
INTRODUCTION
Apoptosis serves as a natural barrier to cancer development, and targeting this cancer hallmark represents an indispensable therapeutic strategy [1] . Apoptosis can be induced via two major pathways, extrinsic and intrinsic, and in acute myelogenic leukemia (AML) the latter can be directly triggered by elevation of cAMP, which acts synergistically with first-line antileukemic agents [2]. This creates a unique situation, where an additional targetable pathway, previously unexploited by traditional chemotherapeutics, may exist in AML cells [2].
The effect of intracellular cAMP (icAMP) elevation is tissue/cell specific. In certain tumors, including pituitary, adrenocortical and thyroid adenomas and carcinomas, the cAMP/ protein kinase A (PKA) pathway provides signals required for tumor development and/or cell survival. In leukemias/lymphomas, cAMP elevation can be pro-apoptotic, whereas in leukocytes/macrophages it is reported to be anti-apoptotic (see Tables 1 and 2 in ref. [3], [4]). Additionally, cAMP can have both pro- and anti-apoptotic activity within the same cell depending upon experimental conditions. icAMP compartmentalization may also contribute to the complexity of signaling [5]. Nonetheless, a significant body of literature suggests that modulating the cAMP pathway provides a number of promising targets for treating leukemia [6].
Table 1: Hit compounds identified in the screen for inhibition of cAMP efflux
Hit compound |
Structure |
Notes |
Artesunate |
|
In 2007, FDA approved investigational new drug protocol #76,725 entitled “Intravenous Artesunate for Treatment of Severe Malaria in the United States” |
Dihydroartemisinin |
|
Artesunate is hydrolysed to its active metabolite dihydroartemisinin. Along with artemisinin currently used as antimalarial drugs in Asia. |
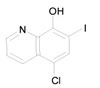
Clioquinol |
|
Clioquinol (Iodochlorhydroxyquin) is the FDA approved antifungal and antiprotozoal drug. A phase I trial for clioquinol in patients with hematologic malignancies has been reported [46] |
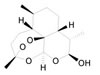
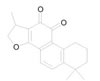
Cryptotanshinone |
|
A major tanshinone isolated from Salvia miltiorrhiza. It was shown to inhibit cancer cell proliferation [47] |
Parthenolide |
|
A sesquiterpene lactone from Tanacetum parthenium. An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells [48] |
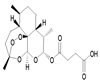
Patulin |
|
A mycotoxin produced from Penicillium and Aspergillus. Patulin-induced apoptosis in human leukemia cells is mediated through the mitochondrial pathway [49] |
AML (IPC-81) and multiple myeloma cells undergo rapid apoptosis after cAMP elevation [7, 8]. In S49 T-cell lymphoma cells, apoptosis can be induced through a cAMP/PKA-dependent pathway [9]. Increasing icAMP by using cAMP analogues [10], adenylyl cyclase (AC) stimulators [11], or phosphodiesterase (PDE) inhibitors [12, 13] has been a focus of cancer therapeutics research [3, 14]. This approach has been supported by the fact that the concentration of cyclic nucleotides is elevated in the plasma and urine of individuals with certain leukemias [15, 16], and in some cases these levels correlate with disease progression [16]. In this report, we hypothesize that one mechanism for malignant cell apoptotic evasion could be active efflux of cAMP [14]. Rather than relying on PDEs to degrade icAMP, active cAMP removal from the cytoplasm can provide a survival advantage. We envision that cAMP efflux prevents an elevation of icAMP that could trigger up-regulation of the Bcl-2 interacting mediator of cell death (Bim/BCL2L11) protein [2, 17], or down-regulation of the myeloid cell leukemia 1 (Mcl-1) protein [8, 18]. Inhibition of cAMP efflux alone should be sufficient to selectively trigger death in cells that rely on this anti-apoptotic mechanism for survival. To test this idea, we decided to identify drug-like compounds that are capable of blocking cAMP efflux.
To identify “inhibitors of cAMP efflux” (ICE), we developed and validated a novel assay for the detection of cAMP efflux (in press), using a well characterized model for AML known to efflux cAMP through ABCC4, the multidrug resistance-associated protein-4 (MRP4) transporter [19]. Next, we screened libraries composed of biologically active substances and off-patent drugs. We validated the “hits” in secondary assays that assessed the compound effects on cell signaling, viability and apoptosis. The ICE were also tested for their effects on cAMP efflux inhibition and viability in B-lineage acute lymphoblastic leukemia (B-ALL) cells, primary B-ALL bone marrow patient samples, and healthy human primary blood mononuclear cells (PBMCs). The most promising compounds showed dose-dependent, selective inhibition of leukemic cells vs. PBMCs. Our hypothesis was further supported by measurements of cAMP-dependent activation of downstream effectors after exposure to ICE compounds. Because several identified ICE are FDA-approved drugs, our studies provide a potential path for drug repurposing against leukemias.
RESULTS
A screen for ICE identifies six potentially active compounds
To identify ICE, we took advantage of a model system where cAMP efflux is well studied: U937 cells, which can actively extrude cAMP into extracellular media [19]. In these cells, a rapid increase of cAMP efflux can be triggered through the elevation of the icAMP concentration using G-alphaS GPCR-specific ligands, blocking PDE-dependent cAMP hydrolysis, and by other pharmacological manipulations. The cAMP efflux is ATP- and MRP inhibitor-dependent, and shRNA knockdown has shown that the cAMP efflux is mediated by MRP4/ABCC4. Moreover, an increase of icAMP was sufficient to induce differentiation of U937 and other AML cell lines [19].
To study cAMP efflux, we loaded U937 cells with fluorescently tagged cAMP (F-cAMP). MK-571, (MRPs selective inhibitor) [19], was used as a positive control. Figure 1 shows that MK-571 down-modulated cAMP efflux in a dose-dependent manner, with EC50 ~30 µM. This value was very close to the IC50 previously reported for MRP4/ABCC4 [20]. Thus, the fluorescent tag (Alexa Fluor®488) did not significantly affect the ability of cells to efflux cAMP nor of MK-571 to block this process.
These experiments served as the basis for a screen to identify compounds that could block cAMP efflux. F-cAMP-loaded U937 cells were screened against compounds from the Prestwick Chemical Library (~1200 previously FDA-approved drugs) and the SPECTRUM Collection (2320 compounds - 60% drugs, 25% natural products, 15% bioactive components). The screen identified 51 hits, which were tested in dose-response to validate their activities (data not shown), yielding 7 potentially active ICE compounds: artemisinin, clioquinol, quinalizarin, harmalol, cryptotanshinone, parthenolide, and patulin. Three additional structurally-related compounds (dihydroartemisinin, artemether, artesunate) were also included for evaluation in secondary assays. Based on further validation, only artesunate, clioquinol, cryptotanshinone, dihydroartemisinin, patulin, and parthenolide were chosen for extended studies (Table 1). The detailed screening data will be published elsewhere (in press).
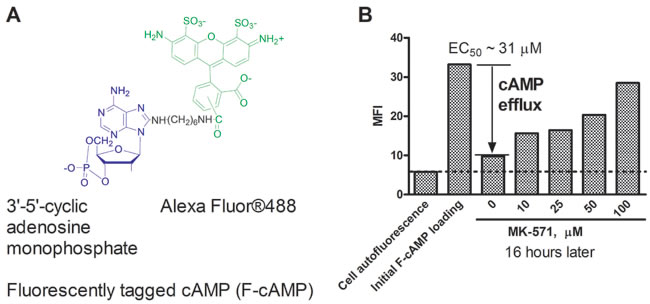
Figure 1: Effect of MK-571 (MRPs selective inhibitor) on efflux of a fluorescently tagged cAMP (F-cAMP). A. Structure of the fluorescent cAMP analog. B. U937 cells loaded with F-cAMP were incubated overnight in the presence of increasing concentrations of MK-571 at 37°C. The EC50 for cAMP efflux blocking was determined using a variable slope sigmoidal dose response equation with “bottom” constraint equal to cell autofluorescence and “top” constraint equal to the specific fluorescence. A representative experiment of three experiments is shown.
Correlation between ICE ability to block F-cAMP efflux and viability loss
Next, we compared ICE efficacy for blocking F-cAMP efflux with their ability to trigger loss of cell viability (Figure 2). A strong positive correlation (r2 = 0.73) has been observed. The values for all six ICE compounds and MK-571 control ranked in the same order and were located within 95% confidence interval. Moreover, the slope of the regression line was equal to 1. This suggests that the relative potency of ICE in affecting cell viability varies in parallel with the ability to block F-cAMP efflux.
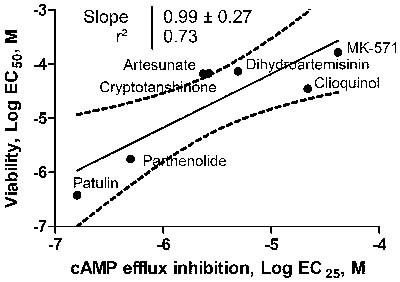
Figure 2: Relationship between ICE ability to block F-cAMP efflux and loss of cell viability. U937 cell were incubated overnight in the presence of increasing concentrations of ICE and MK-571 and cell viability was determined as described in Materials and Methods. Next, the data were fitted to a sigmoidal dose response equation and the EC50 values for cell viability loss were plotted vs. EC25 determined for F-AMP efflux inhibition. The EC25 was equivalent to a two standard deviation cut-off that was used for a primary compound screening hit determination criteria. The data were fitted to a linear regression equation. The 95% confidence interval, a square of Pearson’s correlation coefficient and a slope of the line are shown.
CREB/AFT-1 phosphorylation in response to ICE
Next, to evaluate whether reducing cAMP efflux would result in an elevation of cytoplasmic cAMP-dependent cell signaling, we studied the effects of ICE on phosphorylation of cAMP-responsive element-binding protein (CREB; Ser133) and activating transcription factor-1 (ATF-1; Ser63), classical cAMP effectors that activate target genes through cAMP response elements (CRE). This pathway is also directly implicated in cAMP-induced apoptosis in leukemia [2]. All studied compounds showed increased binding of anti-CREB (pS133) / ATF-1 (pS63) specific antibodies as compared to vehicle control (Figure 3). For two compounds (clioquinol and parthenolide), the binding of antibodies was comparable to the adenylate cyclase stimulator forskolin positive control. Thus, ICE compounds can stimulate CREB/AFT-1 phosphorylation.
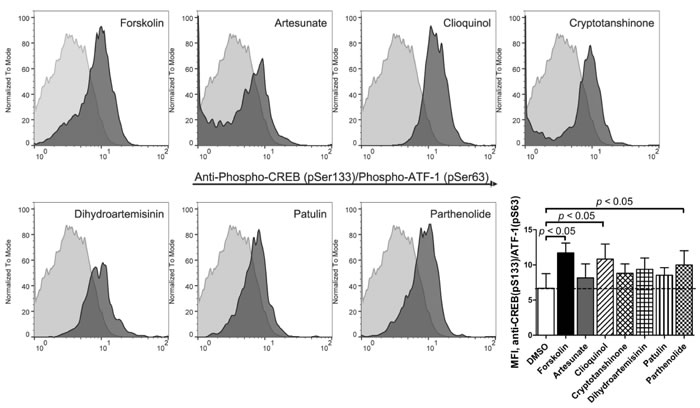
Figure 3: Binding of anti-phospho-CREB/AFT-1-specific antibody in response to ICE. U937 cells were treated for 1 hour with 20 µM ICE compounds or forskolin (positive control), or DMSO (vehicle, negative control). Next, cells were fixed, permeabilized and stained with primary labelled anti-CREB (pSer133) / ATF-1 (pSer63) monoclonal antibody. Histogram overlays from one representative experiment show negative control events (light grey) and compound-treated events (dark grey). Bar graph shows MFI ± SEM (standard error of the mean) for four independent experiments. Statistical significance was determined by one-way ANOVA with repeated measures using a Dunnett post-test to compare treated samples to DMSO control values (p < 0.05).
