
INTRODUCTION
Metastasis is the major cause of mortality from prostate cancer [1, 2] and systemic metastasis often occurs following local therapy failure in high-risk patients, including locally advanced (positive surgical margin) or high grade (Gleason sum score ≥ 8) tumors [3, 4]. Prostate cancers often migrate to extra-prostatic tissue and local lymph nodes before distant organ metastasis. The 10-year progression-free survival probabilities were 79% for organ-confined disease, but only 12% for disease with lymph node metastases. Therefore, suppressing cancer cell invasion at a very early stage would prevent the development of distal metastatic disease and slow down disease progression, improving patient quality of life for prostate cancers.
Dihydropyrimidinase-related protein 3 (DPYSL3), also called collapsin response mediator protein 4 (CRMP4), is a member of DPYSL gene family that encodes five cytosolic phospho-proteins involved in semaphorin/collapsin-induced cellular events [5]. DPYSL gene family (DPYSL1-5) shares about 50-70% sequence homology [6–9]. Previous studies indicated that CRMP1 protein is an invasion suppressor in human glioma and lung cancers [10–12], but CRMP-2 and CRMP-5 proteins were extensively expressed in colorectal cancers and high-grade lung neuro-endocrine carcinomas, respectively [12, 13]. In a search for metastasis-associated proteins using proteomics approach, we previously identified CRMP4 protein as a tumor metastasis suppressor in prostate cancers [14]. Once overexpressed or up-regulated in prostate cancer cells, CRMP4 protein suppressed cell motility or invasion in vitro and reduced tumor metastasis in mouse xenograft models [14–16]. Therefore, up-regulating DPYSL3 gene expression in prostate cancer is expected to suppress tumor metastasis, providing a significant benefit for locally advanced high-risk prostate cancer patients.
Small double-strand activating RNA (saRNA) molecules that are complementary to the gene promoter region have been demonstrated to transcriptionally up-regulate target gene expression [17–19]. This phenomenon is termed as RNA activation (RNAa) and is evolutionarily conserved across species [20]. It has been shown that the saRNAs targeting the promoter region of tumor suppressor genes, such as E-cadherin, p21cip1 and Krüppel-like family of transcription factor-4, inhibited tumor cell growth in vitro and in vivo [21–24]. Thus, we hypothesized that saRNAs with optimal properties can be used to increase the expression of silenced tumor suppressor genes such as DPYSL3 in prostate cancers.
In this study, we screened a series of saRNA molecules targeting DPYSL3 gene promoter region and identified several saRNAs that could effectively enhance DPYSL3 gene expression at the transcription level via a promoter-dependent mechanism. Transfection of these saRNAs into prostate cancer cells significantly reduced cancer cell migration and invasion in vitro. Injection of these saRNAs conjugated with a prostate cancer-homing aptamer molecule dramatically suppressed distal metastasis of prostate cancer in a mouse orthotopic xenograft model.
RESULTS
DPYSL3v2 gene expression is downregulated in metastatic prostate cancers
Human DPYSL3 gene has two transcriptional variants due to distinct promoter usage [25], as illustrated in supplemental Figure S1. These two isoforms of DPYSL3 gene encode two proteins that differ in their N-terminal amino acid sequence of exon 1 region [7, 25]. The isoform-1 has 2055 nt in cDNA nucleotide sequence while isoform-2 is 1713 nt. These isoforms are translated to proteins of CRMP4b (DPYSL3v1, 684 aa, 75 KD) and CRMP4a (DPYSL3v2, 570 aa, 64 KD).
We examined the expression profiles of these two isoforms in human prostate cancers and prostate cancer cell lines. In the online database Oncomine™, 9 out of 14 published datasets showed a significant reduction of DPYSL3 gene expression in malignant tissues compared to the benign tissues (Table 1) and the fold reduction was from 1.705 to 3.325. Analysis of one dataset from publically available Oncomine™ database [26] revealed that DPYSL3 expression was largely reduced in metastatic prostate cancer tissues compared to benign prostatic tissues (about 20-fold) and primary prostate cancers (about 15-fold) (Figure 1A). We also re-analyzed a published cDNA microarray dataset generated from prostate cancer tissues as described previously [27, 28] and identified a clear association of DPYSL3 gene reduction along with disease progression from primary cancer to castration-resistant metastatic cancers (Figure 1B). These data further confirm our previous report [14] that DPYSL3 gene expression is reduced in metastatic prostate cancers.
