
Introduction
Gastric (stomach) cancer (GC) is the fifth most commonly diagnosed cancer (952,000 new cases diagnosed in 2012) and the third leading cause of cancer-related deaths in both sexes worldwide [1]. In the United States, approximately 10,720 people died of GC (6,500 men and 4,220 women) in 2015 [2]. The majority of GC cases (70%) occur in developing countries, and half all cases occur in eastern Asia, mainly in Korea, Mongolia, Japan, and China [1].
Despite ongoing efforts to develop effective treatments, the 5-year survival rate of GC patients is only 29% [2]. As a heterogeneous disease, GC has complex molecular mechanisms for uncontrolled cell growth, which could be caused by promoter methylation [3], deregulated gene expression [4], competing endogenous long non-coding RNAs [5, 6], and/or copy number alteration of tumor-suppressor genes and oncogenes [7]. The majority of GC studies to date have not focused beyond the gene level; thus, they fail to provide the whole picture of tumorigenesis. In this study, we aimed to develop the first literature-based genetic resource with extensive annotations, GCGene. This data resource can also be used to prioritize genes by their GC-associated importance relevance and to identify both the common and unique cellular events at different oncogenic stages.
Results and Discussion
To survey the genetic information related to all GC types, we performed extensive data integration and literature curation. Ultimately, we identified 1,815 non-redundant GC-associated genes for inclusion in this database, and we conducted functional annotation and gene prioritization of these genes (Table S1).
Database construction and content
Web interface
Based on the systematic survey of GC-associated genes in publically available databases and literature, we developed a user-friendly web interface to make this annotated information freely available to all researchers. The database is supported by a web browser that allows researchers to explore all the GC-associated genes using chromosome and coloured KEGG pathway maps (Figure 1). GCGene allows users to conduct quick queries by GeneID or gene symbol and to run BLAST searches against all human sequences. For advanced integrative study, a list of all genes curated in this database is available for download.
Information on the GCGene gene page
To organize information for each gene, we classified our annotation details into seven groups: general information, literature, gene expression, regulation, genetic mutation, homologous gene, and protein-protein interaction. When exploring the annotations, users can click the labels at the top of each web page to reach any specific annotation quickly (Figure 1). On the general information page for each gene, the official gene symbol, alias, biological function, involved biological phenotype, and nucleotide/protein sequences are listed. The cross-references to other public bioinformatics databases such as TSGene [8, 9], NCBI Entrez Gene [10], dbEMT [11], iHOP [12], and MSGene [13] are included. All available literature associated with a gene is highlighted with cancer keywords. A comprehensive gene-expression profile of normal and tumor samples plotted from BioGPS is provided [14]. On the regulation page, the transcription factor, post-translational modification, and methylation are annotated. Homologous sequences from other model species have also been integrated from the NCBI HomoloGene database [15]. On the mutation page, the genetic polymorphisms from the most updated COSMIC database (version 73) have been collected [16]. Finally, the protein-protein interaction data from the PathwayCommons database (version 6) have been grouped into physical interactions, signaling interactions, and metabolic interactions [17].
Browsing the classified genes in GCGene
GCGene supports a variety of ways to browse putative GC genes, including highlighted KEGG maps and chromosome distribution maps. The genomic distribution of all the genes has been plotted on 24 chromosomes with individual charts (Figure 1). Users can browse each chromosome to access all of the GC-associated genes in the region. The number of literature citations for each gene is provided, indicating its relative importance in GC development. To provide access to this information, we have included a browsing function that identifies different gene sets based on the number of literature citations.
Keyword-based search in GCGene
A search function at the top right corner of each web page can be used to conduct quick queries using human gene official symbols or Entrez Gene IDs. Advanced searches can be conducted by typing the gene name or its functional characteristics, including chromosome location, interaction partner, biological process, or disease (Figure 1). In addition, users can search all the curated literature by key words, which is useful in identifying candidate genes for specific biological processes.
Sequence-based search in GCGene
With the BLAST interface, users can evaluate gene sequence similarity by inputting the sequence of interest. The sequence alignment option can be modified with an E-value and an identity score. This database also facilitates bulk downloads of all nucleotide and protein sequences in a FASTA format for advanced local-sequence-based BLAST search (Figure 1).