VLA-4 deactivation in response to ICE
Another signaling pathway that in leukocytes can be triggered by the elevation of cytoplasmic cyclic nucleotides is the conformational deactivation of the Very Late Antigen-4 (VLA-4, alpha4 beta1 integrin), an adhesion molecule implicated in homing and retention of early hematopoietic progenitors in the bone marrow. The elevation of icAMP using G-alphaS GPCR-specific ligands, forskolin and by other pharmacological manipulations results in rapid dissociation of the VLA-4-specific ligand-mimicking probe, LDV-FITC [21]. We studied the effect of ICE on VLA-4 deactivation using the same previously characterized model system (Figure 4). Studied compounds triggered rapid dissociation of LDV-FITC in U937 cells pre-activated through a non-desensitizing mutant of the FPR1. In several experiments, the effects of parthenolide and patulin exceeded the effects of the positive control, forskolin (Figure 4). Cryptotanshinone induced rapid and reversible VLA-4 deactivation that looked similar to the effect of a cell-permeable cyclic nucleotide analog [22]. Thus, ICE compounds were also capable of triggering an integrin de-activation pathway, where the role of elevated cyclic nucleotide concentration is critical [21, 22].
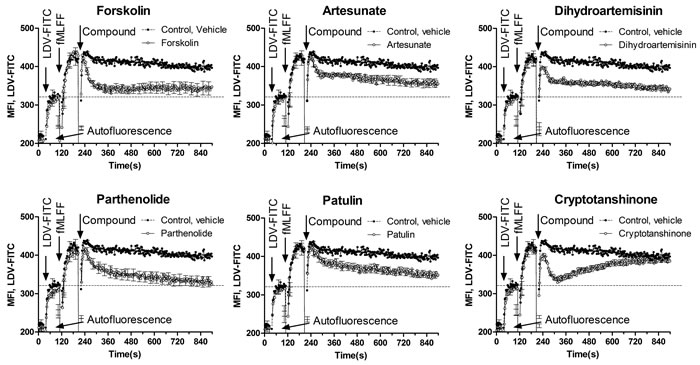
Figure 4: Dissociation of the VLA-4-specific fluorescent ligand in response to ICE. U937 cells transfected with a non-desensitizing mutant of FPR1 were maintained in cRPMI at 37°C while the samples were stirred continuously during data acquisition. Cell suspension was sequentially treated with LDV-FITC (first arrow), FPR1-specific ligand for cell activation (fMLFF, second arrow), and ICE compounds or forskolin positive control (40 µM) or DMSO vehicle (third arrow). The use of the non-desensitizing mutant of FPR1 allows for the maintenance of the high affinity VLA-4 state during the course of the experiment (control, vehicle). This allows for monitoring the real-time deactivation kinetics after compound addition. Each line represents MFI ± SEM of three independent experiments. In each experiment, every experimental point represents MFI of several hundred events acquired every second. Bars representing SEM are shown for every 10 s time point. Dashed line represents binding of the LDV-FITC prior to cell activation.
Adenylyl cyclase inhibition reduces ICE-induced cell viability loss
Because synthesis is a source for icAMP accumulation, and soluble adenylyl cyclase (sAC) is directly implicated in the mitochondrial pathway of apoptosis, we studied how blocking the AC by the selective sAC inhibitor, KH7, influences ICE effects. Based on the data published by Kumar et al., 2009 [23], we expected that blocking cAMP production would reduce ICE potency (Figure 5A). In fact, we detected a significant protective effect of KH7 on the cell viability loss induced by the four most potent ICE compounds (Figure 5B). The lack of effect for less potent ICE could be due to a lesser effect on viability: KH7 displayed the most prominent effect in samples where control viability was far below 50%. Thus, inhibition of sAC activity was sufficient to reduce the effects of the most potent ICE.
Hence, compounds identified in an assay based on blocking cAMP efflux can reduce cell viability, and stimulate two signaling pathways that are each modulated by elevation of icAMP, CREB phosphorylation and VLA-4 deactivation. Moreover, blocking of sAC activity prevented ICE-induced cell viability loss. These data are consistent with an ICE molecular mechanism involving elevation of icAMP. The accumulation of icAMP as the result of decreased cAMP efflux in U937 cells was first demonstrated by Copsel, et al. [19].
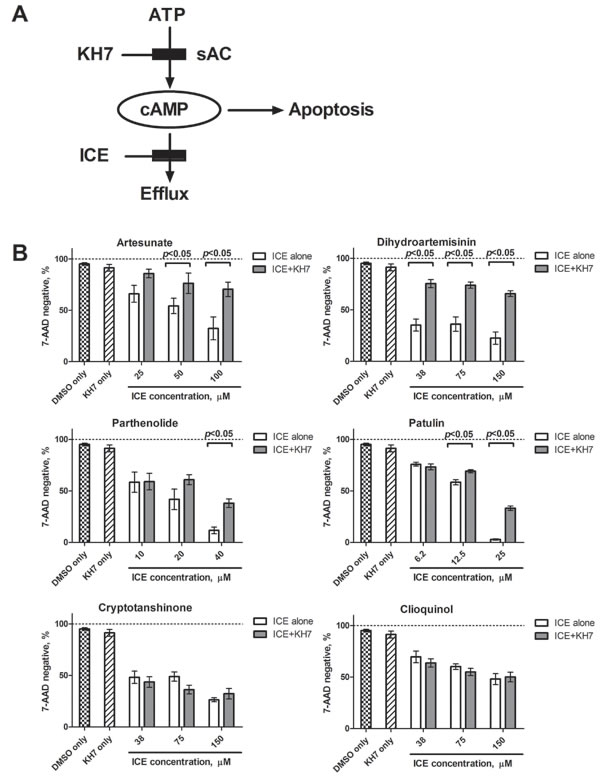
Figure 5: Effect of the selective inhibitor of soluble adenylyl cyclase (KH7) on cell viability loss induced by ICE. A. Schematics of KH7 action. By blocking synthesis of cAMP, KH7 is expected to decrease overall icAMP accumulation and thus, to protect cells from ICE-induced apoptosis. B. U937 cells were treated overnight with increasing concentrations of ICE in the presence or absence of 6 µΜ ΚΗ7. ICE concentrations were chosen based on relative ICE potency. Cell viability was assessed using 7-AAD staining as described in Materials and Methods. Control samples were treated with 6 µΜ ΚΗ7 or equal volume of vehicle (DMSO). Graph shows percentage of 7-AAD negative events ± SEM of two independent experiments. Each bar represents mean of five values (n = 5). Statistical significance was determined using unpaired t-test and significant difference is indicated as (p < 0.05) for each pair of KH7 treated and untreated samples.
Normal primary peripheral blood mononuclear cells (PBMCs) did not significantly efflux cAMP
According to our hypothesis, active cAMP efflux represents a novel apoptosis evasion mechanism that is activated in certain malignant cells, and normal cells should lack this cAMP efflux ability. To test this proposition, we compared F-cAMP efflux in U937 cells and normal PBMCs (Figure 6). After 24 h incubation, PBMCs loaded with F-cAMP retained ~80-90 % of the probe fluorescence when incubated at 37 °C or at 4°C, whereas U937 cells lost ~80 % of the probe fluorescence only at 37 °C. A significant part of this loss was blocked by incubation with efflux inhibitor MK-571, indicating active participation of ABCC family transporters. No decrease in the probe fluorescence was detected at 4oC, suggesting that cell membrane integrity was well preserved and passive probe leak had not occurred. This result suggests that active cAMP removal cannot be detected in normal PBMCs in the same time frame as in the AML model.
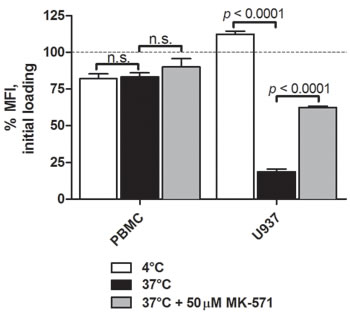
Figure 6: cAMP efflux activity of U937 cells and human PBMCs. Cells were loaded with F-cAMP as described in Materials and Methods. F-cAMP retention after overnight incubation with ICE in samples incubated at 4°C (passive leakage) or 37°C (active removal), and in the presence of cAMP efflux inhibitor control MK-571, was measured by flow cytometry. Cell autofluorescence was subtracted, and the data were normalized to the fluorescence value at initial staining indicated by dashed line. Data shown is MFI ± SEM from one representative independent experiment conducted in triplicate. Statistical significance was determined by a two-tailed Student’s t-test, n.s., non-significant.
ICE induce apoptosis of U937 cells
The effect of ICE on U937 cell apoptosis after overnight incubation has been studied using the MultiCyt 4-Plex Apoptosis kit that reports four different apoptosis endpoints: effector caspases 3 and 7 activation, phosphatidylserine surface expression, mitochondrial membrane depolarization and cell membrane integrity. The ICE induced apoptosis in a dose-dependent manner, and the EC50 values for all four apoptosis endpoints were determined to range from 2-3 µM to several hundred µM (Figure 7). The relative sensitivity of different apoptosis endpoints reflected different consecutive steps of the intrinsic pathway. Mitochondrial depolarization and membrane damage were the most sensitive while the effector caspase activation was the least. Cryptotanshinone showed a very small effect on annexin-V binding and caspase activation, even at the highest ICE concentrations. These EC50s were excluded from further analyses (as indicated by NC).
To simplify analysis, the data were color-coded (heat map) according to the determined EC50 values (Figure 7). The most potent compounds (EC50 values 1-10 µM) in the apoptosis assay were: patulin, parthenolide, and dihydroartemisinin (white color). In addition, the relative ranking was largely independent on the particular endpoint. Mean EC50s calculated for each compound are shown at the bottom of a heat map and indicate relative potency of studied ICE. Thus, U937 cells treated with ICE exhibited a dose-dependent increase in apoptosis.
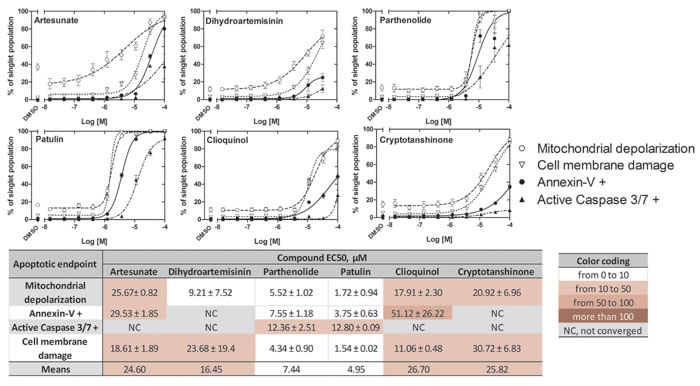
Figure 7: Effects of ICE on apoptosis of U937 cells. Cell mitochondrial depolarization, annexin-V binding, caspase 3/7 activation and cell membrane damage are shown as dose response curves and a heat map. Percentage of U937 cells positive for four different apoptosis endpoints was determined as described in Materials and Methods. Dose response curves for each endpoint (n = 4) after 24 h incubation with ICE display cell viability (%) ± SEM. Data were fit using variable slope sigmoidal dose response equation with no constraints. Heat map shows EC50s ± SEM. NC, not converged.
ICE decrease viability and show cancer cell specificity
Next, we tested the effect of ICE on the viability of U937 cells and B-ALL cell lines (Table 2). On U937 cells, several compounds were more potent than the positive control MK-571 (Figure 8). In general, B-ALL cell lines showed more sensitivity than the U937 cells to several of the selected compounds. However, B-ALL lines showed a decreased sensitivity to the two structurally related drugs: artesunate and dihydroartemisinin. The EC50 values for the ICE with each B-ALL line ranged from low nanomolar to ~300 µM (Figure 8). It can be noted, however, that the compounds, which consistently decreased viability ranked in the same order across all six B-ALL lines: patulin, parthenolide, clioquinol, cryptotanshinone.
Table 2: The hematologic cell lines included in the study, their subtype and genetic rearrangements [50]
Cell line |
Subtype |
Genetic rearrangement |
Fusion gene |
U937 |
AML |
t(10;11)(p12;q14) |
PICALM/MLL10(AF10) |
697 |
B-ALL |
t(1;19)(q23;p13) |
TCF3(E2A)/PBX1* |
Nalm-6 |
B-ALL |
t(5;12)(q33;p13) |
ETV6/PDGRFB |
Sup-B15 |
B-ALL |
t(9;22)(q34;q11) |
P190 BCR/ABL1 |
Reh |
B-ALL |
t(12;21)(q13;q22) |
ETV6(TEL)/RUNX1(AML1) |
RS4;11 |
B-ALL |
t(4;11)(q21;q23) |
MLL/MLLT2(AF4) |
MHH-Call 3 |
B-ALL |
t(1;19)(q23;p13) |
TCF3(E2A)/PBX1* |
* – Two cell lines were shown to have identical genetic rearrangement and a fusion gene.