Table 1: ONCOMINE™ database analysis of DPYSL3 gene expression
Reported studies |
Cases |
Fold change |
P |
|
|---|---|---|---|---|
Normal prostate |
Prostate Cancers |
|||
Welsh et al. Cancer Res 2001 |
9 |
25 |
-3.325 |
9.39E-5 |
Grasso et al. Nature 2012 |
28 |
59 |
-2.199 |
9.57E-9 |
Arredouani et al. Clin Cancer Res 2009 |
8 |
13 |
-2.118 |
5.24E-5 |
Yu et al. J Clin Oncol 2004 |
23 |
65 |
-2.070 |
1.83E-8 |
|
|
|
|
|
Lapointe et al. PNAS 2004 |
41 |
60 |
-1.888 |
1.87E-12 |
Vanaja et al. Cancer Res 2003 |
8 |
27 |
-1.867 |
2.74E-4 |
Liu et al. Cancer Res 2006 |
13 |
44 |
-1.850 |
1.24E-9 |
Luo et al. Mol Carcinog 2002 |
15 |
15 |
-1.808 |
1E-3 |
Taylor et al. Cancer Cell 2010 |
29 |
131 |
-1.705 |
2.148E-8 |
LaTulippe et al. Cancer Res 2002 |
3 |
23 |
-2.776 |
0.058 |
Varambally et al. Cancer Cell 2005 |
6 |
7 |
-1.462 |
0.071 |
Magee et al. Cancer Res 2001 |
4 |
8 |
-1.588 |
0.084 |
Sing et al. Cancer Cell 2002 |
50 |
52 |
-1.193 |
0.314 |
Wallace TA et al. Cancer Res. 2008 |
20 |
69 |
-1.042 |
0.407 |
Expression profiles of DPYSL3 genes in 14 cDNA microarray datasets were extracted from the Oncomine database along with the publication citations, the case numbers, fold induction and the P values. Bold fonts indicate data with statistical significance.
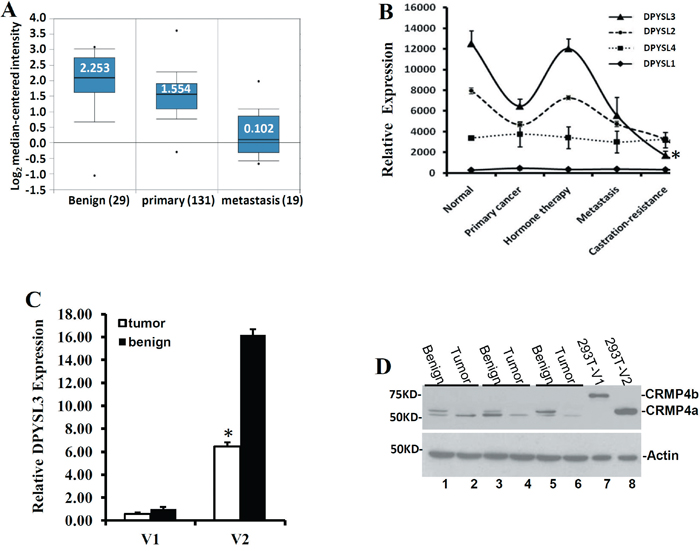
Figure 1: DPYSL3v2/CRMP4a expression is reduced in metastatic prostate cancers. A. Public dataset [26] was extracted from Oncomine™ and graphed in to three groups. The numbers in white indicate the median value and patient case number is indicated on x-axel. B. cDNA microarray dataset from a previously published report [27] was re-analyzed. Data represent the Mean from different patient groups, including benign prostate specimens (normal, n = 5), primary cancers (n = 23), tumors after hormone therapy (n = 17), metastasis (n = 9) and castration-resistant tumors (n = 3). The errors bars indicate the standard error of mean (SEM). The asterisk indicates a significant difference compared to other groups (p < 0.05, student’s t-test). C. Quantitative analysis of DPYSL3 variants in prostate cancers were conducted using total RNAs extracted from frozen tumor specimens and the individually matched nonmalignant compartments, as described [46]. The expression levels of DPYSL3 variants were normalized against the epithelium-specific gene KRT18 before the relative values were calculated. The relative ratio of gene expression level in malignant compared to benign tissues was presented as fold induction. Error bar represents the SEM. The asterisk indicates a significant difference compared to the ‘normal’ group (p < 0.05, student’s t-test). D. Western blot assays were used to evaluate CRMP4 protein expression in 3 pairs of prostate specimens. Cell lysates from 293T cells overexpressing CRMP4a and CRMP4b were used to as positive control. Actin blot served as protein loading control. Data represent two separate experiments.