Gene ranking for all the genes in GCGene
Small-scale studies of GC often focus on verifying specific functions of cancer genes under a certain genetic background or other phenotype characteristic. Because hundreds of genes are associated with GC, it is necessary to systematically prioritize the most informative genes and to systematically construct a large-scale gene map for GC. Using the ToppGene gene-ranking tool [18], we prioritized the relative importance of all 1,815 genes in GCGene. To build a ranking model using ToppGene, we defined a training set with the 18 most commonly studied genes with ≥30 literature citations: CDH1, CXCL8, ERBB2, GSTM1, GSTP1, GSTT1, HIF1A, IL10, IL1B, IL1RN, MTHFR, PTGS2, RUNX3, TNF, TP53, TYMS, VEGFA, and XRCC1. ToppGene utilizes integrated biological annotation data to extract biological features from the training set to rank the remaining genes. Those biological annotations include protein domain, gene ontology evidence, pathway annotations, gene coexpression, sequence features, and other data mined from the literature. Finally, ToppGene was used to combine all the rankings into a global ranking for all the candidate GC genes using order statistics (Table S2). Not surprisingly, those 100 top-ranked genes are enriched in the cancer pathways identified with terms such as “regulation of cell proliferation,” “pathways in cancer,” “PI3K-Akt signaling pathway,” and “proteoglycans in cancer” (Table S3).
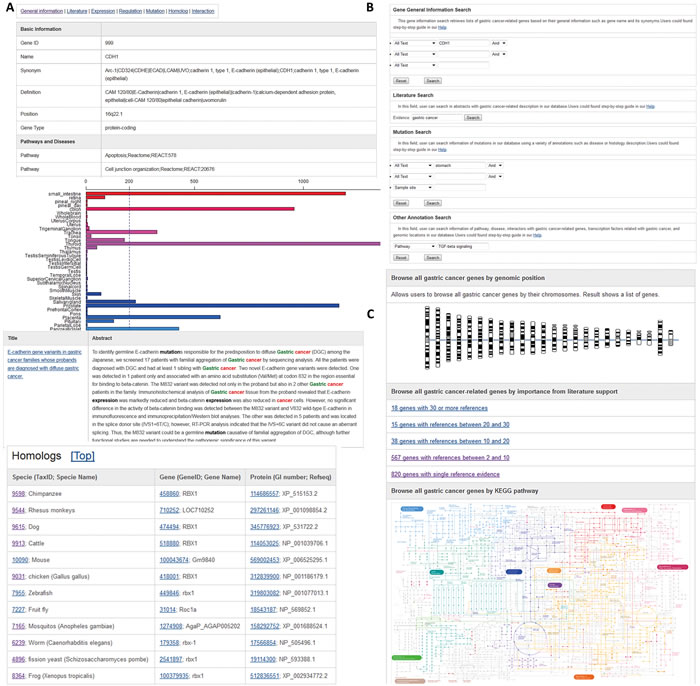
Figure 1: Web interface of GCGene. A. The basic information in each GC-associated gene page; B. Query interface for text search; C. Browsing the genes in GCGene using chromosomes, literature supports, and KEGG pathways.
Mutational patterns of the most relevant genes in GCGene
We systematically examined the 100 top-ranked genes in GCGene according to their somatic mutational patterns in multiple cancers using cBio portal [19]. These patterns are useful for the identification of highly mutated genes in other cancer types for further screening. As shown in Figure 2, a wide variety of genetic alterations (i.e., mutations, deletions, amplifications, and multiple alterations) in the 100 top-ranked genes occur in ≥80% of cases among 12 cancer cohorts. Notably, the majority of genes associated with these 12 cancers have multiple genetic alterations; therefore, some genes have both a single-nucleotide variation and a copy-number variation.
It is not surprising that multiple genetic mutations occur most frequently in cancers with higher alteration frequencies. With unique single-nucleotide mutations, deletion and amplification are more common in cancers with lower alteration frequencies. In our analysis, lung squamous-cell carcinoma had the highest overall gene alteration rate. Of the 176 cases studied, 98.9% showed genetic mutations of the genes; nearly 70% presented multiple alterations. TCGA ovarian serous cystadenocarcinoma and head and neck cancer had very similar rates of alteration. TCGA gastric adenocarcinoma had the fourth highest rate of mutation of these genes, 92.3%. The other two gastric adenocarcinomas have similar mutation frequencies in terms of single-nucleotide mutations. According to the datasets from Pfizer and UHK, stomach adenocarcinoma had 79% and 72.7% mutational frequency in 79 cases and 16 cases, respectively. This relatively lower rate of penetration might be explained by the small sample size. However, these three GC datasets show similar rates of single-nucleotide mutation.
