
INTRODUCTION
Cancer remains one of the leading causes of death [1]. This mostly is due to the capacity of malignant cells to metastasize, the unpredictable spread of tumor cells frequently setting the corner stone for curative therapy [2]. Evidence is accumulating that limitations in cancer therapy can be overcome by attacking a small population of cancer-initiating cells (CIC) that are essential for primary tumor and metastatic growth [3]. CIC are characterized by tumorigenicity, self-renewal and differentiation capacity, anchorage independent growth, longevity and drug resistance [4]. CIC are also defined by sets of surface markers [5], which were repeatedly reported to be of functional relevance [6, 7]. In gastrointestinal cancer evidence was provided that EpC acts as a CIC biomarker, but requires support by claudin7 (cld7) [8–10]. Experimental evidence pointing towards a dominance of the cld7 contribution demanded controlling the genuine cld7 activity [9].
Claudins are a family of four-pass tight junction (TJ) proteins [11–13]. The importance of clds, including cld7, was repeatedly demonstrated by targeted deletion (ko). Cld1ko mice die within one day after birth due to severe defects in the barrier functions of the skin [14]. A cld7ko is lethal within 10 days after birth due to destruction of the intestine [15]. The authors speculate on a missing association with integrins and a striking upregulation of MMP9 contributing to gut destruction [15]. An intestine-specific conditional cld7ko mouse revealed a specific enhancement of paracellular small organic solute flux across the TJ, which included N-formyl-L-methionionyl-L-leucyl-L-phenylalanine (fMLP), a major bacterial product that initiates colonic inflammation [16].
However, clds can also be diffusely distributed in lateral membranes [17–20]. This accounts particularly for cld7 [21–23] and was first described for the localization in kidney tubuli [24]. Claudins are PKA, PKC and MLCK targets [25–29], where cld phosphorylation can prohibit integration into TJ, which is accompanied by loss of epithelial cell polarization [30–32]. We expect that multiple phosphorylation sites need to be affected, as we did not observe relocation, when mutating individual serine residues [33]. Instead, our studies confirmed claudin7 palmitoylation and partitioning into glycolipid-enriched membrane microdomains (GEM) [9, 10, 33, 34]. GEM are known to harbor palmitoylated proteins and due to the particular lipid composition to function as a scaffold creating a platform for signal transduction and, via cytoskeleton linker molecules, for reorganization of the cytoskeleton [35–38]. GEM are additionally prone for internalization [39, 40], where GEM membrane and linked cytosolic molecules are recruited into early endosomes, the GEM complexes being maintained and recovered in exosomes [41–43]. In concern about the contribution of clds to oncogenesis and tumor progression, several reports describe TJ proteins preventing or promoting tumor progression [18, 44–46]. Our data pointing towards functional importance of cld7 in tumor progression [9, 10], we want to mention particularly one report on cld7 expression in triple negative breast cancer. The bulk tumor does not express cld7-associated rab25, but expression is seen in CIC [47]. We interpret these data in the sense that cld7 and palmitoylated cld7 account for distinct, non-overlapping activities such that dependent on the cellular context, the functional engagement in TJ or in GEM are dominating. To circumvent the problem of skewed results, we transfected HEK cells with palmitoylation deficient cld7 (cld7mPalm), which confirmed strong enrichment only of palmitoylation-competent cld7 in GEM [33]. To elaborate palmitoylated cld7 selective activities, we here rescued a cld7kd in a metastatic pancreatic tumor line with cld7mPalm.
EpCAM (EpC) is a CIC marker [48], frequently associated with cld7 [9, 36, 49–52]. The oncogenic and tumor progression supporting activity of EpC is due to EpC interfering with E-cadherin-mediated cell-cell adhesion via disrupting the link between α-catenin and F-actin [53] as well as by its engagement in Wnt/β-catenin signaling [54] and by controlling cell movement via down-regulation of PKC [55] and regulation of MMP7 expression [56, 57]. These activities are promoted by the cytoplasmic tail of EpC (EpICD), which forms a complex with β-catenin, FHL2 (four-and-half-LIM-only) and Lef-1, relocates to the nucleus and initiates c-myc, cyclin A and E transcription [58]. EpICD also initiates transcription of reprogramming genes like Oct4 and Nanog, which is accompanied by epithelial-mesenchymal transition (EMT) with upregulation of vimentin, Snail, Slug and downregulation of E-cadherin in a colon cancer and a hepatoma line [59]. Though not evaluated, it is tempting to speculate on a cld7 contribution as hepatocyte progenitors express EpC and cld7 [60] and in colon and pancreatic cancer, EpC is cld7-associated [50]. Under physiological conditions, too, the EpC-cld7 association appears vital. An EpCko, associated with intestine destruction-promoted death within one week after birth, is due to the missing association of EpC with cld7 [61]. These findings pointed towards a concerted activity of EpC and cld7 in tumor progression, which was confirmed by a cld7kd and an EpCkd in a metastasizing line, which both sufficed to wave metastatic growth [9]. As EpC is one of the dominating partners of cld7, we also generated an EpC rescue with a mutation of the cld7-binding site (EpCmAG) to differentiate not only between non-palmitoylated and palmitoylated cld7, but also on the impact of an EpC association.
We confirm that only palmitoylated cld7 promotes tumor progression by supporting motility and invasion, GEM location-dependent activation of the PI3K/Akt pathway and upregulation of mesenchymal genes. Finally, GEM-located palmitoylated cld7 associates with vesicle transporter complexes. This might have severe consequences on exosome delivery and the communication with neighboring tumor, stroma and hematopoietic cells.
RESULTS
Cld7 belongs to the family of TJ proteins, supposed to inhibit tumor progression. However, cld7 is also recovered outside of TJ. We provided evidence that palmitoylated cld7 is enriched in GEM, cooperating with GEM-, but not TJ-located molecules [33]. Indeed, non-palmitoylated versus palmitoylated cld7 exhibit on non-overlapping and opposing activities.
The model
ASML is a metastasizing pancreatic adenocarcinoma [62], highly expressing cld7 and EpC. At least part of the two molecules are associated via a direct protein-protein interaction [50]. A cld7kd as well as an EpCkd are accompanied by loss in metastatic potential [9]. To control for the impact of cld7 palmitoylation, ASML-cld7kd cells were rescued with a palmitoylation site mutated cld7 (ASML-cld7mPalm). A transient cld7 rescue served as control. As ASML-EpCkd cells also do not metastasize, which could be due to palmitoylated cld7 supporting the generation of the cotranscription factor EpICD [33], we additionally generated an ASML-EpCresc line and a rescue line, where the binding site for cld7 is mutated (ASML-EpCmAG). The latter allows judging, whether functional activity of palmitoylated cld7 is strictly linked to associated EpC.
Cld7 has two palmitoylation sites. A palmitoylation assay revealed that mutating AA184 and AA186 prevents cld7 palmitoylation indicating that predominantly the cld7 C-terminal tail is palmitoylated (Figure 1A). ASML-cld7mPalm express cld7 at a comparable level to ASMLwt cells. This also accounts for EpC expression in ASML-EpCresc and ASML-EpCmAG (Figure 1B). Furthermore, cld7 co-immunoprecipitates with EpC, but not with EpCmAG. Cld7mPalm does not coimmunoprecipitate with EpC. Both EpC and cld7 coimmunoprecipitate with the tetraspanin Tspan8, but coimmunoprecipitation with EpCmAG is impaired (Figure 1C). Confocal microscopy confirmed strong colocalization of cld7 with EpC in ASMLwt and ASML-EpCresc cells, but poor colocalization in ASML-EpCmAG and ASML-cld7mPalm cells (Figure 1D). Furthermore, in ASMLwt and -cld7resc cells cld7 and EpC are enriched in light density (GEM) fractions, but cld7mPalm is shifted towards heavier fractions. Recovery of EpC in light density fractions depends on the association with (palmitoylation competent) cld7. EpC is poorly recovered in light density fractions of ASML-cld7kd and -EpCmAG lysates and is not rescued into light density fractions in ASML-cld7mPalm lysates. Recovery of the constitutively GEM-located tetraspanin Tspan8 is not affected by the cld7kd or cld7mPalm (Figure 1E).
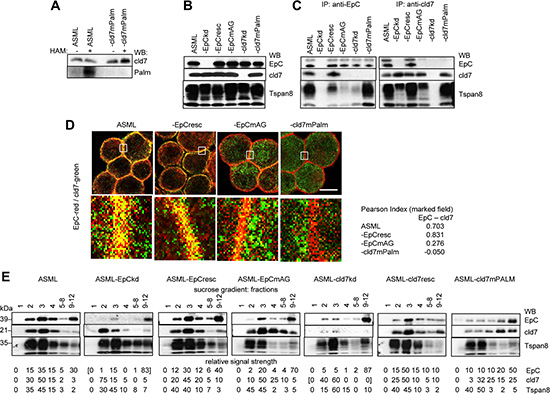
Figure 1: Characterization of ASML-EpC and cld7 knockdown and rescue clones: ASML cells were transfected with EpC- and cld7-shRNA and cloned in selection medium. EpC expression was rescued in ASML-EpCkd clones using primers for wt rescue (EpCresc) or point mutated (position 282 and 279) EpC (EpCmAG); cld7 was rescued in ASML-cld7kd clones with a mutation at the palmitoylation site at AA184 and AA186 or was transiently rescued using primers for wt rescue (cld7resc). (A) Wt, kd and rescue ASML clones were lysed in the presence of N-ethylmaleimide (NEM) to irreversibly block unmodified thiol groups. After incubation with HAM buffer for unmasking palmitoylated cysteine thiol groups, samples where incubated in biotin-BMCC for selective labeling of palmitoylated cysteines. Samples were blotted with streptavidin-HRP and after stripping with anti-cld7; (B) lysates of wt, kd and rescue ASML clones (one representative clone was selected) were separated by SDS-PAGE and blotted with anti-EpC (D5.7), anti-cld7 and anti-Tspan8 (D6.1, control); (C) lysates of wt, kd and rescue ASML clones were precipitated with anti-EpC or anti-cld7. After SDS-PAGE, precipitates were blotted with anti-EpC, anti-cld7 or anti-Tspan8; (D) wt and rescue ASML clones were stained with anti-EpC(red) and anti-cld7 (green); staining was evaluated by confocal microscopy, digital overlays (scale bar: 10 μM). The indicated area (white square) was amplified 10-fold for better discrimination. The Pearson correlation coefficiency is shown for the encircled membrane area; (E) lysates of wt, kd and rescue ASML clones were separated according to density by sucrose gradient centrifugation; 1ml fractions were collected and fractions 5–8 and 9–12 were pooled. After SDS-PAGE, fractions were blotted with anti-EpC, anti-cld7 and anti-Tspan8 (GEM control). EpC and cld7 were efficiently downregulated in kd clones and were recovered in rescue clones. Mutation of the palmitoylation site at AA184 and AA186 prevented cld7 palmitoylation. Cld7 palmitoylation strongly facilitates the association with EpC, the cld7-EpC complex being enriched in GEM.