To understand if DPYSL3 isoforms are differently expressed in prostate cancer tissues, we conducted a real-time PCR analysis of prostate tissues obtained from radical prostatectomy. Quantitative data revealed that DPYSL3v2 transcript was the dominant one with a remarkably higher level than DPYSL3v1 transcript. However, DPYSL3v2 levels were significantly lower in malignant tissues compared to that in case-matched surrounding benign tissues (Figure 1C). These results were consistent with our previous report [14]. At the protein level, only CRMP4a (encoded by DPYSL3v2) but not CRMP4b (encoded by DPYSL3v1) was detected in both benign and malignant tissues (Figure 1D), which is consistent with the low mRNA expression level of DPYSL3v1 gene transcript in prostate tissues. Nonetheless, CRMP4a protein levels were much lower in malignant tissues compared to their benign counterparts, similar to the mRNA expression pattern. Interestingly, CRMP4a protein exerted as a duplet band in benign tissues but as a single band in malignant tissues, indicating a protein modification that is lost in malignant tissue, for example, CRMP4 was reported to be phosphorylated by GSK-3 after CDK5/DYRK2 prime phosphorylation [29].
Promoter-targeted saRNA enhances DPYSL3v2 gene expression
To enhance DPYSL3 gene expression, we utilized a recently invented small activating RNA (saRNA) approach. Multiple saRNA molecules were designed to target DPYSL3 promoters based on the criteria reported previously [17, 20]. The saRNA targeting sites were shown in Figure S2A and S2B, and their sense DNA sequences were listed in Table 2. Four different prostate cancer cell lines were used in screening active saRNAs with quantitative real-time PCR assays. A total of 8 saRNAs targeting the DPYSL3v1 promoter was tested but none of them had any significant enhancing effect on DPYSL3v1 expression (Figure 2A-2E). In contrast, 4 out of 14 saRNAs targeting the DPYSL3v2 promoter actively enhanced gene expression in different cell lines, especially two (saV2-5 and saV2-9) of these 4 saRNAs exerted dramatic enhancing effect in all cell lines tested (Figure 2A-2E). There was no cross effect observed between the promoter-specific saRNAs.