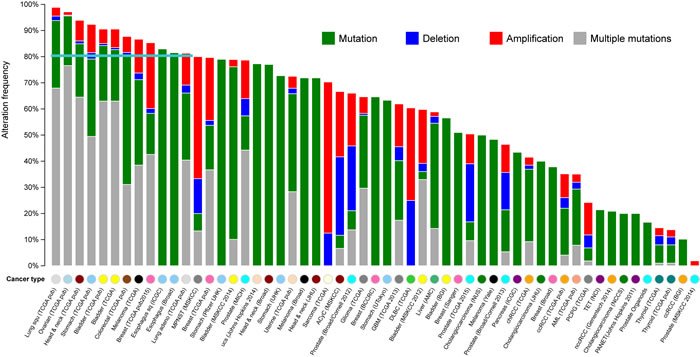
Figure 2: The mutational landscape for the 100 top-ranked GC-associated genes in multiple cancers. The X axis represents the cancer types in TCGA, the Y axis represents the alteration frequencies of top 100 genes that correspond to the cancers on the X axis. The different colors indicate different cancer types based on their original tissues.
Networking the top-ranked genes to identify the hub genes associated with patient survival time
Recent advances in high-throughput technologies have dramatically increased the availability of protein-protein interaction (PPI) data and have stimulated the modelling of pathways to improve our understanding specific cellular events at the systems level. To avoid the high level of noise, sparseness, and highly skewed degree distribution of PPI networks, we utilized only reliable human PPIs summarized in a few popular biological pathway resources such as the KEGG and Reactome databases [20].
Using a search module [21], we extracted a subnetwork from all the human pathway-based interactomes. The reconstructed GC interactome contains 76 genes and 152 gene-gene interactions based on current evidence from known biological pathways (Figure 3A). Of the 76 nodes, 65 are among the 100 top-ranked GC-associated genes. The remaining 11 are genes that may potentially bridge the top-ranked GC-associated gene to fully implement its cellular function. The majority of GC genes are linked to each other in a highly modular structure. This finding not only supports the accuracy of our data but also shows that the GC genes are highly interconnected and form a high-density cellular modulus.
Further network topological analysis also revealed that most molecules in our map are closely connected. The degrees of connection of all nodes in our reconstructed GC map follow a power law distribution P(k)~k-b, where P(k) is the probability that a molecule has connections with other k molecules and b is an exponent with an estimated value of 1.207 (Figure 3B). Thus, our GC map is different from all the human PPI networks in which most nodes are sparsely connected, with an average b value of 2.9 [22]. We developed this feature to map the distribution of the shortest pathways throughout the entire network. This map revealed smaller pathway degrees (2 to 4), which means that majority of the node connections can be reached in only three steps on average (Figure 3C).
With dense interactions, the highly connected nodes in this network may have prominent roles as common connections that mediate information transduction in the short pathways. In total, we identified 9 genes with at least 10 connections: SRC (17), TP53 (15), MYC (14), JAK2 (12), STAT3 (11), FOS (11), RHOA (10), MYB (10), and ITGB1 (10). Interestingly, SRC is the most connected node. Notably, only MYB has not been reported to be involved in GC in these 9 genes.
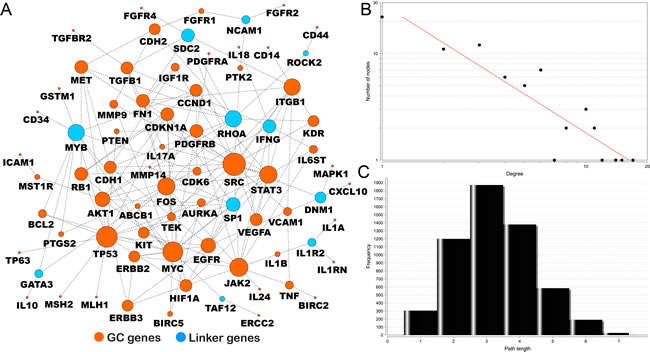
Figure 3: GC interactome using pathway-based protein-protein interaction data. A. The 65 genes in orange are the 100 top-ranked genes in our GCGene; the remaining 11 genes in blue are linker genes that connect the 65 genes. The size of the node represents the number of connections in the network; B. the degree distribution; and C. the short path length frequency.
We performed a survival analysis based on published TCGA mutational data using the cBio portal. Patients with genetic mutations in any of the 9 genes are significantly correlated with overall survival (Figure 4A). Further survival analyses using gene expression data also confirmed the importance of the 9 genes [23]. We found that lower expression of 8 genes (upper quartile vs. remaining samples) except STAT3 is significantly correlated with longer relapse-free survival (P ≤ 0.05) (Figure 4B-4D, Figures S1-S6). In particular, the MYB is mutated in 2% of the TCGA GC cohort, and it is also associated with survival (Figure 4D). Taken together, these results highlight the potential role of MYB in GC progression. In summary, our reconstructed map not only reveals multiple hubs related to survival but also provides a broader context for the previously unconnected GC genes.