Taken together, in ASML cells, which do not form TJ, cld7 is palmitoylated and enriched in GEM. The association with EpC is promoted by cld7 palmitoylation, but is not completely abolished in the absence of (palmitoylated) cld7, possibly due to EpC associating also with other palmitoylated GEM-located molecules, like e.g. Tspan8.
Anchorage independence and metastasis formation require palmitoylation-competent cld7
Anchorage independence and tumor progression are central features of CIC. Soft agar colony formation of ASML-cld7kd and ASML-EpCkd cells is strongly decreased, but is largely regained in ASML-EpCresc and partly in ASML-EpCmAG cells. Soft agar colony formation is not restored in ASML-cld7mPalm cells, which start to form small clusters, but die after 1 wk of culture (Figure 2A).
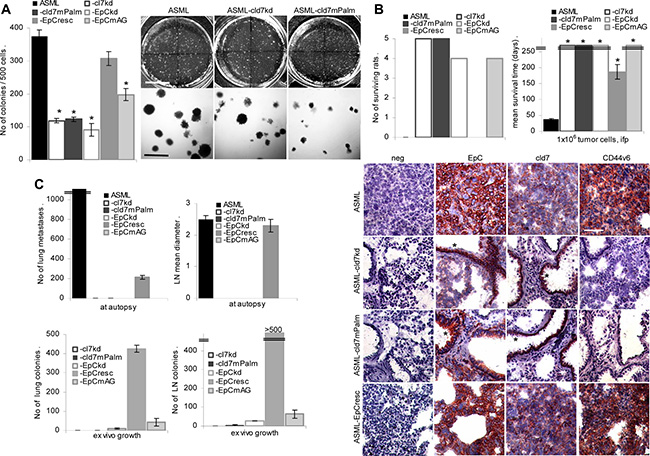
Figure 2: Palmitoylated cld7 supports anchorage independent growth and metastasis formation: (A) Wt, kd and rescue ASML cells (500) were seeded in soft agar in 10 cm Petri-dishes. Colonies were counted after 14 d. The mean number ± SD (triplicates) of colonies and representative examples are shown. (B, C) BDX rats received 1 × 106 wt, kd or rescue ASML cells, ifp. (B) The number of surviving rats and the mean survival time are shown; (C) the number of visible lung metastases, the mean draining lymph node diameter, the number of tumor cell colonies growing in suspended lung and LN tissue was evaluated after 4 wk. (D) Shock frozen lung sections from ASML wt, -cld7kd, cld7mPalm and -EpCresc bearing rats were stained with anti-EpC, -cld7 and -CD44v6 (scale bar: 120 μm). Tumor cells (EpC+, cld7+, CD44v6+) are only seen in the lung of ASMLwt and -EpCresc bearing rats. In the lungs of ASML-cld7kd and -cld7mPalm bearing rats only bronchiolar epithelium (*) is stained by anti-EpC and anti-cld7. Anchorage-independent growth and metastasis are severely impaired in both kd clones, but are regained in ASML-EpCresc and partly ASML-EpCmAG, but not in -cld7mPalm cells, indicating only palmitoylated cld7 supporting metastasis.
ASML-cld7kd cells completely lost the capacity to metastasize via the lymphatic system and metastatic capacity is not rescued in ASML-cld7mPalm cells. None of the rats developed visible metastasis. Very few tumor cell colonies grew in ex vivo cultured lymph node and none in lung suspensions. Instead, ASML-EpCresc cells develop lymph node metastases and a limited number of lung metastases after intrafootpad application. Although with a significant delay, ASML-EpCresc bearing rats become moribund after 154–215 days mostly due to the metastatic lymph node burden. Few ASML-EpCmAG cells were recovered in lymph nodes and lung in ex vivo cultures, but did not form visible metastases. Immunohistology confirmed that ASML and ASML-EpCresc cells displaced the lung tissue with only EpC+/cld7+/CD44v6+ tumor cells being seen in most sections. Instead, no tumor nodules were seen in the lung of rats that received ASML-cld7kd or ASML-cld7mPalm cells, only bronchiolar epithelial cells being stained by anti-EpC and anti-cld7 (Figure 2B, 2C).
Thus, palmitoylated cld7 is indispensable for ASML metastasis formation. There are 3 major, mutually not exclusive features, whereby palmitoylated cld7 could support the metastasis process. (i) Palmitoylated cld7 promotes tumor cell motility by associating with integrins and the cytoskeleton and/or by cooperating with proteases to create space for metastases; (ii) palmitoylated cld7 is engaged in apoptosis resistance and (iii) EMT.
Palmitoylated cld7 and motility
ASML cells do not grow locally, the capacity to leave the injection site and to reach the first lymph node station becoming vital. Transwell migration and wound healing of ASML-cld7kd and -EpCkd cells is significantly reduced. It is restored in ASML-cld7resc and -EpCresc cells, but not in ASML-cld7mPalm and -EpCmAG cells (Figure 3A, 3B). In transwell migration the cld7kd exerted a stronger effect than the EpCkd, which was controlled for the migration of individual cells by videomicroscopy. Distinct to the reduced migration of ASML-cld7kd and -cld7mPalm cells, migration of single ASML-EpCkd cells was increased and migration of -EpCmAG was not affected (Figure 3C). This finding indicates that cld7 actively promotes motility, whereas “free” EpC hampers motility, though to a minor degree.
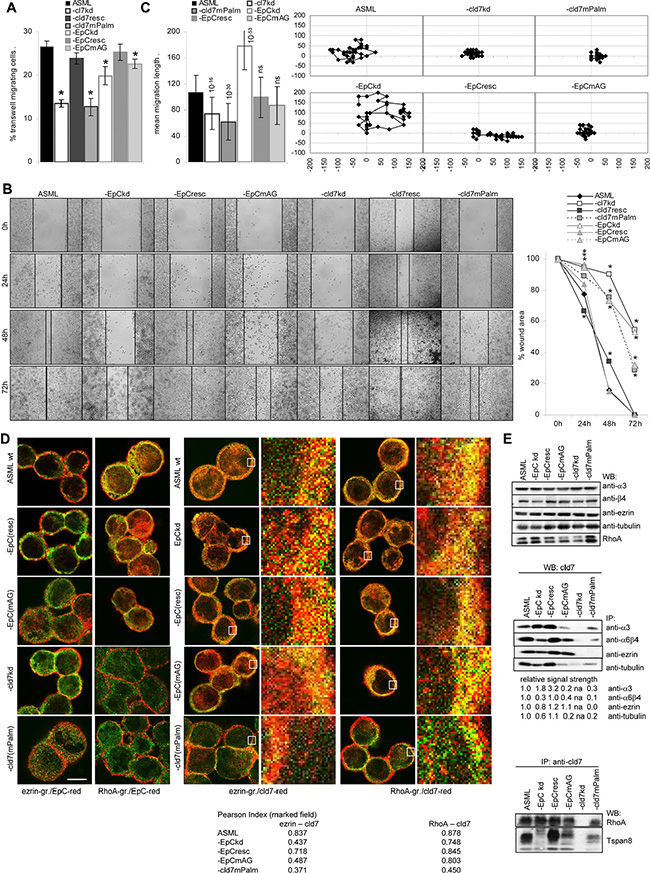
Figure 3: The impact of palmitoylated cld7 on cell motility: (A) Wt, kd and rescue ASML cells (2 × 104 in RPMI/1% BSA) were seeded in the upper part of a Boyden chamber; the lower part, separated by a 0.8 μm pore size membrane contained RPMI/20% FCS. Recovery of cells on the lower membrane site was evaluated after 16 h by crystal violet staining. The percent ± SD of migrating cells compared to the total input are shown. (B) Wt, kd and rescue ASML cells were seeded in 24-well plates. When cultures reached a subconfluent stage, the monolayer was scratched with a pipette tip. Wound healing was followed for 72 h. Examples (scale bar: 250 μm) and the mean percent ± SD of the wound area compared to the 0 time point are shown. (C) Cells as above were seeded in 6-well plates coated with LN111. Pictures were taken every 20 min for 24 h. Migration of 20 individual cells was recorded. An example of migration of a single cell as well as the mean migration ± SD of 20 cells/well is presented. (A–C) Significant differences as compared to ASMLwt cells: *. (D) Wt, kd and rescue ASML cells were stained with anti-ezrin (green) or anti-RhoA (green) and anti-EpC (red) or anti-cld7 (red). Staining was evaluated by confocal microscopy; digital overlays of staining are shown (scale bar: 10 μm). The indicated area (white square) was amplified 10-fold for better discrimination. The Pearson correlation coefficient is shown for the encircled membrane area. (E) Lysates of cells as above were precipitated with anti-α3, -α6β4 (B5.5), -ezrin and -tubulin and were blotted with anti-cld7 or were precipitated with anti-cld7 and blotted with -RhoA and -Tspan8. The relative signal strength of cld7 precipitates is indicated. The strength of the cld7 signal in ASML wt was arbitrarily set as 1.0. WB of α3, β4, ezrin, tubulin and Rho are included as controls. Migration of ASML-cld7kd and -cld7mPalm, but not of -cld7resc cells is severely reduced. Impaired migration is accompanied by reduced association of cld7mPalm with ezrin, and, less pronounced, α3, α6β4, tubulin, RhoA and Tspan8. In the absence of (palmitoylated) cld7, EpC does poorly colocalize/associate with RhoA.