To investigate the potential selectivity of the compounds, we compared ICE effects on cell viability in leukemic and non-malignant cells (PBMCs from healthy volunteers). The EC50 values indicated that for the most potent of the tested compounds to be efficacious, PBMCs required a concentration of at least an order of magnitude higher than was necessary for the leukemic cell lines (Figure 8, notice darker color-coding for PBMCs). This difference was expected, since normal PBMCs do not have an established system for cAMP efflux (Figure 6), and therefore should be less sensitive to ICE.
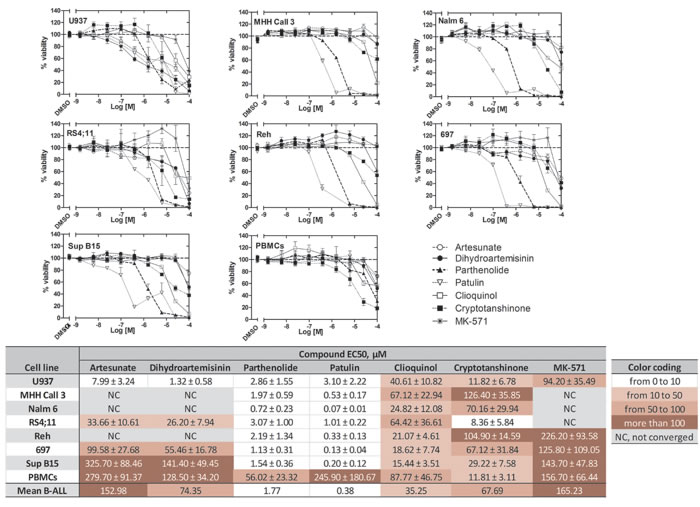
Figure 8: Effects of ICE on cell viability of U937 cells, the B-ALL cell lines 697, Reh, MHH Call 3, RS4;11, Sup B15, and Nalm 6, and normal human PBMCs. Dose response curves for each cell type after overnight incubation with ICE display cell viability (%) ± SEM. For U937, n = 8; for PBMCs and B-ALL cells, n = 6. Data were normalized assuming that negative control (DMSO) was equal to 100% viability. Data were fit using the sigmoidal dose response equation with constraints: top = 100, bottom = 0, hill slope = -1. Table shows EC50s ± SEM. NC, not converged.
Leukemic cells have different abilities to efflux cAMP
To further study cAMP efflux in leukemic cells, the B-ALL cell lines were loaded with F-cAMP, and its efflux from the cells was evaluated. After incubation at 4°C (passive leakage), the cells lost 20-60% of their fluorescence from the initial F-cAMP loading. Incubation at 37˚C (active efflux) resulted in F-cAMP removal that ranged from ~60% to ~90%, with no apparent relationship between 4°C and 37°C samples (Figure 9A). For example, Nalm-6 and 697 cells showed dissimilar passive leakage, but both cell lines exhibited similar active F-cAMP removal abilities.
The cell lines also exhibited varied sensitivity to MK-571, a control for transporter-dependent efflux. We found a strong relationship between levels of F-cAMP remaining in cells at 37°C, both alone and in the presence of MK-571. Based on this parameter, cell lines could be stratified into three groups, which indicated correlations between ability to actively efflux cAMP and MK-571 inhibition of cAMP removal. Group 1: Sup-B15 and MHH-Call3 removed ~60% of the F-cAMP by active efflux, which was blocked completely by MK-571. Group 2: Reh and RS4;11 cells removed 75-80% of the F-cAMP by active efflux, and ~30-40% with MK-571. Group 3: 697 and Nalm-6 cells removed > 90% of the F-cAMP by active efflux, and 60-70% with MK-571 (Figure 9B). Thus, the cell lines that effluxed F-cAMP poorly were better inhibited by MK-571, and those that were efficient in the removal of F-cAMP were less inhibited by MK-571. This suggests that for cell lines in Groups 2 and 3, an additional MK-571-insensitive mechanism participated in the removal of F-cAMP.
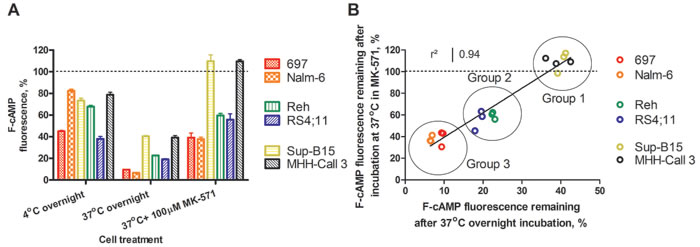
Figure 9: Retention of F-cAMP in hematopoietic cell lines. A. F-cAMP leakage from B-lineage ALL cell lines after overnight incubation at 4°C, or 37°C, and in the presence of the positive control MK-571. Lower values correspond to higher efflux. Data shown are the mean ± SEM from three independent experiments. Initial F-cAMP loading is indicated by a dashed line. B. The correlation between F-cAMP fluorescence remaining in cells incubated at 37°C alone and in the presence of MK-571. See text for details.
The primary cAMP transporter, MRP4/ABCC4, is differentially expressed by leukemic cell lines
cAMP is primarily released by cells via the ABCC4 (MRP4) and ABCC5 (MRP5) transporters [24]. The affinity of ABCC4 for cAMP is ~10-fold greater than that of ABCC5 (km values of 44.5 µM and 379 µM, respectively) [24, 25]. Therefore, we hypothesized that the presence of ABCC4 on leukemic cells would correlate with their ability to efflux F-cAMP. ABCC4 phenotypes were determined for the B-ALL cell lines, and the number of ABCC4-specific binding sites ranged from ~100 on MHH-Call3 to ~104 on RS4;11 cells (Figure 10). However, no apparent correlation between ABCC4 expression and F-cAMP efflux were detected (compare Figure 9 and Figure 10). This suggests that other transporters and/or mechanisms may play additional roles in leukemic cell removal of cAMP.
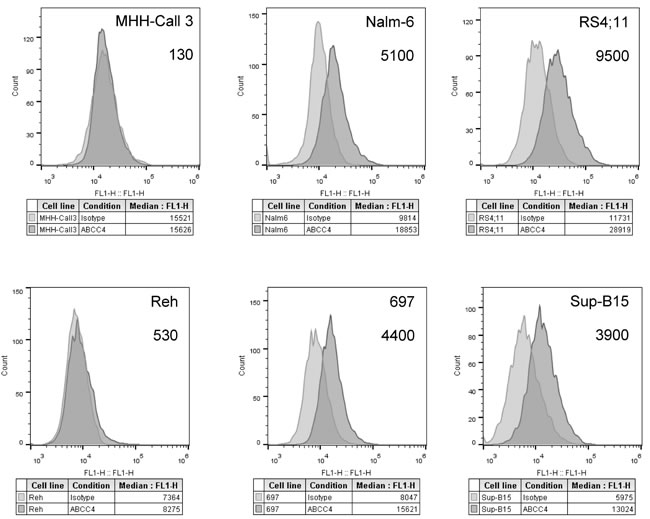
Figure 10: Determination of ATP-binding cassette transporter ABCC4-specific binding sites on B-ALL cell lines. Raw histograms of IgG isotype controls (grey) and ABCC4-specific antibody (dark grey) binding to each cell line. Specific binding sites calculated using Quantum™ Simply Cellular® anti-human IgG calibration beads stained with primary anti-ABCC4-specific and secondary fluorescent antibody. Calibration beads allowed mean fluorescence intensity (MFI) values to be converted into non-specific binding sites. Binding site values for IgG primary antibody-bound isotype control cells were subtracted from the ABCC4 non-specific binding site data to calculate the number of ABCC4-specific binding sites per cell. Estimated site numbers are indicated on each panel. A representative experiment of two experiments is shown.
ICE decrease primary ALL patient sample viability
To further establish the effectiveness of ICE in reducing cancer cell viability, primary cells from six B-ALL patients (Table 3) were tested ex vivo. The results showed dose-dependent decreases in patient sample viability after exposure to the compounds (Figure 11). To verify the relative ranking of ICE, the mean EC50 values for each compound were plotted against the mean EC50 values determined for all B-ALL cell lines. The result indicated a strong correlation for most patient samples (Figure 11B). This result suggests that the data obtained using cell lines adequately reflect the relative compound potency detected in patient samples, and therefore, cell lines can be used to test ICE derivatives in future medicinal chemistry applications.
Table 3: Genotypic and phenotypic profile of B-ALL patient samples
Patient |
Age |
Gender |
WBC |
Ploidy |
Phenotype |
Risk |
Cytogenetics/FISH |
124-12 |
4 |
F |
2.9 |
STD |
46,XX,del(9)(p13),der(19)t(1;19)(q23;p13.3)[10]/46,idem,add(5)(p13),add(9)(p21),del(10)(q23)[2]/46,idem, t(1;13)(p13;q14)[3]/46,XX[5] |
||
238-13* |
18 |
M |
629.2 |
diploid |
CD34+ CD19+ CD22dim |
VHR |
46,XY,t(4;11)(q21;q23)[cp6]/49,XY,+X,+1,t(4;11)(q21;q23),+21[5]/46,X,-Y,t(4;11)(q21;q23),+mar[3] |
329-13* |
26 |
F |
53.1 |
CD10+ CD19+ CD22+ |
VHR |
46,XY,t(4;11)(q21;q23)[cp6]/49,XY,+X,+1,t(4;11)(q21;q23),+21[5]/46,X,-Y,t(4;11)(q21;q23),+mar[3] |
|
017-14 |
22 |
M |
39.5 |
CD10+ CD19+ CD22+ |
VHR |
45,XY,-7,t(9;22)(q34;q11.2)[16]/46,XY[4] complement with translocation t(9;22) and monosomy 7 in sixteen of twenty cells analyzed; BCR/ABL1 transcript ratio: 0.37. Ratios>0.1 |
|
280-13 |
2 |
M |
22.0 |
0.92 hypoploid |
CD10+ CD19+ CD34+ |
STD |
46,XY[4] |
116-13 |
7 |
F |
4.0 |
diploid |
CD10+ CD19+ CD34+ |
STD |
46,XX |
* – have MLL-AF4 rearrangement
Abbreviations: ALL: acute lymphoblastic leukemia; WBC: white blood cell; FISH: fluorescent in situ hybridization; STD: standard risk; VHR: very high risk
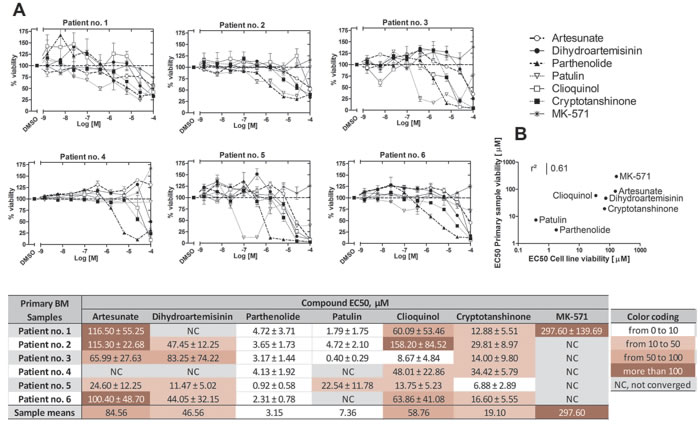
Figure 11: Effects of ICE on cell viability of primary ALL patient samples. A. Compound dose response curves for each patient sample tested in duplicate. Data were normalized assuming that negative control (DMSO) was equal to 100% viability. Data were fit using the sigmoidal dose response equation with constraints: top = 100, bottom = 0, hill slope = -1. Table shows EC50s ± SEM. NC, not converged. B. Correlation between mean compound EC50 values obtained from B-ALL cell line data (Figure 8) and mean EC50 values from primary B-ALL patient samples. The coefficient of determination (r2) was calculated from the Pearson correlation coefficient.