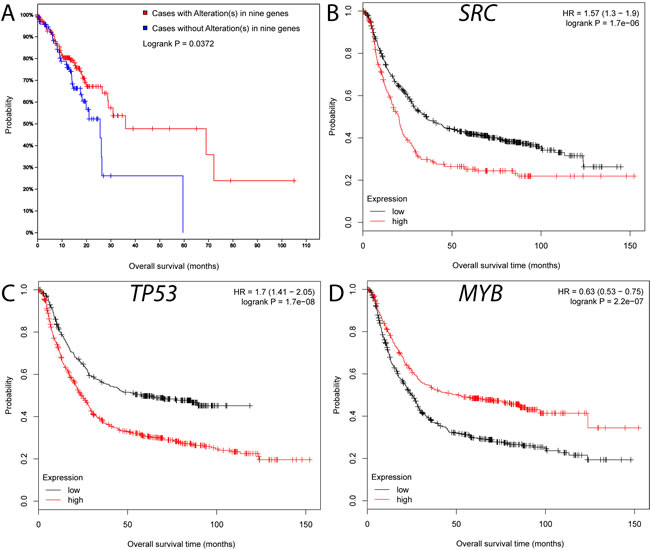
Figure 4: Survival analyses for the hub genes in the GC interactome. A. Survival characteristics of the nine hub genes on the genetic mutation using cBio data portal [19]. B.-D. Survival characteristics of SRC (B), TP53 (C) and MYB (D) based on the integrated GC gene expression using a KM plot [23].
Conclusions
In conclusion, our systematic curation of genetic information related to GC yielded 1,815 putative human genes (1,678 protein-coding and 137 non-coding genes) for inclusion in the GCGene database. A user-friendly web interface was developed to provide access to all the genes, with additional gene annotation and literature information. As the first literature-based gene database for GC, GCGene provides a novel resource for researchers performing high-throughput genetic and clinical tests to identify GC-associated genetic variants.
To facilitate subsequent literature updates, we constructed an automatic literature search scheme using the “My NCBI” tool, which will return the relevant literature every two weeks. We will use the document similarity clustering method in Entrez to group the newly available articles to assist with literature curation. Additionally, to keep pace with the rapid growth of cancer genome data, we have built an automated system capable of importing functional information from various public data sources, which will enable us to integrate more annotations quickly. Once the data is updated, the web page will be updated accordingly on an annual basis.
MATERIALS AND METHODS
Data integration from existing bioinformatics recourses
The gene collection related to GC was mainly based on 10 data sources: the OMIM (Online Mendelian Inheritance in Man, download on January 25, 2015) [24], GAD (The Genetic Association database, latest version updated on August 18, 2014) [25], gene manually curation from GeneRIF (Download on January 25, 2015) [26], genome-wide association studies from GWASCatalog (Download on January 25, 2015) [27], and 6 candidate gene lists produced by a large-scale genome-wide methylation and genetic mutation study on GC [28] (Figure 5A). As the most authoritative compendium of human disease-associated genes, OMIM does not include many genes. We obtained only 4 genes (IL1B, IL1RN, KRAS, and CDH1) associated with hereditary diffuse GC from OMIM. The GAD database is an archive of published human genetic association studies that contains curated information on candidate genes. In total, we collected 279 unique human genes from GAD from 637 published studies. In addition, 11 candidate genes were downloaded from 3 genome-wide association studies in the GWASCatalog database. In 2014, a whole-genome sequencing and comprehensive molecular profiling of GC identified numerous new driver mutations, including recently mutated genes of the microsatellite instability type (91 genes) and microsatellite-stable type (53 genes), Sanger sequencing was used to validate driver mutations (18 genes), genes within driver copy number variation regions (102 genes), as well as genes in hypermethylated (91 genes) and hypomethylated (92 genes) regions. We combined these publically available resources and harvested a list of 590 nonredundant human genes.