In order to define the mechanism(s) underlying palmitoylated cld7-promoted motility, we searched by mass spectrometry for proteins preferentially co-immunoprecipitating after mild lysis, not to destroy GEM complexes, with cld7 and/or cld7mPalm. ASMLwt were precipitated with anti-EpC and anti-cld7. ASML-EpCkd, -EpCmAG and -cld7mPalmlysates were precipitated with anti-cld7. A considerable number of cytoskeletal proteins associate with EpC and cld7. While integrins and some tetraspanins preferentially coimmunoprecipitate with EpC (Supplementary Table S1A), the association requires palmitoylation-competent cld7. This is demonstrated for α6β4 and Tspan8 that do not or poorly colocalize with EpC in ASML-EpCmAG and -cld7mPalm cells and most poorly with cld7 in ASML-cld7mPalm cells (Supplementary Figure S1). Α6β4 and Tspan8 also poorly coimmunoprecipitate with cld7 in ASML-cld7mPalm lysates. Coimmunoprecipitation of α3 with cld7 is less severely affected in ASML-cld7mPalm cells (Figure 3E). Furthermore, the cytoskeletal linker proteins actinin, moesin and RhoA, which are engaged in actin cytoskeleton organization, preferentially associate with palmitoylation-competent cld7. On the contrary, cytoskeletal keratins and myosin associate with EpC and cld7, but more readily with non-palmitoylated cld7 (Supplementary Table S1A). Confocal microscopy confirmed strongly reduced colocalization of ezrin and RhoA with cld7mPalm. EpC poorly colocalizes with ezrin and RhoA in ASML-EpCmAG and -cld7mPalm (Figure 3D). WB of immunoprecipitates confirmed the association of cld7 with ezrin, tubulin and RhoA. However, ezrin does not coimmunoprecipitate with cld7mPalm and coimmunoprecipitation of tubulin and RhoA with cld7 is strongly reduced in ASML-cld7mPalm precipitates. Finally, cld7 poorly coimmunoprecipitates with α3 and tubulin in ASML-EpCmAG and with Tspan8 in ASML-EpCkd and -EpCmAG cells, which indicates a contribution of cld7-associated EpC to complex formation (Figure 3E).
Taken together, multiple associations between cld7, actin linker and actin (re)organizing proteins well explain the reduced motility of ASML-cld7kd cells. Fittingly, palmitoylation-competent cld7 poorly associates with keratins.
Palmitoylated cld7 and invasiveness
Matrigel invasion and penetration of ASML-cld7kd cells is strongly reduced and is not rescued in ASML-cld7mPalm cells. Invasion and penetration of ASML-EpCkd and -EpCmAG cells is also reduced, although less efficiently (Figure 4A). However, the protease profile of ASML cells is not significantly altered in ASML-cld7kd and ASML-EpCkd and rescue clones (Supplementary Figure S2). Instead, particularly MMP9 activity (zymography) is strongly reduced in ASML-cld7kd and ASML-cld7mPalm cells (Figure 4B). Co-immunoprecipitation (mass spectrometric analysis, data not shown), revealed an association with uPAR and CD147 (basigin), the latter recruiting proteases from neighboring cells. These associations and an association with MMP14 were confirmed by WB after co-immunoprecipitation. The association with uPAR depended on cld7 palmitoylation, whereas the association with MMP14 and CD147 was also seen in ASML-cld7mPalm cells. The latter finding is supported by strong colocalization of CD147 with cld7mPalm (Figure 4C, 4D).
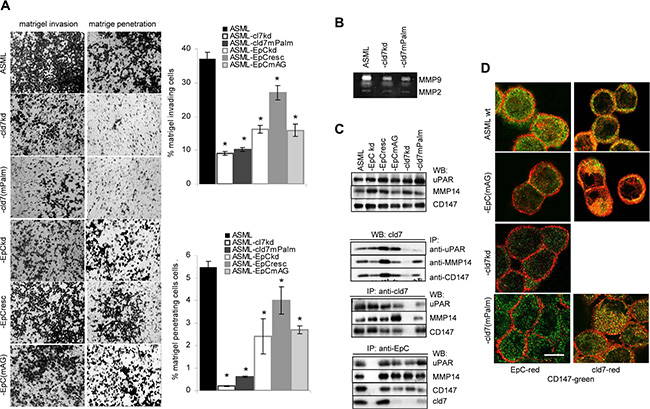
Figure 4: Palmitoylated claudin7 supports invasiveness and protease activity: (A) Wt, kd and rescue ASML cells were seeded on matrigel. Matrigel invasion and penetration was evaluated after 16 h incubation. The mean percent ± SD of invading and penetrating cells and representative examples are shown. (B) Zymography of culture supernatants of ASMLwt, -cld7kd and -cld7mPalm cells; MMP2 and MMP9 bands are indicated. (C) Lysates from cells as in (A) were precipitated with anti-CD147 (EMMPRIN), anti-uPAR and anti-MMP14 and were blotted with anti-cld7 or were precipitated with anti-cld7 or anti-EpC and blotted with anti-uPAR and anti-CD147. WB of lysates with anti-uPAR, -MMP14 and -CD147 are included as controls. (D) Cells as above were stained with anti-CD147 (green) and either anti-EpC (red) or anti-cld7 (red). Digital overlays are shown (scale bar: 10 μm). ASML-cld7kd cells are poorly invasive and invasiveness is not restored in ASML-cld7mPalm cells. This also accounts for reduced MMP9 activity and fits to reduced coimmunoprecipitation of cld7mPalm with MMP14 and uPAR. EpC has a weaker impact on invasiveness and MMP activity.
We demonstrated before that several proteases associate with GEM-located tetraspanins and also with CD44v6 [63, 64]. As invasiveness was not rescued in ASML-cld7mPalm, we suggest that the contribution of cld7 is linked to the integration of palmitoylated cld7 into GEM, rather than to a direct impact of cld7 on protease activity.
Apoptosis resistance, cld7 and PTEN
High drug resistance of ASML cells is strongly reduced in ASML-cld7kd and is not rescued in -cld7mPalm cells. Drug resistance of ASML-EpCkd cells also is reduced, though less severely. Drug resistance is re-established in ASML-EpCresc and partially in -EpCmAG cells. This accounted for AnnV/PI staining (apoptosis), mitochondrial integrity (MTT assay) and proliferation (3H-thymidine incorporation) (Figure 5A, Supplementary Figure S3A).
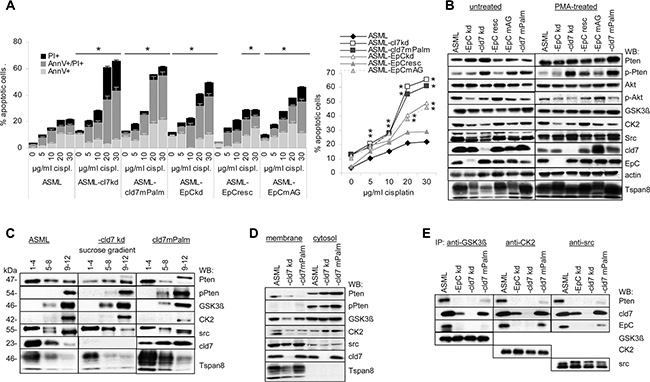
Figure 5: Cld7 and apoptosis resistance: (A) Wt, kd and rescue ASML cells were cultured in the presence of increasing amounts of cisplatin. Flow cytometry analysis of the percent of AnnV+, AnnV+PI+ and PI+ cells; mean ± SD (triplicates) are shown, significant differences to wt cells: *. (B) WB of Akt, Pten, pPten and Pten phosphorylating kinases in untreated and PMA-treated wt, kd and rescue ASML cells; (C) sucrose gradient fractions of the lysates as above and WB with anti-Pten, -pPten, -GSK3β, -CK2, -src, -cld7 and -Tspan8; (D) Membrane and cytosol lysates from cells as above were blotted with anti-GSK3β, -CK2, -src, -pPten and -cld7. (E) WB with anti-Pten, -cld7, -EpC, -GSK3β, -CK2 and -src after precipitation with anti-GSK3β, anti-CK2 and anti-src. Drug resistance of ASML-cld7kd cells is severely impaired and not rescued in ASML-cld7mPalm cells. Upregulation of Pten in PMA-treated ASML-cld7kd and -cld7mPalm cells indicates Pten repression only by palmitoylation-competent cld7. Concomitant pronounced cytoplasmic Pten phosphorylation does not contribute rescuing apoptosis resistance.
In the absence of stress, expression of proteins engaged in receptor-mediated apoptosis or the mitochondrial pathway of apoptosis is unaltered. However, cisplatin-treated ASML-cld7kd and -cld7mPalm cells show increased levels of activated Casp3 and cleaved Casp9 (Supplementary Figure S3B, S3C), reduced phosphorylated PI3K, Akt and BAD as well as Bcl2 and BclXl recovery. Slightly upregulated BID, BAK, BAX and Smac/Diablo expression is not dependent on cld7 palmitoylation (Supplementary Figure S3D). mTOR is downregulated in ASML-cld7kd, -cld7mPalm, -EpCkd and -EpCmAG. Compared to ASML cells, Pten expression is upregulated in ASML-cld7kd and ASML-cld7mPalm cells. However, Pten phosphorylation is also upregulated (Supplementary Figure S3E). The flow-cytometry analysis was confirmed by a signaling protein array and/or by WB (Supplementary Figure S3D–S3F, Figure 5B). Pronounced Pten phosphorylation in ASML-cld7kd and -cld7mPalm opposed expectation, as the cells did not regain apoptosis resistance. Furthermore, expression of GSK3β, CK2 and src, known to be engaged in Pten phosphorylation [65], is not (GSK3β, CK2) or only slightly (src) upregulated in ASML-cld7kd and -cld7mPalm cells (Figure 5B). Mass spectrometry analysis of signaling molecules co-immunoprecipitating with cld7 did not provide hints towards a special reduction of serine threonine kinases co-immunoprecipitating with cld7 in ASML-cld7mPalm lysates (Supplementary Table S1B). Thus, we speculated that by exclusion of palmitoylation-deficient cld7 from GEM, pronounced Pten phosphorylation does not rescue PI3K/Akt pathway activation. WB of pooled sucrose gradient fractions indicated that only src, but not GSK3βand CK2 is recovered in light density fractions. Phosphorylated Pten also is not recovered in light density fractions (Figure 5C). A WB of membrane and cytosolic lysates confirmed recovery of pPten exclusively in the cytosol, the strongest signal being seen in the ASML-cld7mPalm cytosol. CK2 and GSK3β were preferentially recovered in the cytosol, src was enriched in the membrane fraction, cld7 was recovered in the membrane and the cytosolic fraction, whereas Tspan8 was exclusively recovered in the membrane fraction (Figure 5D). Thus, an association of GSK3β, CK2 and src with cytoplasmic cld7mPalm could account for pronounced Pten phosphorylation. Indeed, cld7mPalm coimmunoprecipitates with GSK3β, CK2 and src, which also, albeit very weakly coimmunoprecipitate with Pten (Figure 5E).
The data confirm a dominating role of palmitoylation-competent cld7 in drug resistance due to Pten repression. Cytoplasmic Pten phosphorylation via cld7mPalm recruited GSK3β, CK2 and src obviously does not suffice to restore apoptosis resistance.