Moreover, the overall shape of the dose-response curves obtained for different patient samples revealed an interesting pattern: the shape of the curves was more patient-specific rather than compound-specific. For example, in the samples from patients no. 1 and no. 5, we observed a significant increase in cell viability vs. vehicle treated sample at lower concentrations of dihydroartemisinin, clioquinol or parthenolide (see points above the dashed line, Figure 11A). This behavior was also evident in viability data from B-ALL cell lines (Figure 8). These results are consistent with the report that small or transient increases in icAMP can be anti-apoptotic and therefore, can support cell survival [2, 3]. Under identical conditions, the same compounds elicited no such effect in the samples obtained from patients no. 2 or no. 4. Another difference can be observed when the slopes of the dose-response curves are analyzed. It appears that in some patient samples, the slopes are steeper than in others. These data suggest that certain traits of a particular patient sample play a significant role in the response to different ICE compounds. It might be useful to correlate genotypic and phenotypic profiles of patient samples with these dose-response curve parameters. However, this will require further studies with larger numbers of patient samples with diverse genotypic and phenotypic profiles.
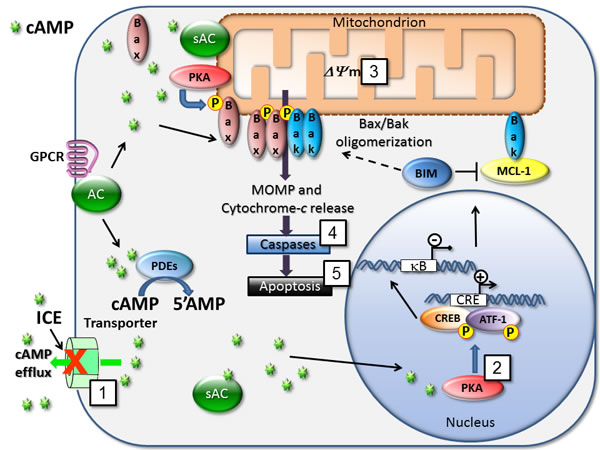
Figure 12: Mechanisms for cAMP-dependent regulation of cell death. cAMP is produced by various pools of soluble and membrane-bound adenylyl cyclases (sAC and AC). Blocking cAMP efflux with ICE (1) triggers several signaling endpoints that are related to the modulation of pro-apoptotic and cell survival pathways. Here, we detected phosphorylation of CREB/ATF-1 (2), mitochondrial depolarization (3), effector caspases 3 and 7 activation (4), and apoptosis (5) in the form of annexin-V binding and cell membrane damage. Blocking adenylyl cyclase activity using selective inhibitor of soluble adenylyl cyclase (KH7) was sufficient to reduce effects of most potent compounds. Various molecular mechanisms are implicated in effects of cAMP on apoptosis induction. In AML cells pro-apoptotic protein Bim/BCL2L11 expression is up-regulated via CRE/CREB in a cAMP-dependent manner [2]. Induction of Bim represents a crucial event in the cAMP-induced apoptosis in murine T-cell lymphoma and human acute leukemia [9]. cAMP is also shown to inhibit MCL-1 protein transcription in human MM cells [8] or human B-precursor cells [18]. Relocalization of the cytosolic sAC pool toward mitochondria leads to PKA activation and phosphorylation of the pro-apoptotic protein Bax that activates mitochondrial apoptotic pathway [44]. Also, cAMP is implicated in a regulation of NF-κB action [32], which is a potential target in AML [45].
DISCUSSION
For many years, researchers have been eager to develop methods to robustly modulate icAMP in cancer cells, and all known steps of the cAMP-related signaling pathway have been targeted [6]. Only recently, researchers have realized that icAMP can be also modulated by cyclic nucleotide efflux transporters. The finding that blocking cAMP efflux or down-regulation of efflux transporters can trigger cAMP elevation and downstream signaling [19] has opened new avenues for developing a novel class of anticancer therapeutics via inhibitors of cAMP efflux. Analysis of the proteins capable of cyclic nucleotide efflux also suggests notable possibilities.
The cAMP transporter ABCC4 has increased expression in blood cancer cells as compared to normal hematopoietic cells [26], and its expression decreases significantly upon cell differentiation [27]. ABC transporters are likewise upregulated in stem-like cells, which may suggest that these cells require active removal of cAMP or other structurally-related compounds in order to remain in a pluripotent state, and that the metabolic specificity of these cells requires active transport of certain metabolites [28]. These cancer stem cells are also associated with higher resistance to typical cancer therapeutics [29]. However, our results for ABCC4-expression did not define any clear correlations between cAMP efflux and ABCC4 expression in the cell lines. It is possible that other transporters capable of cAMP efflux (ABCC5 or ABCC11 [25, 30]) are present, or that there is a difference in the efflux capacity of different transporters. This may indicate that the ability to inhibit cAMP transport is substrate- or condition-dependent.
Of the active compounds identified in our screen for ICE, the majority were sesquiterpene lactones: parthenolide is derived from the feverfew (Tanacetum parthenium) plant; artemisinin and dihydroartemisinin are from Artemisia annua; and artesunate and artemether are semi-synthetic artemisinin derivatives. Cryptotanshinone is also plant-derived (Salvia miotiorrhiza), whereas clioquinol is an antimicrobial hydroxyquinolone derivative, and patulin is an antibiotic mycotoxin produced by Aspergillus and Penicillium. These compounds have been reported to exhibit anticancer activity in a number of model systems (Table 4), and the reported anticancer effects of these compounds were often similar to those seen in our assays (Table 5). However, none of the compounds has previously been reported to modulate cyclic nucleotide efflux.
Table 4: Previously reported anticancer activity of the compounds identified as ICE
Compound |
Tested cancer types |
Patulin |
kidney[51], oral squamous cell carcinoma[52], glioblastoma[53], colon[54-56], colorectal[57], hematological[49,58], liver[59] |
Parthenolide |
melanoma[60,61], cervical[62,63], hematological[48,64,65], breast[66] |
Artesunate |
ovarian[67], cervical[68], Kaposi’s sarcoma[69], squamous carcinoma[70], breast[71,72], liver[73], pancreatic[74,75], hematological[76-80], colorectal[79,81], neuroblatsoma[82], lung[79,83], osteosarcoma[84] |
Dihydroartemisinin |
ovarian[67,85], breast[86], liver[86,87], melanoma[88], pancreatic[89-93], lung[94-96], cervical[68], hematological[97-102], prostate[103], glioma[104], osteosarcoma[105] |
Clioquinol |
hematological[46,106-108], breast[109,110], cervical[111], prostate[112-114] |
Cryptotanshinone |
hematological[115,116], prostate[47,117,118], breast[47,119], melanoma[120,121], lung[121], rhabodomyosarcoma[47] |
Artemisinin, dihydroartemisinin, artesunate and artemether are currently used worldwide for the treatment of malaria. Artemether is a component of the drug Coartem manufactured by Novartis and approved by the US Food and Drug Administration in 2009. Several ongoing clinical trials of artemisinin derivatives to treat hepatocellular carcinoma, breast cancer, cervical intraepithelial neoplasia, as well as a trial evaluating “biological activity of oral clioquinol in patients with relapsed or refractory hematological malignancy” initiated by Dr. Mark Minden from Ontario Cancer Institute Princess Margaret Hospital in Canada, can be found on clinicaltrials.gov. Therefore, these drugs have the highest potential to be repurposed into current treatment regimens. Poor water-solubility and bioavailability of parthenolide prompted medical chemistry efforts to improve these characteristics [31]. However, since these drugs were identified from two relatively small libraries totaling ~3500 compounds, we envision that larger efforts to identify compounds capable of blocking cAMP efflux may lead to better drugs. This is especially important because of the unique signaling role of cAMP in apoptosis.
Table 5: Previously reported molecular/ signaling mechanisms related to anticancer activity of the compounds identified as ICE
Compound |
Cell cycle arrest |
Anticancer effects |
Potential mechanism(s) of action |
Parthenolide |
S [123] |
↓VEGF expression & metastasis [61,124,125]; ↑p53 activation [48]; |
↓NF-κB & AP-1 activation [48,61,124,126,127]; ↓MAPK/ERK signaling [126,127]; |
Dihydroartemisinin |
G1 [90], |
↓VEGF expression & angiogenesis [99,101,102]; ↓cyclin B, CDC25 [87] |
|
Artesunate |
S [81], |
↓metastasis/migration [80,83]; ↓VEGF expression/angiogenesis [70,76,80]; ↓cyclins B & D1, Cdks 2, 4, 6[70]; ↑p21, p27 [70] |
↓NFκB & AP-1 activation [83,130,131]; ↓nitric oxide, cAMP-mediated, Wnt/β-catenin, PI3K/Akt signaling [81,130,131]; |
Cryptotanshinone |
G1 [120], G2 [120] |
↓cyclin D1, Bcl-2 [47,115,116]; ↑p53, Chk1, Chk2 [120] |
↓mTOR, STAT3 signaling [116,117]; ↓eIF-4E [47] |
Clioquinol |
G1 [110] |
↓cyclin D1 [110]; ↑p21, p27, p53 [106] |
|
Patulin |
G1 [132], G2 [57] |
↓ERK1/2 activation [52,133]; ↑UPR[52,133]; ↑intracellular [Ca2+] [134] |
↑ROS, DNA damage [57,59,132,133,135]; ↓glutathione [133,135] |
Abbreviations: VEGF: vascular endothelial growth factor; NF-κB: nuclear factor kappa-B; AP-1: activator protein 1; MAPK: mitogen-activated protein kinase; ERK: extracellular signal-regulated kinases; STAT: signal transducer and activator of transcription; ROS: reactive oxygen species; PI3K: phosphatidylinositol-4,5-bisphosphate 3-kinase; Akt: protein kinase B; mTOR: mammalian target of rapamycin; eIF-4E: eukarytotic initiation factor 4E; UPR: unfolded protein response
As a messenger, cAMP plays significant regulatory roles within cells. Multiple signaling mechanisms critical for leukemogenesis can be down-modulated by cAMP. At least two cAMP/PKA-related pathways can be involved in the induction of cAMP-dependent apoptosis in cancer cells: 1) the mitochondrial-mediated (intrinsic) pathway, and 2) modulation of the NF-κB signaling pathway (Figure 12). The pro-apoptotic intrinsic mechanism promoted by cAMP depends upon PKA [17], which can phosphorylate CREB, and results in transcription of the apoptotic activator Bim/BCL2L11 [2]. cAMP modulation of NF-κB can affect transcription of pro-survival genes [32]. Our data suggest that inhibition of cAMP transport by ICE was able to elicit many of these effects.
Targeting cancer through a “pathway-dependent approach” that consists of different means of elevating cAMP is considered a viable option for novel therapeutic development [6]. It appears that every potential step of the pathway, including cAMP synthesis, degradation, and downstream signaling, has been taken into consideration to stimulate icAMP accumulation [6, 33, 34]. Classically, cyclic nucleotide analogs or other cAMP-elevating agents have been used to treat hematological malignancies by slowing cell growth and differentiating cancer cells [14]. However, while modestly effective, these compounds exhibit toxicity in non-cancerous tissues [35-37]. Our data suggest that targeting cAMP efflux with small molecules could be an efficient way to raise icAMP in certain types of cancer, and this could potentially result in the development of a new class of pathway-specific therapeutics. Because increased cAMP efflux is not a typical trait of healthy cells, the identified ICE exhibited specificity toward leukemic cells. The relatively efficient targeting of cAMP transport in cancers would directly depend on cell- or patient-specific characteristics and efflux ability. We anticipate that identification of drugs that inhibit this transport could allow for selective targeting of cancers that capitalize on cAMP pathway modulation for survival, and this merits further investigation into the effects of ICE on multiple ABC transporters. The fact that the machinery responsible for apoptotic evasion by cAMP efflux can also potentially support the removal of structurally related chemotherapy drugs (e.g., ara-C), and thus may contribute to multidrug resistance [38-40], makes this work unusually promising.