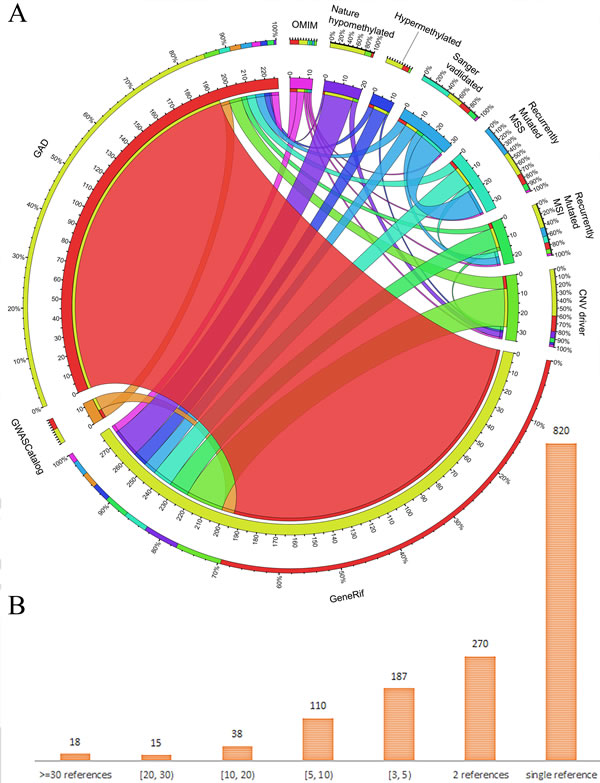
Figure 5: Overlapping of genes from different data sources and statistics. A. Shared GC-associated genes across multiple data sources. The length of circularly arranged segments is proportional to the total genes in each regenerative process group. The ribbons connecting different segments represent the number of shared genes between regenerative process groups. The outer ring has stacked bar plots that represent the relative contribution of other regenerative process group to the regenerative process group totals. Ribbons connecting different segments represent the number of shared genes between regenerative tissues; B. statistics for number of genes with various pieces of literature evidence.
Literature collection and gene curation
To assemble a detailed and precise GC gene resource with literature evidence, we performed an extensive literature query of GeneRIF database on January 10, 2015, using Perl regular expression to identify sentences with both gastric and cancer keywords: [(gastric OR stomach) AND (cancer OR tumor OR carcinoma)]. In total, we retrieved 2,904 PubMed abstracts. GeneRIF (Gene Reference Into Function) is a collection of short descriptions of gene functions in the Entrez Gene database [29]. However, GeneRIF records do not provide full abstracts for further curation. Thus, we downloaded all 2,904 PubMed abstracts in Medline format for manual review.
The curation of GC genes from literature in this study was conducted in three major steps: (1) grouping all 2,904 retrieved abstracts based on their semantic similarity using the “Related Articles” function in Entrez; (2) extracting contents related to GC from grouped abstracts; and (3) manually collecting gene names from the descriptions of the text and mapping the gene names to Entrez gene IDs. These curation steps allowed us to quickly and easily cross-check whether and how the curated abstract was related to GC. To provide a unified functional annotation, we used Entrez gene IDs as the key in all the tables of our GCGene database to cross-link the same genes from different public bioinformatics databases. To ensure the accuracy of our literature evidence, we collected the species information and the gene alias and manually mapped them to the official HUGO gene symbol. For example, in the sentence “Results suggest that the COX-2/microsomal prostaglandin E synthase-1 pathway contributes to the Helicobacter-associated gastric tumorigenesis,” [30] the gene COX-2 was one of the synonyms for the murine gene Ptgs2 in the Entrez gene database. After careful manual cross-checking, we mapped all the curated genes to their corresponding human homologous groups using the NCBI HomoloGene database using the same method we implemented in a previous analysis [9, 31, 32]. In total, we identified 1,369 human homologous genes using Entrez. By integrating 590 genes from other public databases, we consolidated 1,815 human genes, 1,678 protein-coding and 137 non-coding genes (Table S1). The overlapping relationship among different data sources revealed that ~70% of genes from our literature content curation are also recorded in the GAD database (Figure 5A). These comparisons validated the high quality of our literature curation as well as multiple items of supporting evidence. Based on the curated references, we identified 18 genes with ≥30 supporting references (Figure 5B). The majority of the genes from literature curation (820 of 1,369 GC-associated genes; 59.90%) had only a single literature reference (Figure 5B), which may indicate the need for further experimental validation for these 820 candidate genes.