Palmitoylated cld7 and EMT
Metastasis formation requires EMT. The metastasis suppressor E-cadherin is slightly upregulated in ASML-EpCkd, but not in ASML-cld7kd cells. However, EMT-associated N-cadherin, fibronectin (FN) and vimentin expression is reduced in ASML-cld7kd and ASML-EpCkd cells. Expression of N-cadherin and FN is rescued in ASML-EpCresc, but not or less efficiently in ASML-cld7mPalm cells (Figure 6A, 6B). Reduced Oct3/4, Snail, Sox2, Wnt1, Notch and β-catenin in ASML-cld7kd cells also is not rescued in ASML-cld7mPalm cells. Instead, p-β-catenin expression is higher in ASML-cld7kd and slightly higher in -cld7mPalm cells (flow-cytometry) and lysates (WB). Recovery of Oct3/4, Snail, Sox2 as well as of Notch and β-catenin also is reduced in ASML-cld7kd and ASML-cld7mPalm nuclei. ZEB1 is downregulated only in ASML-EpCkd and -cld7kd lysates. Unexpectedly, Slug expression is increased in ASML-cld7kd and -cld7mPalm cells and lysates (Figure 6C–6E). Cld7 does not associate with Oct, Snail and Nanog, but coimmunoprecipitates with Notch, coimmunoprecipitation being reduced in ASML-EpCkd, -EpCmAG and -cld7mPalm lysates (Figure 6F).
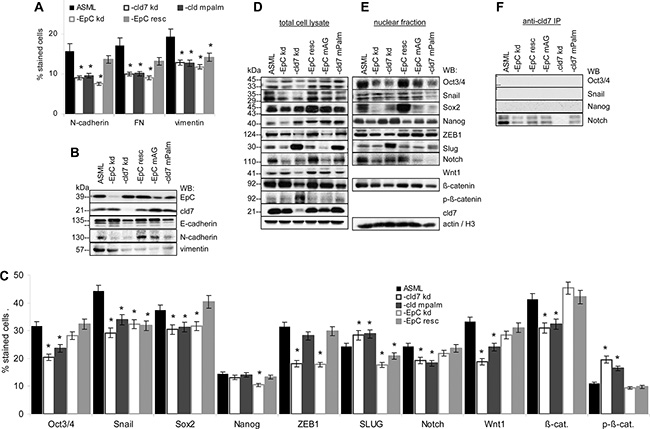
Figure 6: Cld7 palmitoylation and EMT gene expression: (A, C) Wt, kd and rescue ASML cells were stained with antibodies against EMT markers, EMT-related signaling molecules and transcription factors; the mean percent ± SD of stained cells is shown. Significant differences to ASMLwt cells are indicated by *. (B, D) Total cell lysates and (E) the nuclear fraction of the cells as above were separated by SDS-PAGE and blotted with the indicated antibodies; (F) Lysates were precipitated with anti-cld7 and blotted with anti-Oct3/4, -Snail, -Nanog and -Notch. A cld7kd severely affects EMT signaling molecules and transcription factors, with the exception of Slug. Reduced recovery accounts particularly for the nuclear fraction. N-cadherin, FN and vimentin expression is reduced. These changes are not reverted in ASML-cld7mPalm cells. Only an EpCkd is associated with E-cadherin upregulation.
Taken together, palmitoylated cld7 contributes to expression of several EMT-related proteins and transcription factors and hampers β-catenin phosphorylation. The association of palmitoylated cld7 with Notch might be the initial trigger for altered EMT gene expression.
A dominating role of GEM located cld7 in vesicle transport
Metastasis formation is supported by exosomes [66]. Though we focused on cell inherent activities of GEM-located cld7, the strong engagement of palmitoylation-competent cld7 in intracellular vesicle traffic demands mentioning.
Besides co-immunoprecipitating with adhesion molecules (Supplementary Table S1A) and mostly cytoplasmic signaling molecules (Supplementary Table S1B), cld7 also coimmunoprecipitates with soluble carrier and transporter proteins, some of which (aldose reductase, Na,K-ATPase, TM9SF2, transportin and lactadherin 2) preferentially co-immunoprecipitate with cld7mPalm (Supplementary Table S1C).
GEM, including tetraspanin-enriched membrane microdomains (TEM) are prone for internalization [39, 40]. The internalization complex is maintained during intracellular vesicle traffic and vesicle exocytosis [67]. Recruitment of palmitoylation-competent cld7 and cld7-associated EpC into GEM was already demonstrated (Figure 1) as well as the GEM-located cld7 association with integrins and ARP2/3 complex components that are engaged in early endosome formation (Supplementary Table S1A). In addition, chaperons, which are enriched in exosomes, are most abundantly recovered in EpC and cld7 co-immunoprecipitates, where 6 of 28 did not or poorly associate with cld7mPalm (Supplementary Table S1D). Palmitoylation-competent cld7 also associates with several transporter complexes and vesicle transport-associated molecules, particularly myoferlin, rab25 and Sec31a. Several additional rab proteins associated with cld7 independent of palmitoylation. On the opposite, only palmitoylation-deficient cld7 associated with caveolin (Supplementary Table S1E). Mostly palmitoylation-independent, cld7 associated with vesicle transporter complexes, like coatomer complexes, dynein, Vamp family proteins (Supplementary Table S1F) and tubulins (Supplementary Table S1A). Finally, cld7 abundantly associates with proteases of the proteasome complex. These associations partly depend on cld7 palmitoylation; the association with the ribophorin protease complex being only seen with palmitoylation-competent cld7 (Supplementary Table S1G). Colocalization with cld7/palmitoylated cld7 was confirmed for the vesicle transporters rab5, rab7, rab11, Lamp1 as well as for HSP70, the membrane coat clathrin, the internalization complex associated dynamin and the cytoskeletal linker protein tubulin. Notably, caveolin does not colocalize with palmitoylation-competent cld7 (Figure 7A). WB confirmed cld7 palmitoylation-dependent coimmunoprecipitation for rab5 and rab7. The association with Lamp1, CathepsinD, dynamin and AnnII was only partially cld7 palmitoylation-dependent (Figure 7B).
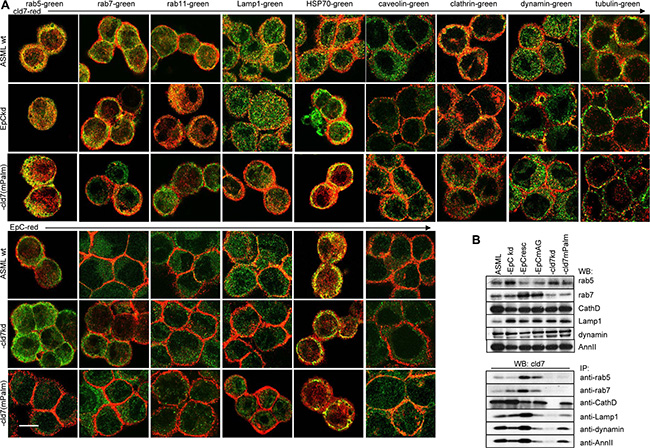
Figure 7: The linkage between palmitoylated cld7, vesicle formation and vesicle transporter proteins: (A) Colocalization of cld7 (red) and EpC (red) with vesicle formation and vesicle transport associated proteins (green) in wt, EpCkd, cld7kd and cld7mPalm ASML cells. Digital overlays of confocal microscopy images are shown (scale bar: 10 μm); (B) WB and coimmunoprecipitation of cld7 with vesicle formation and transport proteins in wt, kd and rescue ASML cells. Only palmitoylation-competent cld7 colocalizes and coimmunoprecipitates with molecules engaged in GEM-supported vesicle formation and vesicle transport. Instead, palmitoylation-competent cld7 does not colocalize with caveolin.
Flow cytometry confirmed high cld7, EpC, Tspan8 and distinct α6β4, ezrin, tubulin, RhoA and caveolin expression in ASML exosomes. However, the percentage of cld7+, EpC+ and RhoA+ exosomes was reduced, whereas the percentage of caveolin+ exosomes was increased in ASML-cld7mPalm exosomes (Figure 8A). Coimmunoprecipitation of exosome lysates confirmed the distinct composition of palmitoylation-competent versus -deficient cld7 exosomes. Cld7 coimmunoprecipitated with the CIC markers EpC, α6β4 and Tspan8 in ASMLwt exosomes. Coimmunoprecipitation of EpC, CD44v6, β4 and Tspan8 was strongly reduced in ASML-cld7mPalm exosomes. Also, anti-EpC, anti-CD44v6 and anti-α6β4 did not coimmunoprecipitate cld7 in ASML-cld7mPalm, but coimmunoprecipitation of anti-CD44v6 with anti-α6β4 and vice versa was not affected in ASML-cld7mPalm (Figure 8B).
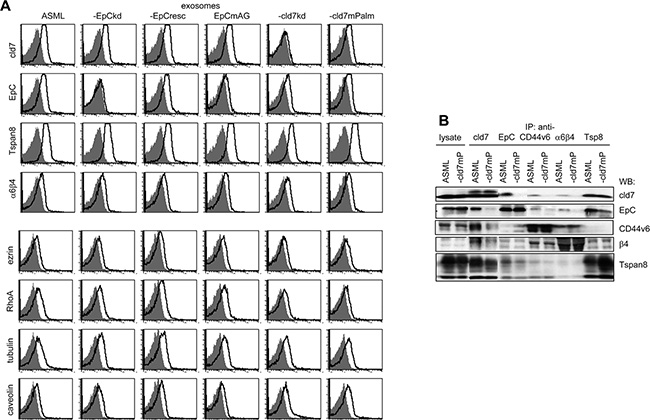
Figure 8: Recovery of cld7 and associated molecules in exosomes: (A) Flow cytometry of latex beads bound wt, kd and rescue ASML exosomes stained with anti-cld7, -EpC, -Tspan8, -α6β4, -ezrin, -RhoA, -tubulin and -caveolin. Overlays of the negative control and stained samples are shown. (B) Lysates of ASMLwt, and -cld7mPalm exosomes were precipitated with anti-cld7, -EpC, -CD44v6, -Tspan8 or -α6β4. Dissolved precipitates were separated by SDS-PAGE and were blotted with the indicated antibodies. The WB of exosome lysates is included as control. ASMLwt exosomes stain brightly with anti-Tspan8, -EpC, and -cld7 and distinctly with anti-α6β4, -ezrin, -RhoA and -tubulin. ASML-cld7mPalm exosomes show reduced cld7+, -EpC+ and RhoA+, but more caveolin+ exosomes. Exosome lysate coimmunoprecipitation confirmed reduced recovery of EpC, CD44v6, α6β4 and Tspan8 in anti-cld7 precipitates of ASML-cld7mPalm than ASMLwt exosomes.