MATERIALS AND METHODS
Ethics
All blood samples from healthy volunteers were obtained with written, informed consent per local institutional research guidelines according to the University of New Mexico Human Research Protections Office protocol 11-225. Bone marrow samples acquired from pediatric B-ALL patients were taken upon written, informed consent according to the University of New Mexico Human Research Protections Office protocol 05-435.
Cells and reagents
Alexa Fluor®488 8-(6-aminohexyl) aminoadenosine 3´,5´-cyclicmonophosphate, bis(triethylammonium) salt (F-cAMP) (Life Technologies, cat. A35775). The VLA-4-specific probe 4-((N’-2-methylphenyl)ureido)-phenylacetyl-L-leucyl-L-aspartyl-L-valyl-L-prolyl-L-alanyl-L-alanyl-L-lysine-FITC (LDV-FITC) was synthesized at AIBioTech. The selective inhibitor of soluble adenylyl cyclase, KH7, was purchased from Cayman Chemical Company (cat. 330676-02-3). All other reagents and hit compounds for secondary assays were from Sigma-Aldrich.
Cell lines, purchased from ATCC and DSMZ, were cultured in RPMI-1640 medium supplemented with 2 mM L-glutamine, 100 U/ml penicillin-streptomycin and 10% heat-inactivated fetal bovine serum (FBS; 20% for MHH-Call3), hereafter referred to as cRPMI, and incubated in a humidified atmosphere with 5% CO2 at 37°C.
PBMCs
Healthy PBMCs were obtained from volunteers. Mononuclear cells were purified using Mono-Poly resolving medium (MP Biomedicals, cat. 091698049) according to manufacturer’s instructions. PBMCs were resuspended in cRPMI and kept on ice prior to use.
Primary ALL patient samples
Bone marrow samples were acquired at diagnosis from pediatric B-ALL patients. Mononuclear cells were enriched by centrifugation in Ficoll-Paque (GE Healthcare, cat. 17-1440-02) and aliquoted for storage in liquid nitrogen until use. Samples were thawed in a 37°C water bath, resuspended in 20% FBS cRPMI, and centrifuged to remove freezing medium. Cells were resuspended in conditioned medium (DMEM, 10% FBS, 50 U/ml penicillin-streptomycin, 2 mM L-glutamine) from HS-5 stromal cell cultures.
cAMP efflux assay
Cells were loaded with F-cAMP as described [41]. Briefly, cells were resuspended in a hypertonic solution containing 10% polyethylene glycol 1000, 0.5 M sucrose, and 250 µM F-cAMP in cRPMI without FBS, incubated 10 min at room temperature, washed, and then resuspended in hypotonic solution (60% cRPMI, 40% sterile water) for 2 min at room temperature to complete F-cAMP loading. Cells were resuspended in cRPMI and equilibrated for 2 h under normal culture conditions.
For testing cAMP efflux, an aliquot of loaded cells was kept at 4°C to serve as control, and the remaining cells were incubated in the presence of vehicle or compounds overnight. Samples were evaluated using Accuri C6 or BD FACScan flow cytometers (Becton Dickinson (BD)).
High throughput screening (HTS)
Liquid handling was accomplished with a Biomek FX Multichannel system (Beckman-Coulter, Fullerton, CA, USA) and/or Biotek Multiflo system (Winooski, VT, USA). F-cAMP-loaded U937 cells (2000/well) were seeded into 384-well polypropylene plates (Greiner Bio-One 784201, Monroe, NC, USA). Cells were treated with compounds (10 µM final concentration) from the Prestwick Chemical Library (Illkirch, France) or SPECTRUM Collection (MicroSource, Gaylordsville, CT, USA) delivered by pintool (V&P Scientific, San Diego, CA), at a final DMSO concentration of 1%. Plates were foil-sealed with AlumaSeal 384™ (Excel Scientific, Victorville, CA, USA), and incubated inverted overnight. Sample plates were analyzed by CyAn flow cytometers (Beckman-Coulter) configured with HyperCyt high-throughput auto-sampler systems (IntelliCyt, Albuquerque, NM, USA) using 488 nm excitation to assess FL-1 (530/40) MFI levels.
Data were analyzed with HyperView software (IntelliCyt) and time-gated to determine data from each well. Wells with ≥ 50 events were evaluated for FL-1 MFI values. Samples with MFI values ≥ 2 standard deviations above the plate mean negative control values were considered “hit” compounds.
Compound validation
To validate sample data and decrease the number of false-positive hits, compounds were assayed in a high-throughput dose response assay. Plates were set up as in the HTS, with the exception that the plate formats contained 10-well dose responses for each hit compound, at final concentrations ranging from 30 µM to 4 nM.
For B-ALL cell lines, tests of hit compound inhibition of cAMP efflux were conducted as in the HTS, with compounds in 10-well dose responses ranging from 100 µM - 1.53 nM, at cell densities of 5000/well.
The dose response data were normalized for percent response based on sample FL-1 MFI values in comparison to untreated F-cAMP-loaded cells kept at 4°C overnight or at time = 0, and were fitted by GraphPad Prism 5 software (GraphPad Software, La Jolla, CA, USA) for sigmoidal dose response with constrained hill slope = 1. Compounds with S-shaped dose response curves and EC50 values less than 3 μM were selected for secondary assays.
Detection of CREB/AFT-1 phosphorylation in response to ICE
Cells were suspended at 106 cells/ml in cRPMI and incubated for 1 hour at 37oC with 20 µM ICE compounds or forskolin (positive control). Negative control samples were treated with DMSO at equal volume. Cells were fixed with 20x volume of pre-warmed 1x solution BD PhosflowTM Lyse/Fix Buffer (cat. 558049), incubated at 37°C for 10 minutes, permeabilized with BD PhosflowTM Perm buffer II (cat. 558052), and incubated on ice for 30 minutes. Permeabilized cells were then washed and stained with BD Phosflow™ Alexa Fluor® 488 Mouse Anti-CREB (pS133) / ATF-1 (pS63) clone J151-21 (cat. 558435) according to the manufacturer’s instructions.
Kinetic analysis of VLA-4 deactivation
Kinetic analysis of the binding and dissociation of the LDV-FITC probe was described previously [42]. Briefly, flow cytometric data were acquired at 37°C while the samples were stirred continuously with a stir bar (Bel-Art Products). First, U937 cells transfected with a non-desensitizing mutant of the Formyl Peptide Receptor FPR1 [43] were analyzed for 30-120 s to establish a baseline. The FPR1 mutant triggers VLA-4 activation, which persists for hundreds of seconds, allowing for integrin deactivation to be detected. Next, the LDV-FITC was added and acquisition was re-established. For cell activation, the high affinity FPR-specific agonist (N-formyl-L-methionyl-L-leucyl-L-phenylalanyl-L-phenylalanine, fMLFF) was added at a saturating concentration (100 nM), and acquisition was re-established. Finally, 40 µM of ICE, forskolin (positive control) or vehicle (negative control) were added. Acquisition was re-established, and data were acquired continuously for up to 1024 s.
The concentration of the LDV-FITC probe used in deactivation experiments (4 nM) was below the dissociation constant (Kd) for its binding to resting VLA-4 (low affinity state, Kd ~12 nM), and above the Kd for physiologically activated VLA-4 (high affinity state, Kd ~1-2 nM). Therefore, the transition from low to high affinity state led to increased binding of the probe. This was detected as an increase in the median fluorescence intensity (MFI). VLA-4 deactivation led to the dissociation of the probe and decreased MFI [21].
Viability
Cell viability was determined with the CellTiter-Glo® Luminescent Cell Viability Assay (Promega). Greiner 655083 had 100 µL cRPMI +/- 4x104 cells/well, and dose response curves of compounds ranged from 100 µM to 1.53 nM (1% DMSO). Wells with DMSO-only served as negative controls. Samples were incubated overnight, and the assay was completed according to the manufacturer’s protocol. Plates were analyzed using a Victor3V™ 1420 Multilabel Counter (Perkin Elmer).
For primary ALL patient samples, media consisted of 50% cRPMI, 50% conditioned DMEM from HS-5 stromal cell cultures and compounds. Cell densities ranged from 28,000-68,000 cells/well, and plates were foil-sealed prior to incubation.
Determination of cell viability by 7-AAD exclusion
Cells treated with ICE and control samples were incubated with 1 µg/ml of 7-AAD for 20-60 minutes at 4°C on a rotator in the dark, and analyzed by flow cytometry according to manufacturer instructions. Gates were set on 7-AAD negative and positive events based on histograms. Percent of 7-AAD negative events is shown.
Apoptosis
Apoptosis was determined using MultiCyt® 4-Plex Apoptosis Screening Kit (IntelliCyt, cat. 90053). The kit provides four different apoptosis endpoints that include effector caspases 3 and 7 activation, phosphatidylserine surface expression, mitochondrial membrane depolarization and cell membrane integrity. The activation of caspases is detected using NucView™ 488 Caspase3/7-specific substrate that exhibits increase in fluorescence upon cleavage by activated enzyme. Surface phosphatidylserine is detected by the binding of labelled annexin-V. Another fluorescent dye accumulates in intact mitochondria and upon mitochondrial membrane depolarization it leaks into the cytoplasm and exhibits a decrease in fluorescence. Cell membrane damage is detected by the uptake of a proprietary DNA intercalating agent analogous to 7-aminoactinomycin D.
384-well plates (Greiner 784201), pre-loaded with 5 µL cRPMI were stamped using a pintool with 100 nl of DMSO-solubilized compounds from the 100-fold concentrated stock solution. Next, 5 µL U937 cells (2 x 106/ml in cRPMI) were added to wells and mixed, and plates were foil-sealed. Compounds (Table 1) were tested in dose response at final concentrations ranging from 15.2 nM to 100 µM (1% DMSO final). Vehicle (DMSO) was used as a negative control. After 24 h incubation under culture conditions, the IntelliCyt MultiCyt 4-Plex Apoptosis Screening Kit was used according to manufacturer’s protocol. The data were acquired using iQue Screener Platform (IntelliCyt). Singlet cell populations were analyzed for individual measures of apoptosis, and gates were set based on control histograms.
Quantitation of ABCC4
ABCC4 transporters in fixed B-ALL cell lines were enumerated by flow cytometry using: primary mouse anti-human ABCC4 or IgG1 negative isotype-matched control (Abcam®, Cambridge, MA, USA), FITC-conjugated AffiniPure F(ab’)2 of goat anti-mouse IgG secondary antibody (Jackson Laboratories, West Grove, PA, USA), and Quantum™ Simply Cellular® anti-human IgG calibration beads (Bangs Laboratories, Fishers, IN, USA) according to manufacturers’ protocols. Post-labeling, all samples were analyzed for FL-1 MFI. The calibration beads were used to generate a linear regression, which associated MFI to the antibody-binding capacity of the beads. This regression was used to calculate the number of antibody binding sites (ABS) per cell sample. For each cell type, the calculated ABS for isotype control samples were subtracted from ABS of the ABCC4 samples to determine specific binding sites.
Data analysis
The kinetic data were converted to MFI versus time using FCSQuery software developed by Dr. Bruce Edwards (University of New Mexico). Analysis of apoptosis was done using ForeCyt software (IntelliCyt). Curve fits and statistics were done using GraphPad Prism software version 5.01 (GraphPad Software), as described in figure captions.
ACKNOWLEDGMENTS
We would like to thanks Dr. Bruce Edwards (University of New Mexico) for providing FCSQuery and HyperView software and Dr. Anna Waller for thoughtful input and discussions.
CONFLICTS OF INTEREST
Larry A. Sklar is a co-inventor of HyperCyt, a high throughput flow cytometry platform used for drug identification utilized in the current manuscript, and is a co-founder of IntelliCyt, a company that manufactures HyperCyt. Other authors declare no conflicts of interest.
GRANT SUPPORT
This work was supported by The Oxnard Foundation (to A.C. and L.A.S.), University of New Mexico Clinical & Translational Science Center Pilot Award 1UL1RR031977 (to A.C.), CCSG P30 CA118100 (Willman), and New Mexico Cancer Nanotechnology Training Center grant R25CA153825 (to D.R.P).
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. Elsevier Inc.; 2011; 144: 646-74.