Biological functional annotations
Information regarding comprehensive biological functional was retrieved from public resources for the annotation of the 1,815 human GC-associated genes in our database. The basic gene information and sequences were collected for each gene from the following databases: NCBI Entrez gene (downloaded on February 28, 2015) [33], UniProt (released February 2015) [34], Ensembl (version 78) [35], and Gene Ontology (downloaded on February 28, 2015) [36]. From BioGPS (downloaded on February 28, 2015) [14], the mRNA expression profiling data from both normal and tumor tissues were acquired from BioGPS (downloaded on February 28, 2015) [14]. The comprehensive pathway-associated information was annotated for GC-associated genes from the following databases: the transporter substrate database (version 1.0) [37], BioCyc (downloaded on February 28, 2015) [38], KEGG Pathway (downloaded on February 28, 2015) [39], the rate-limiting enzyme database (version 1.0) [40], PANTHER (downloaded on February 28, 2015) [41], PID Curated (downloaded on February 28, 2015) [42], the pathway localization database (version 1.0) [43], and Reactome (downloaded on February 28, 2015) [44, 45]. Disease information was imported from GAD (gene association database), KEGG Disease, Fundo (downloaded on February 28, 2015) [46, 47], NHGIR (downloaded on February 28, 2015) [48], and OMIM [33].
The original published GC-associated articles in PubMed were hyperlinked to their respective genes. Using the Perl Script and Swiss knife modules, functional information was integrated from Gene annotations [49], Gene Ontology annotations [36], HPRD/BIND/BioGRID interaction annotations, KEGG LIGAND/BioCarta (downloaded on February 28, 2015) signaling event annotations [50, 51], and OMIM annotations.
Gene set enrichment analysis
The functional enrichment analysis of disease, pathways, and other functional annotations for each gene was accomplished using ToppFun [18]. In these analyses, the encoding genes of all human proteins were used as background, and the statistical significance of enriched annotations was calculated using the hypergeometric model. Based on the Benjamini-Hochberg multiple correction method in ToppFun, the corrected P-values for enriched annotations were calculated. Finally, the enriched annotations with corrected P-values < 0.01 were identified as over-representative annotations for each gene set. The resulting enriched gene ontology terms were further summarized and visualized by the REVIGO online server [52].
Gene ranking using ToppGene and cancer mutation landscape
Using the ToppGene gene ranking tool [18], we prioritized the relative importance of each of the 1,815 GC-associated genes. ToppGene integrates the following biological annotation data to rank the input genes: protein domain, gene ontology evidence, pathway annotations, gene co-expression, sequence features, and data mined from the literature. First, ToppGene requires a training set, which includes most commonly studied genes associated with the biological processes of interest. In the present study, the training set consisted of 18 genes, each with ≥30 literature citations. This training set was used to extract features shared by all GC-associated genes.
Next, based on the extracted biological features from the training set, ToppGene builds a ranking model. The ranking model that contains multiple dimensional data is then used to prioritize the remaining 1,797 genes. Finally, the ToppGene ranking model combines all of the rankings into a global ranking for the 1,815 GC-associated genes using order statistics (Table S2). In the present study, the 100 top-ranked GC-associated genes were then input into the cBio portal to obtain a mutation pattern across multiple cancers.
Construction of protein-protein interactome for the 100 top-ranked GC-associated genes
To study the potential biological mechanisms related to GC-associated genes, we extracted protein-protein interactions between the 100 top-ranked GC-associated genes and other human genes. To accomplish this task, we first collected a list of non-redundant pathway-based human interactomes from the PathwayCommons database, which includes several biological pathway databases such as KEGG and Reactome. We then extracted a subnetwork containing the 100 top-ranked GC-associated genes from the human interactome using an approach similar to the one implemented in our previous study [21]. All of the input genes were mapped into the human interactome using the proposed algorithm, and the subnetwork was extracted according to the shortest pathways between the input genes and other genes.
If the function of genes is systematically studied from the point of view of the network, the complexity and interconnectedness of the biological network is revealed. In general, biological networks tend to follow some simple rules, and the topological properties of the networks may be closely related to their function [53]. Therefore, we used the NetworkAnalyzer plug-in in Cytoscape 2.8 [54] (Figure 3B and 3C) to analyze the extracted subnetworks of GC-associated genes. We used degree to represent the sum of the number of connections for each node in a network [53], and the shortest path represented by the least number of steps from one node to another [53]. Cytoscape 2.8 was used to visualize the network.
Abbreviations
TCGA: The Cancer Genome Atlas; GEO: Gene Expression Omnibus; GCGene: Gastric Cancer Gene database.
Grant Support
This work was supported by the National Natural Science Foundation of China (No. 31171270) and a research start-up fellowship from the University of the Sunshine Coast to M.Z.
Conflicts of Interest
The authors declare that they have no competing interests.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015; 136:E359-386.
2. Siegel RL, Miller KD and Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015; 65:5-29.
3. Qu Y, Dang S and Hou P. Gene methylation in gastric cancer. Clin Chim Acta. 2013; 424:53-65.
4. Zhang Z, Li Z, Li Y and Zang A. MicroRNA and signaling pathways in gastric cancer. Cancer Gene Ther. 2014; 21:305-316.