Taken together, (i) solute carrier expression is affected by a cld7kd; (ii) abundant associations of cld7 with stress response modulators (chaperones) could be important with respect to apoptosis resistance; (iii) the unexpectedly strong association of palmitoylated cld7 with the vesicle transporter machinery, including the deviation from the proteasome, highlights cld7 as a so far unrecognized exosome organizer. This was confirmed by the distinct composition of ASML versus ASML-cld7mPalm exosomes, which also displayed different association profiles. A central role of palmitoylated cld7 in vesicle traffic and exosome biogenesis could well be a major factor in the contribution of cld7 to the CIC phenotype.
In brief, distinct to non-palmitoylated cld7 that in epithelial cells acts as a TJ protein, palmitoylated cld7 is GEM-associated and contributes in this particular environment to tumor cell motility, invasiveness, EMT and exosome biogenesis.
DISCUSSION
Claudins were first described as TJ components that are engaged in sealing, formation of ion channels and organization of paracellular small organic solute flux [16, 68]. However, claudins are also found outside of TJ [17–24], where their functions are still disputed. We described that in tumor cells palmitoylated cld7 is recovered in GEM [9], special microdomains serving as signaling platforms [35–38] and being prone for internalization [39–41]. There was evidence that GEM-located cld7 supports motility [33] and apoptosis resistance [9]. Finally, GEM-located cld7 is associated with EpC [9, 33], the GEM-located cld7-EpC complex promoting EMT [9, 10]. Starting from this point, we evaluated the importance of cld7 palmitoylation on CIC activities in a rat metastasizing tumor model, where an EpCkd was rescued with a point mutated EpC that cannot bind cld7 (EpCmAG). A cld7kd was rescued with a mutation in the C-terminal palmitoylation site (cld7mPalm) that suffices preventing cld7 palmitoylation [33]. This model allowed deciphering palmitoylation-dependent activities of cld7 as well as differentiating between genuine versus driver activities of cld7. Both rescue lines confirmed a dominant role of palmitoylated cld7 in promoting motility and apoptosis resistance and pointed towards an involvement of cld7 in exosome biogenesis.
Metastasis supporting activity of cld7 depends on palmitoylation
ASMLwt bearing rats die with lymph node and lung metastases after 5–6 wk. However, ASML-cld7kd and -cld7mPalm cells did not grow locally, did not reach the draining lymph node or the lung and 5/5 rats were tumor free after > 26 wk. Nonetheless, ASML-cld7mPalm differ from ASML-cld7kd cells starting to divide in soft agar at a comparable frequency to ASMLwt cells, but cells died after 1 wk. As the proliferation rate of ASMLwt, -cld7kd and -cld7mPalm cells does not differ (data not shown), the finding points towards impaired apoptosis resistance of ASML-cld7mPalm.
Taken together, only palmitoylated cld7 supports tumor progression. Depending on the tumor type, the failure of non-palmitoylated cld7 to support metastatic growth could be due to integration in TJ. This could explain the discrepant findings on cld7 prohibiting [69, 70] vs. promoting metastasis [9, 10, 71–75].
Cld7 palmitoylation, motility, invasiveness and the crosstalk with the cytoskeleton
Mass spectrometry of molecules co-immunoprecipitating with cld7 in ASMLwt cells confirmed the association with tetraspanins, CD44, the integrins α3(β1) and α6β4 and the association with cytoskeletal linker proteins [76]. The repeatedly described association with β1 [15, 77] is accompanied by a strong basolateral distribution, a reduction in pFAK and poor adhesion [77]. These reports are in line with our findings, which additionally point towards the requirement for cld7 palmitoylation to associate with integrins and downstream signaling cascades. Furthermore, cld7 amply associates with actin binding/organizing and actin polymerizing molecules. The associations with actin linker proteins mostly depend on cld7 palmitoylation, whereas the association with myosin and cytokeratins frequently is hampered by cld7 palmitoylation. This has consequences on cell motility, which is strongly reduced in ASML-cld7kd and ASML-cld7mPalm cells. We suggest the impact of palmitoylated cld7 on motility is supported by the association with tetraspanins and, possibly via Tspan8, with integrins [39–41].
Cld7 also associates with MMPs and uPAR [15, 78, 79], which contribute to motility by matrix protein degradation. Coimmunoprecipitation confirmed the association with uPAR and a weak association with MMP14. The uPAR association is exclusively observed with palmitoylation-competent cld7, i.e. depends on the GEM localization. Likely via the association with MMP14, MMP9 becomes recruited and activated, the gelatinolytic activity of ASMLwt cells significantly exceeding that of ASML-cld7kd and ASML-cld7mPalm cells. GEM-located palmitoylated cld7 also associates with CD147, which binds MMPs on neighboring cells [80] thereby promoting MMP activity. Corresponding changes being seen in ASML-Tspan8kd and ASML-CD44vkd cells [63, 64], we argue that the engagement of cld7 in invasiveness, similar to its contribution to motility, relies on associated GEM-located molecules.
Taken together, GEM-located palmitoylated cld7 supports motility and invasion by associating with integrins, tetraspanins, cytoskeletal linker proteins and proteases.
Tumor cells motility is frequently associated with EMT [81, 82]. Expression of several EMT-related genes is impaired in ASML-cld7kd and -cld7mPalm cells. Weak E-cadherin expression in ASMLwt cells is not affected. However, vimentin, FN and N-cadherin expression is reduced. Wnt1 is slightly downregulated in ASML-cld7kd and, less pronounced, -cld7mPalm, but unaltered in -EpCkd cells. In line with this finding is the downregulation of β-catenin and the pronounced β-catenin phosphorylation in ASML-cld7kd and -cld7mPalm. Reduced recovery of nuclear β-catenin could account for impaired FN, Snail1, Snail2 and Twist expression [83]. However, only Snail1 recovery is impaired, whereas Slug expression is upregulated. Thus, in ASML cells Snail transcription [84] does not or not exclusively rely on β-catenin. Activation of EMT-related transcription factors can also proceed via integrins, ILK activation leading to NFkB nuclear translocation [86] or via activation of the src/p38MAPK pathway [87, 88]. uPAR, too, can induce EMT through activation of the PI3K/Akt pathway, src kinases, ERK/MAPK and myosin light chain kinase [89]. These alternative pathways of EMT transcription factor induction could be impaired in ASML-cld7mPalm due to the reduced association with α3, α6β4 and uPAR. Finally, by not yet defined mechanisms, cld7 is engaged in Pten repression. High Pten expression in ASML-cld7kd and -cld7mPalm can account for downregulation of Snail, ZEB and Twist transcription [90].
Further explorations are required to precisely define the pathway(s), whereby palmitoylated cld7 supports transcription and/or nuclear translocation of EMT transcription factors, where the association with Notch could be the initial trigger. Nonetheless, the loss of EMT features of ASML-cld7mPalm cells strongly argues for an engagement of palmitoylated cld7 in EMT.
The engagement of cld7 in stress resistance
ASML cells are strikingly apoptosis resistant [62]. This relies on concerted activities of CD44v6 promoting activation of the MAPK and the PI3K/Akt pathway [63], on Tspan8 engaged in PI3K/Akt activation [64] and on cld7. There is no evidence for a direct engagement of cld7 in MAPK or JNK pathway activation. Instead, apoptosis resistance promoted by cld7 is accompanied by low level Pten expression [9]. Furthermore, there is evidence for palmitoylation-competent cld7 recruiting Pten repressing miRNA (unpublished finding). We now defined that only palmitoylated, GEM-located cld7 contributes to apoptosis resistance. Unexpectedly, apoptosis resistance was not rescued in ASML-cld7mPalm cells, despite upregulated Pten phosphorylation. There are several possible explanations. First, Pten phosphorylation in ASML-cld7mPalm cells is ineffective as Pten is not recruited into GEM. Mostly cytoplasmic cld7mPalm associates with GSK3β and CK2, prominent Pten phosphorylating kinases [65]. Alternatively, not mutually exclusive, GSK3β and CK2 can also contribute to Pten stabilization [91, 92].
Taken together the contribution of cld7 to apoptosis resistance relies on Pten repression, supported by Pten inactivation. The inefficacy of cld7mPalm to restore apoptosis resistance, despite pronounced Pten phosphorylation, may rely on the failure to recruit phosphorylated Pten towards GEM. Taking into account the multiple activities of Pten and the abundance of pathways regulating Pten transcription and activity [93] additional contributions of cld7 to Pten regulation cannot be excluded.
Outlook: Cld7 and exosome biogenesis
Tetraspanins, located in so called tetraspanin-enriched microdomains, which are similar to GEM, play a major role in membrane invagination, the generation of early endosomes and their traffic through MVB towards the release as exosomes [67]. These findings fit to tetraspanins being constitutive exosome components [94]. We now described similar features for palmitoylated, GEM-located cld7. As cld7 and Tspan8 are loosely attached, are both located in GEM and are palmitoylated, we cannot sharply decipher, whether the two molecules work in concert or independently. Both cld7 and Tspan8 share the abundant association with chaperons [95], the association being mostly cld7 palmitoylation-independent. Furthermore, both - but Tspan8 more intensely - associate with integrins and membrane-integrated proteases [67, 73, 94]. Instead, cld7 has a strong affinity for proteasomal proteases, which might drive at least part of intraluminal vesicles into the degradation pathway rather than towards release. Tspan8 and cld7 abundantly associate with vesicle transporters of the rab family, some of these associations depending on cld7 palmitoylation, e.g. anti-rab5 and -rab7 coimmunoprecipitate cld7, but not cld7mPalm. On the other hand, Tspan8 and palmitoylation-competent cld7 do not or poorly associate with caveolin, which strengthens the assumption of a caveolin-independent internalization route. In concern about molecular complexes engaged in scission, fission and vesicle transport [96, 97], cld7 shares with tetraspanins [67] the association with clathrin, dynein and Snare proteins. Notably, too, colon cancer organoids deliver two types of exosomes. Only apical exosomes express tetraspanins, cld7 and EpC [22]. Coimmunoprecipitation of ASMLwt and -cld7mPalm exosome lysates confirmed that the exosomal CIC markers EpC, CD44v6 and α6β4 are abundant in cld7 precipitates of ASMLwt, but not -cld7mPalm exosomes. Though it remains to be answered, whether palmitoylated cld7 actively contributes to exosome biogenesis, by its recruitment into exosomes the possibility should be taken into account that palmitoylated, GEM-located cld7 supports tumor progression also via exosomes.