2. Huseby S, Gausdal G, Keen TJ, Kjærland E, Krakstad C, Myhren L, Brønstad K, Kunick C, Schwede F, Genieser H-G, Kleppe R, Døskeland SO. Cyclic AMP induces IPC leukemia cell apoptosis via CRE-and CDK-dependent Bim transcription. Cell Death Dis. 2011; 2: e237.
3. Insel P, Zhang L, Murray F, Yokouchi H, Zambon A. Cyclic AMP is both a pro-apoptotic and anti-apoptotic second messenger. Acta Physiol (Oxf). 2012; 204: 277-87.
4. García-Bermejo L, Pérez C, Vilaboa NE, de Blas E, Aller P. cAMP increasing agents attenuate the generation of apoptosis by etoposide in promonocytic leukemia cells. J Cell Sci. 1998; 111 ( Pt 5: 637-44).
5. Conti M, Mika D, Richter W. Cyclic AMP compartments and signaling specificity: role of cyclic nucleotide phosphodiesterases. J Gen Physiol. 2014; 143: 29-38.
6. Murray F, Insel PA. Targeting cAMP in chronic lymphocytic leukemia: a pathway-dependent approach for the treatment of leukemia and lymphoma. Expert Opin Ther Targets. Informa UK, Ltd.; 2013; 17: 937-49.
7. Gjertsen BT, Cressey LI, Ruchaud S, Houge G, Lanotte M, Døskeland SO. Multiple apoptotic death types triggered through activation of separate pathways by cAMP and inhibitors of protein phosphatases in one (IPC leukemia) cell line. J Cell Sci. 1994; 107 (Pt 1: 3363-77).
8. Follin-Arbelet V, Torgersen ML, Naderi EH, Misund K, Sundan A, Blomhoff HK. Death of multiple myeloma cells induced by cAMP-signaling involves downregulation of Mcl-1 via the JAK/STAT pathway. Cancer Lett. Elsevier Ireland Ltd; 2013; 335: 323-31.
9. Zhang L, Insel PA. The pro-apoptotic protein Bim is a convergence point for cAMP/protein kinase A- and glucocorticoid-promoted apoptosis of lymphoid cells. J Biol Chem. 2004; 279: 20858-65.
10. Coffino P, Bourne HR, Tomkins GM. Mechanism of lymphoma cell death induced by cyclic AMP. Am J Pathol. 1975; 81: 199-204.
11. Conley JM, Brand CS, Bogard AS, Pratt EPS, Xu R, Hockerman GH, Ostrom RS, Dessauer CW, Watts VJ. Development of a high-throughput screening paradigm for the discovery of small-molecule modulators of adenylyl cyclase: identification of an adenylyl cyclase 2 inhibitor. J Pharmacol Exp Ther. 2013; 347: 276-87.
12. Lerner A, Epstein PM. Cyclic nucleotide phosphodiesterases as targets for treatment of haematological malignancies. Biochem J. 2006; 393: 21-41.
13. Meyers JA, Su DW, Lerner A. Chronic lymphocytic leukemia and B and T cells differ in their response to cyclic nucleotide phosphodiesterase inhibitors. J Immunol. 2009; 182: 5400-11.
14. Dou A, Wang X. Cyclic adenosine monophosphate signal pathway in targeted therapy of lymphoma. Chin Med J (Engl). 2010; 123: 95-9.
15. Scavennec J, Carcassonne Y, Gastaut J, Scavennec J. Relationship between the levels of cyclic cytidine 3′:5′ cyclic adenosine 3′:5′-monophosphate in urines and leukocytes and the type of human leukemias. Cancer Res. 1981; 41: 3222-7.
16. Peracchi M, Lombardi L, Maiolo A, Bamonti-Catena F, Toschi V, Chiorboli O, Mozzana R, Polli E. Plasma and urine cyclic nucleotide levels in patients with acute and chronic leukemia. Blood. 1983; 61: 429-34.
17. Zambon AC, Wilderman A, Ho A, Insel PA. Increased expression of the pro-apoptotic protein BIM, a mechanism for cAMP/protein kinase A (PKA)-induced apoptosis of immature T cells. J Biol Chem. 2011; 286: 33260-7.
18. Myklebust JH, Josefsen D, Blomhoff HK, Levy FO, Naderi S, Reed JC, Smeland EB. Activation of the cAMP signaling pathway increases apoptosis in human B- precursor cells and is associated with downregulation of Mcl-1 expression. J Cell Physiol. 1999; 180: 71-80.
19. Copsel S, Garcia C, Diez F, Vermeulem M, Baldi A, Bianciotti LG, Russel FGM, Shayo C, Davio C. Multidrug resistance protein 4 (MRP4/ABCC4) regulates cAMP cellular levels and controls human leukemia cell proliferation and differentiation. J Biol Chem. 2011; 286: 6979-88.
20. Russel FGM, Koenderink JB, Masereeuw R. Multidrug resistance protein 4 (MRP4/ABCC4): a versatile efflux transporter for drugs and signalling molecules. Trends Pharmacol Sci. 2008; 29: 200-7.
21. Chigaev A, Waller A, Amit O, Sklar LA. Galphas-coupled receptor signaling actively down-regulates alpha4beta1-integrin affinity: a possible mechanism for cell de-adhesion. BMC Immunol. 2008; 9: 26.
22. Chigaev A, Smagley Y, Sklar LA. Nitric oxide/cGMP pathway signaling actively down-regulates α4β1-integrin affinity: an unexpected mechanism for inducing cell de-adhesion. BMC Immunol. BioMed Central Ltd; 2011; 12: 28.
23. Kumar S, Kostin S, Flacke JP, Reusch HP, Ladilov Y. Soluble adenylyl cyclase controls mitochondria-dependent apoptosis in coronary endothelial cells. J Biol Chem. 2009; 284: 14760-8.
24. Morrissey KM, Wen CC, Johns SJ, Zhang L, Huang S-M, Giacomini KM. The UCSF-FDA TransPortal: a public drug transporter database. Clin Pharmacol Ther. 2012; 92: 545-6.
25. Borst P, de Wolf C, van de Wetering K. Multidrug resistance-associated proteins 3, 4, and 5. Pflugers Arch. 2007; 453: 661-73.
26. Takeuchi K, Shibata M, Kashiyama E, Umehara K. Expression levels of multidrug resistance-associated protein 4 (MRP4) in human leukemia and lymphoma cell lines, and the inhibitory effects of the MRP-specific inhibitor MK-571 on methotrexate distribution in rats. Exp Ther Med. 2012; 4: 524-32.
27. Oevermann L, Scheitz J, Starke K, Köck K, Kiefer T, Dölken G, Niessen J, Greinacher A, Siegmund W, Zygmunt M, Kroemer HK, Jedlitschky G, Ritter CA. Hematopoietic stem cell differentiation affects expression and function of MRP4 (ABCC4), a transport protein for signaling molecules and drugs. Int J Cancer. 2009; 124: 2303-11.
28. Baumann K. Stem cells: A metabolic switch. Nat Rev Mol Cell Biol. Nature Publishing Group; 2013; 14: 64-5.
29. Felipe Rico J, Hassane DC, Guzman ML. Acute myelogenous leukemia stem cells: From Bench to Bedside. Cancer Lett. Elsevier Ireland Ltd; 2013; 338: 4-9.
30. Chen ZS, Tiwari AK. Multidrug resistance proteins (MRPs/ABCCs) in cancer chemotherapy and genetic diseases. FEBS J. 2011; 278: 3226-45.
31. Neelakantan S, Nasim S, Guzman ML, Jordan CT, Crooks PA. Aminoparthenolides as novel anti-leukemic agents: Discovery of the NF-kappaB inhibitor, DMAPT (LC-1). Bioorg Med Chem Lett. Elsevier Ltd; 2009; 19: 4346-9.
32. Gerlo S, Kooijman R, Beck IM, Kolmus K, Spooren A, Haegeman G. Cyclic AMP: a selective modulator of NF-κB action. Cell Mol Life Sci. 2011; 68: 3823-41.
33. Naviglio S, Caraglia M, Abbruzzese A, Chiosi E, Di Gesto D, Marra M, Romano M, Sorrentino A, Sorvillo L, Spina A, Illiano G. Protein kinase A as a biological target in cancer therapy. Expert Opin Ther Targets. 2009; 13: 83-92.
34. Savai R, Pullamsetti SS, Banat GA, Weissmann N, Ghofrani HA, Grimminger F, Schermuly RT. Targeting cancer with phosphodiesterase inhibitors. Expert Opin Investig Drugs. Informa UK Ltd London, UK; 2010; 19: 117-31.
35. Guillemin MC, Raffoux E, Vitoux D, Kogan S, Soilihi H, Lallemand-Breitenbach V, et al. In vivo activation of cAMP signaling induces growth arrest and differentiation in acute promyelocytic leukemia. J Exp Med. 2002; 196: 1373-80.
36. Propper DJ, Saunders MP, Salisbury AJ, Long L, Byrne KJO, Braybrooke JP, Dowsett M, Taylor M, Talbot DC, Ganesan TS, Harris AL. Phase I Study of the Novel Cyclic AMP ( cAMP ) Analogue 8-Chloro- cAMP in Patients with Cancer : Toxicity , Hormonal , and Immunological Effects. 1999; 5: 1682-9.
37. Schwede F, Maronde E, Genieser H, Jastorff B. Cyclic nucleotide analogs as biochemical tools and prospective drugs. 2000; 87: 199-226.
38. Adema AD, Floor K, Smid K, Honeywell RJ, Scheffer GL, Jansen G, Peters GJ. Overexpression of MRP4 (ABCC4) and MRP5 (ABCC5) confer resistance to the nucleoside analogs cytarabine and troxacitabine, but not gemcitabine. Springerplus. 2014; 3: 732.
39. Keppler D. Drug Transporters. Fromm MF, Kim RB, editors. Berlin, Heidelberg: Springer Berlin Heidelberg; 2011; 201.
40. Koczor CA, Torres RA, Lewis W. The role of transporters in the toxicity of nucleoside and nucleotide analogs. Expert Opin Drug Metab Toxicol. 2012; 8: 665-76.
41. Okada CY, Rechsteiner M. Introduction of macromolecules into cultured mammalian cells by osmotic lysis of pinocytic vesicles. Cell. 1982; 29: 33-41.
42. Chigaev A, Sklar LA. Integrin and Cell Adhesion Molecules. Methods Mol Biol. 2012; 757: 55-71.
43. Prossnitz ER. Desensitization of N -Formylpeptide Receptor-mediated Activation Is Dependent upon Receptor Phosphorylation Desensitization of N -Formylpeptide Receptor-mediated Activation Is Dependent upon Receptor Phosphorylation. J Biol Chem. 1997; 272: 15213-9.
44. Ladilov Y, Appukuttan A. Role of soluble adenylyl cyclase in cell death and growth. Biochim Biophys Acta - Mol Basis Dis. Elsevier B.V.; 2014; 1842: 2646-55.
45. Guzman ML, Allan JN. Concise review: leukemia stem cells in personalized medicine. Stem Cells. 2014; 32: 844-51.
46. Schimmer AD, Jitkova Y, Gronda M, Wang Z, Brandwein J, Chen C, Gupta V, Schuh A, Yee K, Chen J, Ackloo S, Booth T, Keays S, Minden MD. A phase I study of the metal ionophore clioquinol in patients with advanced hematologic malignancies. Clin Lymphoma Myeloma Leuk. Elsevier; 2012; 12: 330-6.
47. Chen W, Luo Y, Liu L, Zhou H, Xu B, Han X, Shen T, Liu Z, Lu Y, Huang S. Cryptotanshinone inhibits cancer cell proliferation by suppressing Mammalian target of rapamycin-mediated cyclin D1 expression and Rb phosphorylation. Cancer Prev Res (Phila). 2010; 3: 1015-25.
48. Guzman ML, Rossi RM, Neelakantan S, Li X, Corbett CA, Hassane DC, et al. An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood. 2007; 110: 4427-35.
49. Wu TS, Liao YC, Yu FY, Chang CH, Liu BH. Mechanism of patulin-induced apoptosis in human leukemia cells (HL-60). Toxicol Lett. 2008; 183: 105-11.