5. Fang XY, Pan HF, Leng RX and Ye DQ. Long noncoding RNAs: novel insights into gastric cancer. Cancer Lett. 2015; 356:357-366.
6. Xia T, Liao Q, Jiang X, Shao Y, Xiao B, Xi Y and Guo J. Long noncoding RNA associated-competing endogenous RNAs in gastric cancer. Sci Rep. 2014; 4:6088.
7. Tsukamoto Y, Uchida T, Karnan S, Noguchi T, Nguyen LT, Tanigawa M, Takeuchi I, Matsuura K, Hijiya N, Nakada C, Kishida T, Kawahara K, Ito H, Murakami K, Fujioka T, Seto M, et al. Genome-wide analysis of DNA copy number alterations and gene expression in gastric cancer. J Pathol. 2008; 216:471-482.
8. Zhao M, Kim P, Mitra R, Zhao J and Zhao Z. TSGene 2.0: an updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res. 2015.
9. Zhao M, Sun J and Zhao Z. TSGene: a web resource for tumor suppressor genes. Nucleic Acids Res. 2013; 41:D970-976.
10. Maglott D, Ostell J, Pruitt KD and Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2011; 39:D52-57.
11. Zhao M, Kong L, Liu Y and Qu H. dbEMT: an epithelial-mesenchymal transition associated gene resource. Sci Rep. 2015; 5:11459.
12. Hoffmann R. Using the iHOP information resource to mine the biomedical literature on genes, proteins, and chemical compounds. Curr Protoc Bioinformatics. 2007; Chapter 1:Unit1 16.
13. Zhao M, Li Z and Qu H. An evidence-based knowledgebase of metastasis suppressors to identify key pathways relevant to cancer metastasis. Sci Rep. 2015; 5:15478.
14. Wu C, Jin X, Tsueng G, Afrasiabi C and Su AI. BioGPS: building your own mash-up of gene annotations and expression profiles. Nucleic Acids Res. 2015.
15. Coordinators NR. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2015; 43:D6-17.
16. Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, Teague JW, Stratton MR, McDermott U, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015; 43:D805-811.
17. Blinov ML, Schaff JC, Ruebenacker O, Wei X, Vasilescu D, Gao F, Morgan F, Ye L, Lakshminarayana A, Moraru, II and Loew LM. Pathway Commons at virtual cell: use of pathway data for mathematical modeling. Bioinformatics. 2014; 30:292-294.
18. Aerts S, Lambrechts D, Maity S, Van Loo P, Coessens B, De Smet F, Tranchevent LC, De Moor B, Marynen P, Hassan B, Carmeliet P and Moreau Y. Gene prioritization through genomic data fusion. Nat Biotechnol. 2006; 24:537-544.
19. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C and Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013; 6:pl1.
20. Cerami EG, Gross BE, Demir E, Rodchenkov I, Babur O, Anwar N, Schultz N, Bader GD and Sander C. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2011; 39:D685-690.
21. Zhao M, Li X and Qu H. EDdb: a web resource for eating disorder and its application to identify an extended adipocytokine signaling pathway related to eating disorder. Sci China Life Sci. 2013; 56:1086-1096.
22. Jin Y, Turaev D, Weinmaier T, Rattei T and Makse HA. The evolutionary dynamics of protein-protein interaction networks inferred from the reconstruction of ancient networks. PloS one. 2013; 8:e58134.
23. Gyorffy B, Surowiak P, Budczies J and Lanczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One. 2013; 8:e82241.
24. Amberger J, Bocchini CA, Scott AF and Hamosh A. McKusick’s Online Mendelian Inheritance in Man (OMIM). Nucleic Acids Res. 2009; 37:D793-D796.
25. Becker KG, Barnes KC, Bright TJ and Wang SA. The genetic association database. Nat Genet. 2004; 36:431-432.
26. Aronson AR, Mork JG, Gay CW, Humphrey SM and Rogers WJ. The NLM Indexing Initiative’s Medical Text Indexer. Stud Health Technol Inform. 2004; 107:268-272.
27. Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L and Parkinson H. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014; 42:D1001-1006.
28. Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST, Siu HC, Deng S, Chu KM, Law S, Chan KH, Chan AS, Tsui WY, Ho SL, Chan AK, Man JL, et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet. 2014; 46:573-582.