As summarized in Figure 9, distinct to non-palmitoylated cld7, which is enriched in TJ complexes, palmitoylated cld7 is recruited into GEM, where it associates with EpC, tetraspanins, integrins, proteases, cytoskeletal components and signal transductions molecules. It is prone for internalization and release in exosomes. The multiple interactions of GEM-located protein complexes prohibit assigning activities as exclusively being palmitoylated cld7- versus GEM complexes-dependent. However, palmitoylated cld7 hardly shares activities with non-palmitoylated cld7 and only palmitoylated cld7 promotes metastasis.
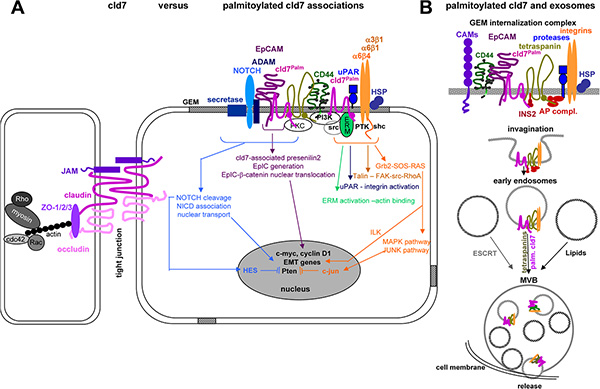
Figure 9: Overview of molecules associating and cooperating with palmitoylated cld7: (A) Only palmitoylated cld7 is enriched in GEM, where it associates in a direct protein-protein interaction with EpC. Interactions with additional transmembrane proteins, predominantly tetraspanins, integrins, CD44v6, transmembrane metalloproteases, ADAMs, uPAR and NOTCH likely are indirect and promoted by the special lipid composition of GEM and the catcher activity of tetraspanins, which may be first order partners for some of the listed membrane molecules. The majority of associations with cytoplamic molecules are indirect and promoted by the lipid composition of GEM. Thus, the multiple deficiencies associated with cld7mPalm in migration, invasion, apoptosis resistance and EMT are due to exclusion from GEM (or recruitment into TJ) and persistence in the cytosol, the latter being demonstrated for Pten phosphorylation. (B) Cld7 is recovered in exosomes. There is evidence that only palmitoylated cld7 is recovered in those exosomes, where biogenesis/vesicle transport depends on tetraspanins rather than ESCRT complexes and lipids. Whether cld7 actively contributes to the biogenesis of these “tetraspanin-dependent” exosomes remains to be explored. Taken together, there is strong evidence that TJ-integrated cld7 and palmitoylated GEM-integrated cld7 fulfill non-overlapping activities. Whether cld7 palmitoylation is linked to the metastatic phenotype or is also observed in non-transformed cells and under which conditions remains to be explored.
MATERIALS AND METHODS
Cell lines
ASML [62], ASML-EpCkd, ASML-cld7kd cells [9] were maintained in RPMI 1640/10% FCS w/wo 0.5 μg/ml G418. ASML-EpCkd cells were transfected with pcDNΑ3.1(+)hygromycin plasmid containing EpC (ASML-EpCresc) or EpC mutated at G282 and A279 (ASML EpCmAG) or ASML-cld7kd cells were transfected with cld7 mutated at AA184 and AA186 (palmitoylation site) using the pcDNΑ3.1(+)hygromycin plasmid (ASML- cld7mPalm). Where indicated, ASML-cld7kd cells were transiently transfected with wt cld7 using the pcDNΑ3.1(+)hygromycin plasmid (ASML-cld7resc). Primers are listed in Supplementary Table S2. Stable rescue clones were established by single cell cloning and were cultured in RPMI 1640/10% FCS/0.5 μg/ml G418/120 μg/ml hygromycin.
Antibodies and chemicals
See Supplementary Table S3.
Exosome preparation
Cells were cultured (48 h) in serum-free medium. Cleared supernatants (2 × 10 min, 500 g, 1 × 20 min, 2000 g, 1 × 30 min, 10000 g) were centrifuged (90 min, 100000 g) and washed (PBS, 90 min, 100000 g). The resuspended pellet was purified by sucrose gradient centrifugation [67].
Sucrose density gradient centrifugation
Cell lysates and exosomes in 2.5 M sucrose were overlaid by a continuous sucrose gradient (0.25 M–2 M) and centrifuged (15 h, 150000 g), collecting twelve 1 ml fractions.
Palmitoylation assay
Palmitoylation of cld7 was determined using the IP-ABE method [98].
IP, Western blot (WB):
Cells and exosomes were lysed for 30 min at 4°C with HEPES buffer, 1% Lubrol, 1 mM PMSF, 1 mM NaVO4, 10 mM NaF, protease inhibitor mix. During mild lysis with Lubrol protein complexes not relying on direct protein-protein interactions are not destroyed. Lysates were centrifuged (13000 g, 10 min, 4°C), mixed with antibody (1 h, 4°C) and incubated with ProteinG-Sepharose (1 h). Washed complexes/lysates, dissolved in Laemmli buffer, were resolved on 10%–12% SDS-PAGE. After protein transfer, blocking, blotting with antibodies, blots were developed with ECL.
Protein identification
After SDS-PAGE, gels were stained with Coomassie-blue. Protein digestion, sample preparation, mass spectrometeric analysis by nanoLC-ESI-MS/MS on an LTQ orbitrap and database searches were performed as described [99].
Flow-cytometry followed routine procedures. Where indicated, cells or latex beads bound exosomes [67] were fixed and permeabilized. Samples were analyzed in a FACSCalibur using the CellQuest program.
Zymography
Culture supernatant of ASML, -cld7kd and -cld7mPalm cells, starved for 24 h, was centrifuged (15 min, 15000 g). Aliquots of supernatant were incubated with Laemmli buffer (15 min, 37oC) and separated in a 10% acrylamide gel containing 1 mg/ml gelatin. After washing (2.5% Triton), gels were incubated in developing buffer (37oC, 48 h) and stained with Coomassie-blue.
Confocal microscopy
Cells on glass-slides were fixed (4% paraformaldehyde, 20 min on ice), permeabilized (1% Triton-X100, 4 min, on ice), blocked (PBS/1% gelatin, 30 min, on ice), incubated with primary antibody (60 min, on ice), washed, incubated with fluorochrome-conjugated secondary antibody (60 min, on ice), blocked (IgG with irrelevant specificity of the same species as the primary antibody), incubated with a second, dye-labeled primary antibody and washed. Slides were mounted in Elvanol. Digitized images were generated using a Leica LMS780 microscope and the Carl Zeiss Vision software for evaluation. The Z-stack offers 30 positions through the depth of the cell. All pictures were taken at Z-stack 14–16. Depending on the quality of the antibody and the density of marker expression, the intensity for the green channel varied between 700–900 master gain values and for the red channel between 500–750 master gain values. The photosystem automatically generates the single fluorescence and overlay pictures. Only overlays are shown at a 50% reduction compared to the original size. Where indicated, selected fields were amplified 10-fold (5-fold compared to original). From the amplified field the membrane or the cytoplasm were encircled for evaluation of the Pearson correlation coefficient between the red and green channel using Image J (Rasband WS, ImageJ, US National Institute of Health, Bethesda, Maryland, USA http;//imagej.nih.gov/ij/) [100].
Histology
Snap frozen sections (5 μm) were fixed, incubated with antibodies, washed, exposed to biotinylated secondary antibodies and alkaline phosphatase conjugated avidin-biotin solution. Sections were counter-stained with H&E. Digitized images were generated using a Leica DMRBE microscope.
Migration
Cells, in the upper part of a Boyden chamber (RPMI/0.1% BSA), were separated from the lower part (RPMI/20% FCS) by 8 μm pore size polycarbonate-membranes. After 16 h the lower membrane side was stained (crystal-violet), measuring OD595 after lysis. Migration is presented as % input cells. In an in vitro wound healing assay, a subconfluent monolayer was scratched with a pipette tip. Wound closure was controlled by light microscopy. For videomicroscopy, 5 × 104 cells were seeded on laminin (LN)332-coated 24-well plates. Plates were placed under an Olympus IX81 inverse microscope with a Hg/Xe lamp, an incubation chamber (37oC, 5% CO2), a CCD camera (Hamamatsu) and a ScanR acquisition soft ware (Olympus, Hamburg, Germany). Two pictures (20-fold magnification)/chamber (2 ms exposure) were taken every 20 min for 12 h. Migration was quantified according to Manual_tracking plugin running in the open-source software Image J for 20 cells per well.
Apoptosis
Cells (1 × 105) were grown for 48 h in RPMI/10% FCS containing cisplatin. Survival was monitored by annexinV-APC/PI staining, MTT assay and 3H-thymidine uptake.
Soft agar assay
Tumor cells in 0.3% agar were seeded on a preformed 1% agar layer counting colonies after 3 wk.
In vivo assays
BDX rats received 1 × 106 tumor cells intrafootpad (ifp). Rats were controlled weekly for local and draining lymph node tumor growth, short breathing or weight loss. Animals were sacrificed when draining nodes reached 2 cm diameter, rats lost > 10% weight or latest after 240 d. Animal experiments were Government-approved (Baden-Wuerttemberg, Germany).
Statistics
P values < 0.05 (two-tailed Student’s t-test, Kruskal-Wallis test) were considered significant.
Abbreviations
ASML: BSp73ASML, CIC: cancer initiating cells, cld7: claudin-7, cld7mPalm: cld7 with a mutation in the palmitoylation site, EMT: epithelial-mesenchymal transition, EpC: EpCAM, EpCmAG: EpC with a AG point mutation, EpCresc: wt EpC rescue, ESI-MS/MS: electrospray ionization tandem mass spectrometry, FN: fibronectin, GEM: glycolipid-enriched membrane microdomains, ICD: intracellular domain, ifp: intrafootpad, IP: immunoprecipitation, kd: knockdown, LN: laminin, m: mutated, resc: rescue, TJ: tight junction, WB: Western blot.
ACKNOWLEDGMENTS AND FUNDING
This investigation was supported by the Deutsche Krebshilfe and the Wilhelm Sander Stiftung (MZ). We greatly appreciate the help by Angela Frank in animal experiments, immunohistology and flow cytometry and cordially thank Dr. W. Gross, University Hospital of Surgery, Heidelberg, for help with the Image J evaluation of confocal microscopy.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
Authors’ contributions
SH and FT performed and analyzed experiments. MS performed the proteome analysis. MZ helped with experiments, planned experiments and wrote the manuscript, which was discussed with SH, FT and MS.