50. Andersson A, Edén P, Lindgren D, Nilsson J, Lassen C, Heldrup J, Fontes M, Borg A, Mitelman F, Johansson B, Höglund M, Fioretos T. Gene expression profiling of leukemic cell lines reveals conserved molecular signatures among subtypes with specific genetic aberrations. Leukemia. 2005; 19: 1042-50.
51. Wu TS, Yu FY, Su CC, Kan JC, Chung CP, Liu BH. Activation of ERK mitogen-activated protein kinase in human cells by the mycotoxin patulin. Toxicol Appl Pharmacol. 2005; 207: 103-11.
52. Fribley AM, Cruz PG, Miller JR, Callaghan MU, Cai P, Narula N, Neubig RR, Showalter HD, Larsen SD, Kirchhoff PD, Larsen MJ, Burr DA, Schultz PJ. Complementary cell-based high-throughput screens identify novel modulators of the unfolded protein response. J Biomol Screen. 2011; 16: 825-35.
53. Hothi P, Martins TJ, Chen L, Deleyrolle L, Reynolds B, Foltz G. High-Throughput Chemical Screens Identify Disulfiram as an Inhibitor of Human Glioblastoma Stem Cells Abstract : Oncotarget. 2012; 3: 1124-36. doi:10.18632/oncotarget.707.
54. Kawauchiya T, Takumi R, Kudo Y, Takamori A, Sasagawa T, Takahashi K, Kikuchi H. Correlation between the destruction of tight junction by patulin treatment and increase of phosphorylation of ZO-1 in Caco-2 human colon cancer cells. Toxicol Lett. Elsevier Ireland Ltd; 2011; 205: 196-202.
55. Mohan HM, Collins D, Maher S, Walsh EG, Winter DC, O’Brien PJ, Brayden DJ, Baird a W. The mycotoxin patulin increases colonic epithelial permeability in vitro. Food Chem Toxicol. Elsevier Ltd; 2012; 50: 4097-102.
56. McLaughlin J, Lambert D, Padfield PJ, Burt JPH, O’Neill CA. The mycotoxin patulin, modulates tight junctions in caco-2 cells. Toxicol In Vitro. Elsevier Ltd; 2009; 23: 83-9.
57. Kwon O, Soung NK, Thimmegowda NR, Jeong SJ, Jang JH, Moon DO, Chung JK, Lee KS, Kwon YT, Erikson RL, Ahn JS, Kim BY. Patulin induces colorectal cancer cells apoptosis through EGR-1 dependent ATF3 up-regulation. Cell Signal. Elsevier Inc.; 2012; 24: 943-50.
58. Liu BH, Wu TS, Yu FY, Su CC. Induction of oxidative stress response by the mycotoxin patulin in mammalian cells. Toxicol Sci. 2007; 95: 340-7.
59. Zhou S, Jiang L, Geng C, Cao J, Zhong L. Patulin-induced oxidative DNA damage and p53 modulation in HepG2 cells. Toxicon. Elsevier Ltd; 2010; 55: 390-5.
60. Czyz M, Koprowska K, Sztiller-Sikorska M. Parthenolide reduces the frequency of ABCB5-positive cells and clonogenic capacity of melanoma cells from anchorage independent melanospheres. Cancer Biol Ther. 2013; 14: 135-45.
61. Czyz M, Lesiak-Mieczkowska K, Koprowska K, Szulawska-Mroczek A, Wozniak M. Cell context-dependent activities of parthenolide in primary and metastatic melanoma cells. Br J Pharmacol. 2010; 160: 1144-57.
62. Hehner SP, Hofmann TG, Dröge W, Schmitz ML. The antiinflammatory sesquiterpene lactone parthenolide inhibits NF-kappa B by targeting the I kappa B kinase complex. J Immunol. 1999; 163: 5617-23.
63. Kwok BHB, Koh B, Ndubuisi MI, Elofsson M, Crews CM. The anti-inflammatory natural product parthenolide from the medicinal herb Feverfew directly binds to and inhibits IkappaB kinase. Chem Biol. 2001; 8: 759-66.
64. Dai Y, Guzman ML, Chen S, Wang L, Yeung SK, Pei XY, Dent P, Jordan CT, Grant S. The NF (Nuclear factor)-κB inhibitor parthenolide interacts with histone deacetylase inhibitors to induce MKK7/JNK1-dependent apoptosis in human acute myeloid leukaemia cells. Br J Haematol. 2010; 151: 70-83.
65. Steele AJ, Jones DT, Ganeshaguru K, Duke VM, Yogashangary BC, North JM, Lowdell MW, Kottaridis PD, Mehta AB, Prentice AG, Hoffbrand A V, Wickremasinghe RG. The sesquiterpene lactone parthenolide induces selective apoptosis of B-chronic lymphocytic leukemia cells in vitro. Leukemia. 2006; 20: 1073-9.
66. Zhou J, Zhang H, Gu P, Bai J, Margolick JB, Zhang Y. NF-kappaB pathway inhibitors preferentially inhibit breast cancer stem-like cells. Breast Cancer Res Treat. 2008; 111: 419-27.
67. Chen T, Li M, Zhang R, Wang H. Dihydroartemisinin induces apoptosis and sensitizes human ovarian cancer cells to carboplatin therapy. J Cell Mol Med. 2009; 13: 1358-70.
68. Disbrow GL, Baege AC, Kierpiec KA, Yuan H, Centeno JA, Thibodeaux CA, Hartmann D, Schlegel R. Dihydroartemisinin is cytotoxic to papillomavirus-expressing epithelial cells in vitro and in vivo. Cancer Res. 2005; 65: 10854-61.
69. Dell’Eva R, Pfeffer U, Vené R, Anfosso L, Forlani A, Albini A, Efferth T. Inhibition of angiogenesis in vivo and growth of Kaposi’s sarcoma xenograft tumors by the anti-malarial artesunate. Biochem Pharmacol. 2004; 68: 2359-66.
70. Jiang Z, Chai J, Chuang HHF, Li S, Wang T, Cheng Y, Chen W, Zhou D. Artesunate induces G0/G1 cell cycle arrest and iron-mediated mitochondrial apoptosis in A431 human epidermoid carcinoma cells. Anticancer Drugs. 2012; 23: 606-13.
71. Bachmeier B, Fichtner I, Killian PH, Kronski E, Pfeffer U, Efferth T. Development of resistance towards artesunate in MDA-MB-231 human breast cancer cells. PLoS One. 2011; 6: e20550.
72. Hamacher-Brady A, Stein HA, Turschner S, Toegel I, Mora R, Jennewein N, Efferth T, Eils R, Brady NR. Artesunate activates mitochondrial apoptosis in breast cancer cells via iron-catalyzed lysosomal reactive oxygen species production. J Biol Chem. 2011; 286: 6587-601.
73. Hou J, Wang D, Zhang R, Wang H. Experimental therapy of hepatoma with artemisinin and its derivatives: in vitro and in vivo activity, chemosensitization, and mechanisms of action. Clin Cancer Res. 2008; 14: 5519-30.
74. Du JH, Zhang H De, Ma ZJ, Ji KM. Artesunate induces oncosis-like cell death in vitro and has antitumor activity against pancreatic cancer xenografts in vivo. Cancer Chemother Pharmacol. 2010; 65: 895-902.
75. Youns M, Efferth T, Reichling J, Fellenberg K, Bauer A, Hoheisel JD. Gene expression profiling identifies novel key players involved in the cytotoxic effect of Artesunate on pancreatic cancer cells. Biochem Pharmacol. 2009; 78: 273-83.
76. Chen H, Shi L, Yang X, Li S, Guo X, Pan L. Artesunate inhibiting angiogenesis induced by human myeloma RPMI8226 cells. Int J Hematol. 2010; 92: 587-97.
77. Efferth T, Davey M, Olbrich A, Rücker G, Gebhart E, Davey R. Activity of drugs from traditional chinese medicine toward sensitive and MDR1- or MRP1-overexpressing multidrug-resistant human CCRF-CEM leukemia cells. Blood Cells, Mol Dis. 2002; 28: 160-8.
78. Efferth T, Giaisi M, Merling A, Krammer PH, Li-Weber M. Artesunate induces ROS-mediated apoptosis in doxorubicin-resistant T leukemia cells. PLoS One. 2007; 2: e693.
79. Steinbrück L, Pereira G, Efferth T. Effects of artesunate on cytokinesis and G2/M cell cycle progression of tumour cells and budding yeast. Cancer Genomics Proteomics. 2010; 7: 337-46.
80. Zhou HJ, Wang WQ, Wu GD, Lee J, Li A. Artesunate inhibits angiogenesis and downregulates vascular endothelial growth factor expression in chronic myeloid leukemia K562 cells. Vascul Pharmacol. 2007; 47: 131-8.
81. Li LN, Zhang HD, Yuan SJ, Yang DX, Wang L, Sun ZX. Differential sensitivity of colorectal cancer cell lines to artesunate is associated with expression of beta-catenin and E-cadherin. Eur J Pharmacol. 2008; 588: 1-8.
82. Michaelis M, Kleinschmidt MC, Barth S, Rothweiler F, Geiler J, Breitling R, et al. Anti-cancer effects of artesunate in a panel of chemoresistant neuroblastoma cell lines. Biochem Pharmacol. 2010; 79: 130-6.
83. Rasheed SAK, Efferth T, Asangani IA, Allgayer H. First evidence that the antimalarial drug artesunate inhibits invasion and in vivo metastasis in lung cancer by targeting essential extracellular proteases. Int J Cancer. 2010; 127: 1475-85.
84. Xu Q, Li ZX, Peng HQ, Sun ZW, Cheng RL, Ye ZM, Li WX. Artesunate inhibits growth and induces apoptosis in human osteosarcoma HOS cell line in vitro and in vivo. J Zhejiang Univ Sci B. 2011; 12: 247-55.
85. Jiao Y, Ge C, Meng Q, Cao J, Tong J, Fan S. Dihydroartemisinin is an inhibitor of ovarian cancer cell growth. Acta Pharmacol Sin. 2007; 28: 1045-56.
86. Ba Q, Zhou N, Duan J, Chen T, Hao M, Yang X, Li J, Yin J, Chu R, Wang H. Dihydroartemisinin exerts its anticancer activity through depleting cellular iron via transferrin receptor-1. PLoS One. 2012; 7: e42703.
87. Zhang CZ, Zhang H, Yun J, Chen GG, Lai PBS. Dihydroartemisinin exhibits antitumor activity toward hepatocellular carcinoma in vitro and in vivo. Biochem Pharmacol. Elsevier Inc.; 2012; 83: 1278-89.
88. Cabello CM, Lamore SD, Bair WB, Qiao S, Azimian S, Lesson JL, Wondrak GT. The redox antimalarial dihydroartemisinin targets human metastatic melanoma cells but not primary melanocytes with induction of NOXA-dependent apoptosis. Invest New Drugs. 2012; 30: 1289-301.
89. Chen H, Sun B, Pan S, Jiang H, Sun X. Dihydroartemisinin inhibits growth of pancreatic cancer cells in vitro and in vivo. Anticancer Drugs. 2009; 20: 131-40.
90. Chen H, Sun B, Wang S, Pan S, Gao Y, Bai X, Xue D. Growth inhibitory effects of dihydroartemisinin on pancreatic cancer cells: involvement of cell cycle arrest and inactivation of nuclear factor-kappaB. J Cancer Res Clin Oncol. 2010; 136: 897-903.
91. Kong R, Jia G, Cheng Z, Wang Y, Mu M, Wang S, Pan S, Gao Y, Jiang H, Dong D, Sun B. Dihydroartemisinin enhances Apo2L/TRAIL-mediated apoptosis in pancreatic cancer cells via ROS-mediated up-regulation of death receptor 5. PLoS One. 2012; 7: e37222.
92. Wang SJ, Gao Y, Chen H, Kong R, Jiang HC, Pan SH, Xue D-B, Bai X-W, Sun B. Dihydroartemisinin inactivates NF-kappaB and potentiates the anti-tumor effect of gemcitabine on pancreatic cancer both in vitro and in vivo. Cancer Lett. Elsevier Ireland Ltd; 2010; 293: 99-108.