29. Lu Z, Cohen KB and Hunter L. GeneRIF quality assurance as summary revision. Pac Symp Biocomput. 2007:269-280.
30. Oshima H, Oshima M, Inaba K and Taketo MM. Hyperplastic gastric tumors induced by activated macrophages in COX-2/mPGES-1 transgenic mice. EMBO J. 2004; 23:1669-1678.
31. Brown GR, Hem V, Katz KS, Ovetsky M, Wallin C, Ermolaeva O, Tolstoy I, Tatusova T, Pruitt KD, Maglott DR and Murphy TD. Gene: a gene-centered information resource at NCBI. Nucleic Acids Res. 2015; 43:D36-42.
32. Kong L, Cheng L, Fan LY, Zhao M and Qu H. IQdb: an intelligence quotient score-associated gene resource for human intelligence. Database (Oxford). 2013; 2013:bat063.
33. Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Federhen S, Feolo M, Fingerman IM, Geer LY, Helmberg W, Kapustin Y, Landsman D, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2011; 39:D38-51.
34. Magrane M and Consortium U. UniProt Knowledgebase: a hub of integrated protein data. Database (Oxford). 2011; 2011:bar009.
35. Flicek P, Amode MR, Barrell D, Beal K, Brent S, Chen Y, Clapham P, Coates G, Fairley S, Fitzgerald S, Gordon L, Hendrix M, Hourlier T, Johnson N, Kahari A, Keefe D, et al. Ensembl 2011. Nucleic Acids Res. 2011; 39:D800-806.
36. Gene Ontology Consortium. The Gene Ontology in 2010: extensions and refinements. Nucleic Acids Res. 2010; 38:D331-335.
37. Zhao M, Chen Y, Qu D and Qu H. TSdb: a database of transporter substrates linking metabolic pathways and transporter systems on a genome scale via their shared substrates. Sci China Life Sci. 2011; 54:60-64.
38. Karp PD, Ouzounis CA, Moore-Kochlacs C, Goldovsky L, Kaipa P, Ahren D, Tsoka S, Darzentas N, Kunin V and Lopez-Bigas N. Expansion of the BioCyc collection of pathway/genome databases to 160 genomes. Nucleic Acids Res. 2005; 33:6083-6089.
39. Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T and Yamanishi Y. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008; 36:D480-484.
40. Zhao M, Chen X, Gao G, Tao L and Wei L. RLEdb: a database of rate-limiting enzymes and their regulation in human, rat, mouse, yeast and E. coli. Cell Res. 2009; 19:793-795.
41. Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A and Narechania A. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003; 13:2129-2141.
42. Schaefer CF, Anthony K, Krupa S, Buchoff J, Day M, Hannay T and Buetow KH. PID: the Pathway Interaction Database. Nucleic Acids Res. 2009; 37:D674-679.
43. Zhao M and Qu H. PathLocdb: a comprehensive database for the subcellular localization of metabolic pathways and its application to multiple localization analysis. BMC Genomics. 2010; 11 Suppl 4:S13.
44. Croft D, O’Kelly G, Wu G, Haw R, Gillespie M, Matthews L, Caudy M, Garapati P, Gopinath G, Jassal B, Jupe S, Kalatskaya I, Mahajan S, May B, Ndegwa N, Schmidt E, et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 2011; 39:D691-697.
45. Matthews L, Gopinath G, Gillespie M, Caudy M, Croft D, de Bono B, Garapati P, Hemish J, Hermjakob H, Jassal B, Kanapin A, Lewis S, Mahajan S, May B, Schmidt E, Vastrik I, et al. Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res. 2009; 37:D619-622.
46. Osborne JD, Flatow J, Holko M, Lin SM, Kibbe WA, Zhu LJ, Danila MI, Feng G and Chisholm RL. Annotating the human genome with Disease Ontology. BMC Genomics. 2009; 10 Suppl 1:S6.
47. Du P, Feng G, Flatow J, Song J, Holko M, Kibbe WA and Lin SM. From disease ontology to disease-ontology lite: statistical methods to adapt a general-purpose ontology for the test of gene-ontology associations. Bioinformatics. 2009; 25:i63-68.
48. Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS and Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009; 106:9362-9367.
49. Maglott D, Ostell J, Pruitt KD and Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2011; 39:D52-D57.
50. Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M and Hirakawa M. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006; 34:D354-357.
51. D N. BioCarta. Biotech Software & Internet Report. 2001; 2:117-120.
52. Supek F, Bosnjak M, Skunca N and Smuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011; 6:e21800.
53. Barabasi AL and Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004; 5:101-113.
54. Smoot ME, Ono K, Ruscheinski J, Wang PL and Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011; 27:431-432.