REFERENCES
1. Bosetti C, Bertuccio P, Malvezzi M, Levi F, Chatenoud L, Negri E, La Vecchia C. Cancer mortality in Europe, 2005–2009, and an overview of trends since 1980. Ann Oncol. 2013; 24:2657–2671.
2. Michl P, Gress TM. Current concepts and novel targets in advanced pancreatic cancer. Gut. 2013; 62:317–326.
3. Mimeault M, Batra SK. Altered gene products involved in the malignant reprogramming of cancer stem/progenitor cells and multitargeted therapies. Mol Aspects Med. 2014; 39:3–32.
4. Chen SY, Huang YC, Liu SP, Tsai FJ, Shyu WC, Lin SZ. An overview of concepts for cancer stem cells. Cell Transplant. 2011; 20:113–120.
5. Kim K, Lee KH, Lee J, Choi J. Overview of current standpoints in profiling of circulating tumor cells. Arch Pharm Res. 2014; 37:88–95.
6. Zöller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. 2011; 11:254–267.
7. Maccalli C, De Maria R. Cancer stem cells: perspectives for therapeutic targeting. Cancer Immunol Immunother. 2015; 64:91–97.
8. Gires O. Lessons from common markers of tumor-initiating cells in solid cancers. Cell Mol Life Sci. 2011; 68:4009–4022.
9. Thuma F, Zöller M. EpCAM-associated claudin-7 supports lymphatic spread and drug resistance in rat pancreatic cancer. Int J Cancer. 2013; 133:855–866.
10. Philip R, Heiler S, Mu W, Büchler MW, Zöller M, Thuma F. Claudin-7 promotes the epithelial-mesenchymal transition in human colorectal cancer. Oncotarget. 2015; 6:2046–2063. doi: 10.18632/oncotarget.2858.
11. Tsukita S, Furuse M. Overcoming barriers in the study of tight junction functions: from occludin to claudin. Genes Cells. 1998; 3:569–573.
12. Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol. 2004; 286: C1213–28.
13. Schulzke JD, Günzel D, John LJ, Fromm M. Perspectives on tight junction research. Ann NY Acad Sci. 2012; 1257:1–19.
14. Furuse M, Hata M, Furuse K, Yoshida Y, Haratake A, Sugitani Y, Noda T, Kubo A, Tsukita S. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J Cell Biol. 2002; 156:1099–1111.
15. Ding L, Lu Z, Foreman O, Tatum R, Lu Q, Renegar R, Cao J, Chen YH. Inflammation and disruption of the mucosal architecture in claudin-7-deficient mice. Gastroenterology. 2012; 142; 305–315.
16. Tanaka H, Takechi M, Kiyonari H, Shioi G, Tamura A, Tsukita S. Intestinal deletion of Claudin-7 enhances paracellular organic solute flux and initiates colonic inflammation in mice. Gut. 2015; 64:1529–1538.
17. Lal-Nag M, Morin PJ. The claudins. Genome Biol. 2009; 10:235.
18. Ding L, Lu Z, Lu Q, Chen YH. The claudin family of proteins in human malignancy: a clinical perspective. Cancer Manag Res. 2013; 5:367–375.
19. Van Itallie CM, Anderson JM. Claudin interactions in and out of the tight junction. Tissue Barriers. 2013; 1: e25247.
20. Findley MK, Koval M. Regulation and roles for claudin-family tight junction proteins. IUBMB Life. 2009; 61:431–437.
21. Gonzalez-Mariscal L, Namorado Mdel C, Martin D, Sierra G, Reyes JL. The tight junction proteins claudin-7 and -8 display a different subcellular localization at Henle’s loops and collecting ducts of rabbit kidney. Nephrol Dial Transplant. 2006; 21:2391–2398.
22. Tauro BJ, Greening DW, Mathias RA, Mathivanan S, Ji H, Simpson RJ. Two distinct populations of exosomes are released from LIM1863 colon carcinoma cell-derived organoids. Mol Cell Proteomics. 2013; 12:587–598.
23. Lu Z, Kim do H, Fan J, Lu Q, Verbanac K, Ding L, Renegar R, Chen YH. A non-tight junction function of claudin-7-Interaction with integrin signaling in suppressing lung cancer cell proliferation and detachment. Mol Cancer. 2015; 14:120.
24. Li WY, Huey CL, Yu AS. Expression of claudin-7 and -8 along the mouse nephron. Am J Physiol Renal Physiol. 2004; 286:F1063–1071.
25. D’Souza T, Agarwal R, Morin PJ. Phosphorylation of claudin-3 at threonine 192 by cAMP-dependent protein kinase regulates tight junction barrier function in ovarian cancer cells. J Biol Chem. 2005; 280:26233–26240.
26. French AD, Fiori JL, Camilli TC, Leotlela PD, O’Connell MP, Frank BP, Subaran S, Indig FE, Taub DD, Weeraratna AT. PKC and PKA phosphorylation affect the subcellular localization of claudin-1 in melanoma cells. Int J Med Sci. 2009; 6:93–101.
27. Chen C, Wang P, Su Q, Wang S, Wang F. Myosin light chain kinase mediates intestinal barrier disruption following burn injury. PLoS One. 2012; 7:e34946.
28. Nishida M, Yoshida M, Nishiumi S, Furuse M, Azuma T. Claudin-2 regulates colorectal inflammation via myosin light chain kinase-dependent signaling. Dig Dis Sci. 2013; 58:1546–1559.
29. Ikari A, Ito M, Okude C, Sawada H, Harada H, Degawa M, Sakai H, Takahashi T, Sugatani J, Miwa M. Claudin-16 is directly phosphorylated by protein kinase A independently of a vasodilator-stimulated phosphoprotein-mediated pathway. J Cell Physiol. 2008; 214:221–229.
30. Li X, Akhtar S, Choudhry MA. Alteration in intestine tight junction protein phosphorylation and apoptosis is associated with increase in IL-18 levels following alcohol intoxication and burn injury. Biochim Biophys Acta. 2012; 1822:196–203.
31. Shen L. Tight junctions on the move: molecular mechanisms for epithelial barrier regulation. Ann N Y Acad Sci. 2012; 1258:9–18.
32. Sjö A, Magnusson KE, Peterson KH. Protein kinase C activation has distinct effects on the localization, phosphorylation and detergent solubility of the claudin protein family in tight and leaky epithelial cells. J Membr Biol. 2010; 236:181–189.
33. Heiler S, Mu W, Zöller M, Thuma F. The importance of claudin-7 palmitoylation on membrane subdomain localization and metastasis-promoting activities. Cell Commun Signal. 2015; 13:29.
34. Nübel T, Preobraschenski J, Tuncay H, Weiss T, Kuhn S, Ladwein M, Langbein L, Zöller M. Claudin-7 regulates EpCAM-mediated functions in tumor progression. Mol Cancer Res. 2009; 7:285–299.
35. Levental I, Grzybek M, Simons K. Greasing their way: lipid modifications determine protein association with membrane rafts. Biochemistry. 2010; 49:6305–6316.
36. Stepanek O, Draber P, Horejsi V. Palmitoylated transmembrane adaptor proteins in leukocyte signaling. Cell Signal. 2014; 26:895–902.
37. Head BP, Patel HH, Insel PA. Interaction of membrane/lipid rafts with the cytoskeleton: impact on signaling and function: membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim Biophys Acta. 2014; 1838:532–545.
38. Mollinedo F, Gajate C. Lipid rafts as major platforms for signaling regulation in cancer. Adv Biol Regul. 2015 ; 57:130–146.
39. Lajoie P, Nabi IR. Lipid rafts, caveolae, and their endocytosis. Int Rev Cell Mol Biol. 2010; 282:135–163.
40. Ewers H, Helenius A. Lipid-mediated endocytosis. Cold Spring Harb Perspect Biol. 2011; 3:a004721.
41. Zöller M. Tetraspanins: push and pull in suppressing and promoting metastasis. Nat Rev Cancer. 2009; 9:40–55.
42. Nicolson GL. Cell membrane fluid-mosaic structure and cancer metastasis. Cancer Res. 2015; 75:1169–1176.
43. Andreu Z, Yáñez-Mó M. Tetraspanins in extracellular vesicle formation and function. Front Immunol. 2014; 5:442.
44. Singh AB, Sharma A, Dhawan P. Claudin family of proteins and cancer: an overview. J Oncol. 2010; 2010:541957.
45. Kwon MJ. Emerging roles of claudins in human cancer. Int J Mol Sci. 2013; 14:18148–18180.
46. Escudero-Esparza A, Jiang WG, Martin TA. The Claudin family and its role in cancer and metastasis. Front Biosci. 2011; 16:1069–1083.
47. Cheng JM, Volk L, Janaki DK, Vyakaranam S, Ran S, Rao KA. Tumor suppressor function of Rab25 in triple-negative breast cancer. Int J Cancer. 2010; 126:2799–2812.
48. Imrich S, Hachmeister M, Gires O. EpCAM and its potential role in tumor-initiating cells. Cell Adh Migr. 2012; 6:30–38.
49. Kuhn S, Koch M, Nübel T, Ladwein M, Antolovic D, Klingbeil P, Hildebrand D, Moldenhauer G, Langbein L, Franke WW, Weitz J, Zöller M. A complex of EpCAM, claudin-7, CD44 variant isoforms, and tetraspanins promotes colorectal cancer progression. Mol Cancer Res. 2007; 5:553–567.
50. Ladwein M, Pape UF, Schmidt DS, Schnölzer M, Fiedler S, Langbein L, Franke WW, Moldenhauer G, Zöller M. The cell-cell adhesion molecule EpCAM interacts directly with the tight junction protein claudin-7. Exp Cell Res. 2005; 309:345–357.
51. Wu CJ, Mannan P, Lu M, Udey MC. Epithelial cell adhesion molecule (EpCAM) regulates claudin dynamics and tight junctions. J Biol Chem. 2013; 288:12253–12268.
52. Okada T, Nakamura T, Watanabe T, Onoda N, Ashida A, Okuyama R, Ito K. Coexpression of EpCAM, CD44 variant isoforms and claudin-7 in anaplastic thyroid carcinoma. PLoS One. 2014; 9:e94487.
53. Winter MJ, Nagelkerken B, Mertens AE, Rees-Bakker HA, Brtiaire-deBruijn IH, Litvinov SV. Expression of EpCAM shifts the state of cadherin-mediated adhesions from strong to weak. Exp Cell Res. 2003; 285:50–58.