93. Wang SJ, Sun B, Cheng ZX, Zhou HX, Gao Y, Kong R, Chen H, Jiang HC, Pan SH, Xue DB, Bai XW. Dihydroartemisinin inhibits angiogenesis in pancreatic cancer by targeting the NF-κB pathway. Cancer Chemother Pharmacol. 2011; 68: 1421-30.
94. Chen M, Chen T, Lu Y, Liu C, Qu J. Dihydroarteminsin-induced apoptosis is not dependent on the translocation of Bim to the endoplasmic reticulum in human lung adenocarcinoma cells. Pathol Oncol Res. 2012; 18: 809-16.
95. Lu YY, Chen TS, Wang XP, Li L. Single-cell analysis of dihydroartemisinin-induced apoptosis through reactive oxygen species-mediated caspase-8 activation and mitochondrial pathway in ASTC-a-1 cells using fluorescence imaging techniques. J Biomed Opt. 2013; 15: 046028.
96. Zhou HJ, Zhang JL, Li A, Wang Z, Lou XE. Dihydroartemisinin improves the efficiency of chemotherapeutics in lung carcinomas in vivo and inhibits murine Lewis lung carcinoma cell line growth in vitro. Cancer Chemother Pharmacol. 2010; 66: 21-9.
97. Gao N, Budhraja A, Cheng S, Liu E-H, Huang C, Chen J, Yang Z, Chen D, Zhang Z, Shi X. Interruption of the MEK/ERK signaling cascade promotes dihydroartemisinin-induced apoptosis in vitro and in vivo. Apoptosis. 2011; 16: 511-23.
98. Handrick R, Ontikatze T, Bauer K-D, Freier F, Rübel A, Dürig J, Belka C, Jendrossek V. Dihydroartemisinin induces apoptosis by a Bak-dependent intrinsic pathway. Mol Cancer Ther. 2010; 9: 2497-510.
99. Lee J, Zhou HJ, Wu XH. Dihydroartemisinin downregulates vascular endothelial growth factor expression and induces apoptosis in chronic myeloid leukemia K562 cells. Cancer Chemother Pharmacol. 2006; 57: 213-20.
100. Lu J, Meng L, Cai Y, Chen Q, Tong L, Lin L, Ding J. Dihydroartemisinin induces apoptosis in HL-60 leukemia cells dependent of iron and p38 mitogen-activated protein kinase activation but independent of reactive oxygen species. Cancer Biol Ther. 2008; 7: 1017-23.
101. Wang Z, Hu W, Zhang JL, Wu XH, Zhou HJ. Dihydroartemisinin induces autophagy and inhibits the growth of iron-loaded human myeloid leukemia K562 cells via ROS toxicity. FEBS Open Bio. Federation of European Biochemical Societies; 2012; 2: 103-12.
102. Wu XH, Zhou HJ, Lee J. Dihydroartemisinin inhibits angiogenesis induced by multiple myeloma RPMI8226 cells under hypoxic conditions via downregulation of vascular endothelial growth factor expression and suppression of vascular endothelial growth factor secretion. Anticancer Drugs. 2006; 17: 839-48.
103. He Q, Shi J, Shen XL, An J, Sun H, Wang L, Hu YJ, Sun Q, Fu LC, Sheikh MS, Huang Y. Dihydroartemisinin upregulates death receptor 5 expression and cooperates with TRAIL to induce apoptosis in human prostate cancer cells. Cancer Biol Ther. 2010; 9: 819-24.
104. Huang XJ, Ma ZQ, Zhang WP, Lu YB, Wei EQ. Dihydroartemisinin exerts cytotoxic effects and inhibits hypoxia inducible factor-1alpha activation in C6 glioma cells. J Pharm Pharmacol. 2007; 59: 849-56.
105. Ji Y, Zhang YC, Pei LB, Shi LL, Yan JL, Ma XH. Anti-tumor effects of dihydroartemisinin on human osteosarcoma. Mol Cell Biochem. 2011; 351: 99-108.
106. Cao B, Li J, Zhu J, Shen M, Han K, Zhang Z, Yu Y, Wang Y, Wu D, Chen S, Sun A, Tang X, Zhao Y, Qiao C, Hou T, Mao X. The antiparasitic clioquinol induces apoptosis in leukemia and myeloma cells by inhibiting histone deacetylase activity. J Biol Chem. 2013; 288: 34181-9.
107. Mao X, Li X, Sprangers R, Wang X, Venugopal a, Wood T, Zhang Y, Kuntz DA, Coe E, Trudel S, Rose D, Batey RA, Kay LE, Schimmer AD. Clioquinol inhibits the proteasome and displays preclinical activity in leukemia and myeloma. Leukemia. 2009; 23: 585-90.
108. Ding W, Liu B, Vaught JL, Yamauchi H, Lind SE. Anticancer activity of the antibiotic clioquinol. Cancer Res. 2005; 65: 3389-95.
109. Daniel KG, Chen D, Orlu S, Cui QC, Miller FR, Dou QP. Clioquinol and pyrrolidine dithiocarbamate complex with copper to form proteasome inhibitors and apoptosis inducers in human breast cancer cells. Breast Cancer Res. 2005; 7: R897-908.
110. Zheng J, Benbrook DM, Yu H, Ding WQ. Clioquinol suppresses cyclin D1 gene expression through transcriptional and post-transcriptional mechanisms. Anticancer Res. 2011; 31: 2739-47.
111. Du T, Filiz G, Caragounis A, Crouch PJ, White AR. Clioquinol promotes cancer cell toxicity through tumor necrosis factor alpha release from macrophages. J Pharmacol Exp Ther. 2008; 324: 360-7.
112. Yu H, Zhou Y, Lind SE, Ding WQ. Clioquinol targets zinc to lysosomes in human cancer cells. Biochem J. 2009; 417: 133-9.
113. Yu H, Lou JR, Ding WQ. Clioquinol independently targets NF-kappaB and lysosome pathways in human cancer cells. Anticancer Res. 2010; 30: 2087-92.
114. Chen D, Cui QC, Yang H, Barrea RA, Sarkar FH, Sheng S, Yan B, Reddy GPV, Dou QP. Clioquinol, a therapeutic agent for Alzheimer’s disease, has proteasome-inhibitory, androgen receptor-suppressing, apoptosis-inducing, and antitumor activities in human prostate cancer cells and xenografts. Cancer Res. 2007; 67: 1636-44.
115. Ge Y, Cheng R, Zhou Y, Shen J, Peng L, Xu X, Dai Q, Liu P, Wang H, Ma X, Jia J, Chen Z. Cryptotanshinone induces cell cycle arrest and apoptosis of multidrug resistant human chronic myeloid leukemia cells by inhibiting the activity of eukaryotic initiation factor 4E. Mol Cell Biochem. 2012; 368: 17-25.
116. Jung JH, Kwon TR, Jeong SJ, Kim EO, Sohn EJ, Yun M, Kim SH. Apoptosis induced by tanshinone IIA and cryptotanshinone is mediated by distinct JAK/STAT3/5 and SHP1/2 signaling in chronic myeloid leukemia K562 Cells. Evid Based Complement Alternat Med. 2013; 2013: 805639.
117. Shin DS, Kim HN, Shin KD, Yoon YJ, Kim SJ, Han DC, Kwon BM. Cryptotanshinone inhibits constitutive signal transducer and activator of transcription 3 function through blocking the dimerization in DU145 prostate cancer cells. Cancer Res. 2009; 69: 193-202.
118. Wu CY, Hsieh CY, Huang KE, Chang C, Kang HY. Cryptotanshinone down-regulates androgen receptor signaling by modulating lysine-specific demethylase 1 function. Int J Cancer. 2012; 131: 1423-34.
119. Nizamutdinova IT, Lee GW, Son KH, Jeon SJ, Kang SS, Kim YS, Lee JH, Seo HG, Chang KC, Kim HJ. Tanshinone I effectively induces apoptosis in estrogen receptor- positive (MCF-7) and estrogen receptor-negative (MDA-MB-231) breast cancer cells. Int J Oncol. 2008; 33: 485-91.
120. Chen L, Zheng S, Sun Z, Wang A, Huang C, Punchard N a, Huang S, Gao X, Lu Y. Cryptotanshinone has diverse effects on cell cycle events in melanoma cell lines with different metastatic capacity. Cancer Chemother Pharmacol. 2011; 68: 17-27.
121. Tse AKW, Chow KY, Cao HH, Cheng CY, Kwan HY, Yu H, Zhu GY, Wu YC, Fong WF, Yu ZL. The herbal compound cryptotanshinone restores sensitivity in cancer cells that are resistant to the tumor necrosis factor-related apoptosis-inducing ligand. J Biol Chem. 2013; 288: 29923-33.
122. Cheng G, Xie L. Parthenolide induces apoptosis and cell cycle arrest of human 5637 bladder cancer cells in vitro. Molecules. 2011; 16: 6758-68.
123. Zunino SJ, Storms DH, Ducore JM. Parthenolide treatment activates stress signaling proteins in high-risk acute lymphoblastic leukemia cells with chromosomal translocation t(4; 11). Int J Oncol. 2010; 37: 1307-13.
124. Kishida Y, Yoshikawa H, Myoui A. Parthenolide, a natural inhibitor of Nuclear Factor-kappaB, inhibits lung colonization of murine osteosarcoma cells. Clin Cancer Res. 2007; 13: 59-67.
125. Liu JW, Cai MX, Xin Y, Wu QS, Ma J, Yang P, Xie HY, Huang DS. Parthenolide induces proliferation inhibition and apoptosis of pancreatic cancer cells in vitro. J Exp Clin Cancer Res. 2010; 29: 108.
126. Kreuger MRO, Grootjans S, Biavatti MW, Vandenabeele P, D’Herde K. Sesquiterpene lactones as drugs with multiple targets in cancer treatment: focus on parthenolide. Anticancer Drugs. 2012; 23: 883-96.
127. Saadane A, Eastman J, Berger M, Bonfield TL. Parthenolide inhibits ERK and AP-1 which are dysregulated and contribute to excessive IL-8 expression and secretion in cystic fibrosis cells. J Inflamm (Lond). BioMed Central Ltd; 2011; 8: 26.
128. Mathema VB, Koh YS, Thakuri BC, Sillanpää M. Parthenolide, a sesquiterpene lactone, expresses multiple anti-cancer and anti-inflammatory activities. Inflammation. 2012; 35: 560-5.
129. Efferth T, Sauerbrey A, Olbrich A, Gebhart E, Rauch P, Weber HO, Hengstler JG, Halatsch M-E, Volm M, Tew KD, Ross DD, Funk JO. Molecular modes of action of artesunate in tumor cell lines. Mol Pharmacol. 2003; 64: 382-94.
130. Konkimalla VB, Blunder M, Korn B, Soomro SA, Jansen H, Chang W, Posner GH, Bauer R, Efferth T. Effect of artemisinins and other endoperoxides on nitric oxide-related signaling pathway in RAW 264.7 mouse macrophage cells. Nitric Oxide. 2008; 19: 184-91.
131. Cheng C, Ho WE, Goh FY, Guan SP, Kong LR, Lai WQ, Leung BP, Wong WSF. Anti-malarial drug artesunate attenuates experimental allergic asthma via inhibition of the phosphoinositide 3-kinase/Akt pathway. PLoS One. 2011; 6: e20932.
132. Saxena N, Ansari KM, Kumar R, Dhawan A, Dwivedi PD, Das M. Patulin causes DNA damage leading to cell cycle arrest and apoptosis through modulation of Bax, p(53) and p(21/WAF1) proteins in skin of mice. Toxicol Appl Pharmacol. Elsevier Inc.; 2009; 234: 192-201.
133. Guo X, Dong Y, Yin S, Zhao C, Huo Y, Fan L, Hu H. Patulin induces pro-survival functions via autophagy inhibition and p62 accumulation. Cell Death Dis. 2013; 4: e822.
134. Lupescu A, Jilani K, Zbidah M, Lang F. Patulin-induced suicidal erythrocyte death. Cell Physiol Biochem. 2013; 32: 291-9.
135. de Melo FT, de Oliveira IM, Greggio S, Dacosta JC, Guecheva TN, Saffi J, Henriques JAP, Rosa RM. DNA damage in organs of mice treated acutely with patulin, a known mycotoxin. Food Chem Toxicol. 2012; 50: 3548-55.