in cancer with MYC copy-number increase. Nature. 2014; 512:82–86.
29. Yao J, Zhou B, Zhang J, Geng P, Liu K, Zhu Y, Zhu W. A new tumor suppressor LncRNA ADAMTS9-AS2 is regulated by DNMT1 and inhibits migration of glioma cells. Tumour Biol. 2014; 35:7935–7944.
30. Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, Lagarde J, Veeravalli L, Ruan X, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome research. 2012; 22:1775–1789.
31. Xie C, Yuan J, Li H, Li M, Zhao G, Bu D, Zhu W, Wu W, Chen R, Zhao Y. NONCODEv4: exploring the world of long non-coding RNA genes. Nucleic Acids Res. 2014; 42:D98–103.
32. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005; 102:15545–15550.
33. Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003; 34:267–273.
34. Ebisuya M, Yamamoto T, Nakajima M, Nishida E. Ripples from neighbouring transcription. Nat Cell Biol. 2008; 10:1106–1113.
35. Hung T, Wang Y, Lin MF, Koegel AK, Kotake Y, Grant GD, Horlings HM, Shah N, Umbricht C, Wang P, Kong B, Langerod A, Borresen-Dale AL, et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet. 2011; 43:621–629.
36. Ponjavic J, Oliver PL, Lunter G, Ponting CP. Genomic and transcriptional co-localization of protein-coding and long non-coding RNA pairs in the developing brain. PLoS genetics. 2009; 5:e1000617.
37. Mancini-Dinardo D, Steele SJ, Levorse JM, Ingram RS, Tilghman SM. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev. 2006; 20:1268–1282.
38. Fitzpatrick GV, Soloway PD, Higgins MJ. Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat Genet. 2002; 32:426–431.
39. Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, Tramontano A, Bozzoni I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011; 147:358–369.
40. Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010; 465:1033–1038.
41. Wang J, Liu X, Wu H, Ni P, Gu Z, Qiao Y, Chen N, Sun F, Fan Q. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic acids research. 2010; 38:5366–5383.
42. Franco-Zorrilla JM, Valli A, Todesco M, Mateos I, Puga MI, Rubio-Somoza I, Leyva A, Weigel D, Garcia JA, Paz-Ares J. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat Genet. 2007; 39:1033–1037.
43. John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004; 2:e363.
44. Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003; 5:R1.
45. Luo Y, Kaz AM, Kanngurn S, Welsch P, Morris SM, Wang J, Lutterbaugh JD, Markowitz SD, Grady WM. NTRK3 is a potential tumor suppressor gene commonly inactivated by epigenetic mechanisms in colorectal cancer. PLoS Genet. 2013; 9:e1003552.
46. Krzeszinski JY, Wei W, Huynh H, Jin Z, Wang X, Chang TC, Xie XJ, He L, Mangala LS, Lopez-Berestein G, Sood AK, Mendell JT, Wan Y. miR-34a blocks osteoporosis and bone metastasis by inhibiting osteoclastogenesis and Tgif2. Nature. 2014; 512:431–435.
47. Lai M, Du G, Shi R, Yao J, Yang G, Wei Y, Zhang D, Xu Z, Zhang R, Li Y, Li Z, Wang L. miR-34a inhibits migration and invasion by regulating the SIRT1/p53 pathway in human SW480 cells. Molecular medicine reports. 2015; 11:3301–3307.
48. Thor T, Kunkele A, Pajtler KW, Wefers AK, Stephan H, Mestdagh P, Heukamp L, Hartmann W, Vandesompele J, Sadowski N, Becker L, Garrett L, Holter SM, et al. MiR- 34a deficiency accelerates medulloblastoma formation in vivo. Int J Cancer. 2015; 136:2293–2303.
49. McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010; 28:495–501.
50. Prensner JR, Iyer MK, Sahu A, Asangani IA, Cao Q, Patel L, Vergara IA, Davicioni E, Erho N, Ghadessi M, Jenkins RB, Triche TJ, Malik R, et al. The long noncoding RNA SChLAP1 promotes aggressive prostate cancer and antagonizes the SWI/SNF complex. Nat Genet. 2013; 45:1392–1398.
51. Mantione KJ, Kream RM, Kuzelova H, Ptacek R, Raboch J, Samuel JM, Stefano GB. Comparing bioinformatic gene expression profiling methods: microarray and RNA-Seq. Medical science monitor basic research. 2014; 20:138–142.
52. Kertesz M, Wan Y, Mazor E, Rinn JL, Nutter RC, Chang HY, Segal E. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010; 467:103–107.
53. Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang X, Darnell JC, Darnell RB. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008; 456:464–469.
54. Yamasaki C, Murakami K, Takeda J, Sato Y, Noda A, Sakate R, Habara T, Nakaoka H, Todokoro F, Matsuya A, Imanishi T, Gojobori T. H-InvDB in 2009: extended database and data mining resources for human genes and transcripts. Nucleic Acids Res. 2010; 38:D626–632.
55. Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, Gil L, Gordon L, Hendrix M, et al. Ensembl 2012. Nucleic Acids Res. 2012; 40:D84–90.
56. Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes & development. 2011; 25:1915–1927.
57. Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998; 95:14863–14868.
58. Saito R, Smoot ME, Ono K, Ruscheinski J, Wang PL, Lotia S, Pico AR, Bader GD, Ideker T. A travel guide to Cytoscape plugins. Nature methods. 2012; 9:1069–1076.
59. Huang da W, Sherman BT and Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009; 37:1–13.