54. Yamashita T, Budhu A, Forgues M, Wang XW. Activation of hepatic stem cell marker EpCAM by Wnt-beta-catenin signaling in hepatocellular carcinoma. Cancer Res. 2007; 67:10831–10839.
55. Maghzal N, Vogt E, Reintsch W, Fraser JS, Fagotto F. The tumor-associated EpCAM regulates morphogenetic movements through intracellular signaling. J Cell Biol. 2010; 191:645–659.
56. Denzel S, Mack B, Eggert C, Massoner P, Stöcklein N, Kemming D, Harréus U, Gires O. MMP7 is a target of the tumour-associated antigen EpCAM. Int J Exp Pathol. 2012; 93:341–353.
57. Sankpal NV, Willman MW, Fleming TP, Mayfield JD, Gillanders WE. Transcriptional repression of epithelial cell adhesion molecule contributes to p53 control of breast cancer invasion. Cancer Res. 2009; 69:753–757.
58. Maetzel D, Denzel S, Mack B, Eggert C, Bärr G, Gires O. Nuclear signalling by tumour-associated antigen EpCAM. Nat Cell Biol. 2009; 11:162–171.
59. Lin CW, Liao MY, Lin WW, Wang YP, Lu TY, Wu HC. Epithelial Cell Adhesion Molecule Regulates Tumor Initiation and Tumorigenesis via Activating Reprogramming Factors and Epithelial-Mesenchymal Transition Genes Expression in Colon Cancer. J Biol Chem. 2012; 287: 39449–39459.
60. Yovchev MI, Grozdanov PN, Zhou H, Racherla H, Guha C, Dabeva MD. Identification of adult hepatic progenitor cells capable of repopulating injured rat liver. Hepatology. 2008; 47:636–647.
61. Lei Z, Maeda T, Tamura A, Nakamura T, Yamazaki Y, Shiratori H, Yashiro K, Tsukita S, Hamada H. EpCAM contributes to formation of functional tight junction in the intestinal epithelium by recruiting claudin proteins. Dev Biol. 2012; 371:136–145.
62. Matzku S, Komitowski D, Mildenberger M, Zöller M. Characterization of BSp73, a spontaneous rat tumor and its in vivo selected variants showing different metastasizing capacities. Invasion Metastasis. 1983; 3:109–123.
63. Jung T, Gross W, Zöller M. CD44v6 coordinates tumor matrix-triggered motility and apoptosis resistance. J Biol Chem. 2011; 286:15862–15874.
64. Yue S, Mu W, Zöller M. Tspan8 and CD151 promote metastasis by distinct mechanisms. Eur J Cancer. 2013; 49:2934–2948.
65. Fragoso R, Barata JT. Kinases, tails and more: regulation of PTEN function by phosphorylation. Methods. 2015; 77–78:75–81.
66. Thuma F, Zöller M. Outsmart tumor exosomes to steal the cancer initiating cell its niche. Semin Cancer Biol. 2014; 28:39–50.
67. Rana S, Yue S, Stadel D, Zöller M. Toward tailored exosomes: the exosomal tetraspanin web contributes to target cell selection. Int J Biochem Cell Biol. 2012; 44:1574–1584.
68. Krug SM, Schulzke JD, Fromm M. Tight junction, selective permeability, and related diseases. Semin Cell Dev Biol. 2014; 36:166–176.
69. Alikanoglu AS, Gunduz S, Demirpence O, Suren D, Gunduz UR, Sezer C, Yildiz M, Yildirim M. Expression pattern and prognostic significance of claudin 1, 4 and 7 in pancreatic cancer. Asian Pac J Cancer Prev. 2015; 16:4387–4392.
70. Cunniffe C, Brankin B, Lambkin H, Ryan F. The role of claudin-1 and claudin-7 in cervical tumorigenesis. Anticancer Res. 2014; 34:2851–2857.
71. Cao C, Wang W, Ma C, Jiang P. Computational analysis identifies invasion-associated genes in pituitary adenomas. Mol Med Rep. 2015; 12:1977–1982.
72. Bouchagier KA, Assimakopoulos SF, Karavias DD, Maroulis I, Tzelepi V, Kalofonos H, Karavias DD, Kardamakis D, Scopa CD, Tsamandas AC. Expression of claudins-1, -4, -5, -7 and occludin in hepatocellular carcinoma and their relation with classic clinicopathological features and patients’ survival. In Vivo. 2014; 28:315–326.
73. Okada T, Nakamura T, Watanabe T, Onoda N, Ashida A, Okuyama R, Ito K. Coexpression of EpCAM, CD44 variant isoforms and claudin-7 in anaplastic thyroid carcinoma. PLoS One. 2014; 9:e94487.
74. Holczbauer Á, Gyöngyösi B, Lotz G, Törzsök P, Kaposi-Novák P, Szijártó A, Tátrai P, Kupcsulik P, Schaff Z, Kiss A. Increased expression of claudin-1 and claudin-7 in liver cirrhosis and hepatocellular carcinoma. Pathol Oncol Res. 2014; 20:493–502.
75. Jun KH, Kim JH, Jung JH, Choi HJ, Chin HM. Expression of claudin-7 and loss of claudin-18 correlate with poor prognosis in gastric cancer. Int J Surg. 2014; 12:156–162.
76. Shen L. Tight junctions on the move: molecular mechanisms for epithelial barrier regulation. Ann N Y Acad Sci. 2012; 1258:9–18.
77. Lu Z, Kim do H, Fan J, Lu Q, Verbanac K, Ding L, Renegar R, Chen YH. A non-tight junction function of claudin-7-Interaction with integrin signaling in suppressing lung cancer cell proliferation and detachment. Mol Cancer. 2015; 14:120.
78. Vilen ST, Suojanen J, Salas F, Risteli J, Ylipalosaari M, Itkonen O, Koistinen H, Baumann M, Stenman UH, Sorsa T, Salo T, Nyberg P. Trypsin-2 enhances carcinoma invasion by processing tight junctions and activating ProMT1-MMP. Cancer Invest. 2012; 30:583–592.
79. Gurbuz N, Ashour AA, Alpay SN, Ozpolat B. Down-regulation of 5-HT1B and 5-HT1D receptors inhibits proliferation, clonogenicity and invasion of human pancreatic cancer cells. PLoS One. 2014; 9: e110067.
80. Joghetaei N, Stein A, Byrne RA, Schulz C, King L, May AE, Schmidt R. The Extracellular Matrix Metalloproteinase Inducer (EMMPRIN, CD147) - a potential novel target in atherothrombosis prevention? Thromb Res. 2013; 131:474–480.
81. Tiwari N, Gheldof A, Tatari M, Christofori G. EMT as the ultimate survival mechanism of cancer cells. Semin Cancer Biol. 2012; 22:194–207.
82. Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal. 2014; 7:re8.
83. Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009; 28:151–166.
84. Sánchez-Tilló E, Liu Y, de Barrios O, Siles L, Fanlo L, Cuatrecasas M, Darling DS, Dean DC, Castells A, Postigo A. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell Mol Life Sci. 2012; 69:3429–3456.
85. IV Santaliz-Ruiz LE, Xie X, Old M, Teknos TN, Pan Q. Emerging role of nanog in tumorigenesis and cancer stem cells. Int J Cancer. 2014; 135:2741–2748.
86. McDonald PC, Fielding AB, Dedhar S. Integrin-linked kinase—essential roles in physiology and cancer biology. J Cell Sci. 2008; 121:3121–3132.
87. Lv ZM, Wang Q, Wan Q, Lin JG, Hu MS, Liu YX, Wang R. The role of the p38 MAPK signaling pathway in high glucose-induced epithelial-mesenchymal transition of cultured human renal tubular epithelial cells. PLoS One. 2011; 6:e22806.
88. Li NY, Weber CE, Wai PY, Cuevas BD, Zhang J, Kuo PC, Mi Z. An MAPK-dependent pathway induces epithelial-mesenchymal transition via Twist activation in human breast cancer cell lines. Surgery. 2013; 154:404–410.
89. Jo M, Lester RD, Montel V, Eastman B, Takimoto S, Gonias SL. Reversibility of epithelial-mesenchymal transition (EMT) induced in breast cancer cells by activation of urokinase receptor-dependent cell signaling. J Biol Chem. 2009; 284:22825–22833.
90. Bertrand FE, McCubrey JA, Angus CW, Nutter JM, Sigounas G. NOTCH and PTEN in prostate cancer. Adv Biol Regul. 2014; 56:51–65.
91. Patsoukis N, Li L, Sari D, Petkova V, Boussiotis VA. PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol Cell Biol. 2013; 33:3091–3098.
92. Cordier F, Chaffotte A, Terrien E, Préhaud C, Theillet FX, Delepierre M, Lafon M, Buc H, Wolff N. Ordered phosphorylation events in two independent cascades of the PTEN C-tail revealed by NMR. J Am Chem Soc. 2012; 134:20533–20543.
93. Milella M, Falcone I, Conciatori F, Cesta Incani U, Del Curatolo A, Inzerilli N, Nuzzo CM, Vaccaro V, Vari S, Cognetti F, Ciuffreda L. PTEN: Multiple Functions in Human Malignant Tumors. Front Oncol. 2015; 5:24.
94. Kowal J, Tkach M, Théry C. Biogenesis and secretion of exosomes. Curr Opin Cell Biol. 2014; 29:116–125.
95. De Maio A. Extracellular heat shock proteins, cellular export vesicles, and the Stress Observation System: a form of communication during injury, infection, and cell damage. It is never known how far a controversial finding will go! Dedicated to Ferruccio Ritossa. Cell Stress Chaperones. 2011; 16:235–249.
96. Jackson LP, Kümmel D, Reinisch KM, Owen DJ. Structures and mechanisms of vesicle coat components and multisubunit tethering complexes. Curr Opin Cell Biol. 2012; 24:475–483.
97. Khan AR. Oligomerization of rab/effector complexes in the regulation of vesicle trafficking. Prog Mol Biol Transl Sci. 2013; 117:579–614.
98. Brigidi GS, Bamji SX. Detection of protein palmitoylation in cultured Hippocampal neurons by immunoprecipitation and acyl-biotin exchange (ABE). J Vis Exp. 2013; 72:e50031.
99. Junwei L, Bonifati S, Hristov G, Martilla T, Valmary-Degano S, Stanzel S, Schnölzer M, Mougin C, Aprahamian M, Grekova S, Raykov Z, Rommelaere J, Marchini A. Synergistic combination of valproic acid and oncolytic parvovirus H-1PV as a potential therapy against cervical and pancreatic carcinomas. EMBO Mol Med. 2013; 5:1537–1555.
100. Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci. 1992; 103:857–862.