Table 2: DPYSL3 saRNA target sequence and genomic location on Chromosome 5
saRNA# |
start |
end |
sense |
promoter site |
|---|---|---|---|---|
V1-1 |
147510470 |
147510452 |
GGATTGTGAGTCTGTCGTA |
V1p-414/-396 |
V1-2 |
147510652 |
147510634 |
GGGCTTTTGTGACTCTCAA |
V1p-596/-578 |
V1-3 |
147510738 |
147510720 |
GTGATCTGAAATGAGGCAA |
V1p-698/-664 |
V1-4 |
147510793 |
147510775 |
GAGTGCTTTGCAAGGCAAA |
V1p-737/-719 |
V1-5 |
147510816 |
147510798 |
ATGAGGGACAGAGCATCAA |
V1p-760/-742 |
V1-6 |
147510852 |
147510834 |
TGTCAGTGTGTGAGATTAA |
V1p-796/-778 |
V1-7 |
147510853 |
147510835 |
GTGTCAGTGTGTGAGATTA |
V1p-797/-779 |
V1-8 |
147510909 |
147510891 |
ATCTGGGCTTAGGTGTGAA |
V1p-853/-835 |
|
|
|
|
|
V2-1 |
147453872 |
147453854 |
AGCTGGCGCAGCAAAAGAA |
V2p-178/-160 |
V2-2 |
147453945 |
147453927 |
GGAAAATCAATAGGGATAA |
V2p-248/-230 |
V2-3 |
147453958 |
147453940 |
GCAGATGCAAAGAGGAAAA |
V2p-261/-243 |
V2-4 |
147453959 |
147453941 |
GGCAGATGCAAAGAGGAAA |
V2p-262/-244 |
V2-5 |
147453960 |
147453942 |
AGGCAGATGCAAAGAGGAA |
V2p-263/-245 |
V2-6 |
147454014 |
147453996 |
CAGCACTCGCGAATCAGAA |
V2p-317/-299 |
V2-7 |
147454430 |
147454412 |
CACGGCTTTCCATTTTCTA |
V2p-733/3715 |
V2-8 |
147454534 |
147454516 |
AGGCAGTGGAGTTTCTTTA |
V2p-837/-819 |
V2-9 |
147454573 |
147454555 |
GCAGCATTCATGTTCTTTC |
V2p-876/-858 |
V2-10 |
147454717 |
147454699 |
ACTCGGGATTGTGAGCAGT |
V2p-1022/-1002 |
V2-11 |
147454722 |
147454704 |
AGTAAACTCGGGATTGTGA |
V2p-1027/-1007 |
V2-12 |
147454795 |
147454777 |
TGTATTCCATCGCCGAAGG |
V2p-1100-1080 |
V2-13 |
147455020 |
147455002 |
GATTGACAATTGGGGAGCG |
V2p-1325/-1305 |
V2-14 |
147455219 |
147455201 |
AGCCCAAGCCGGAGTATTC |
V2p-1524/-1504 |
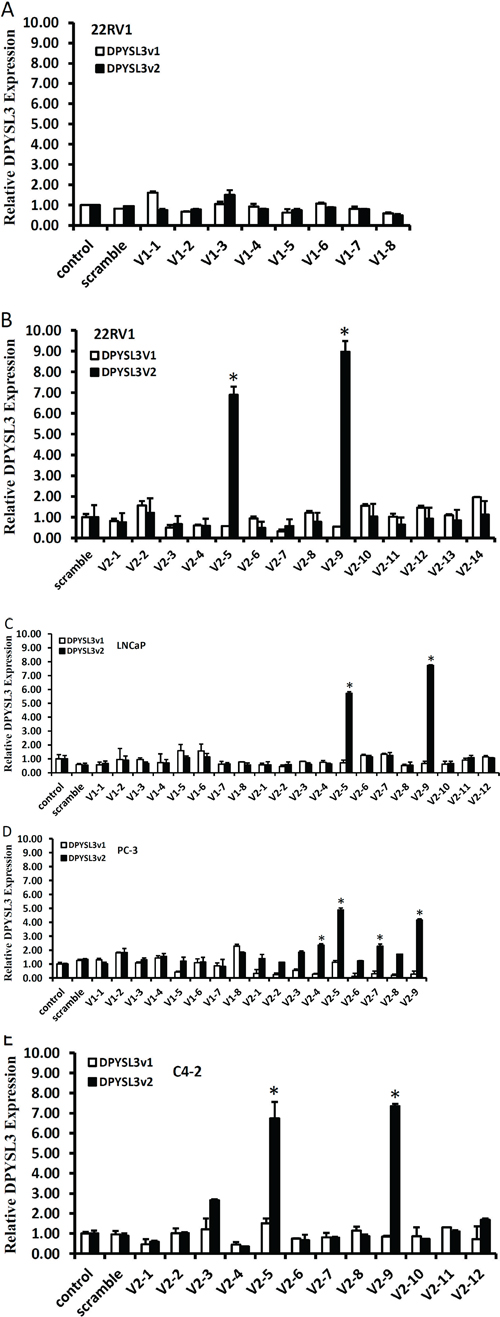
Figure 2: Promoter-targeting saRNAs enhance DPYSL3v2 expression in prostate cancer cells. A total of 8 saRNAs for DPYSL3v1 promoter and 14 saRNAs for DPYSL3v2 were transfected into prostate cancer 22RV1 panel A and B. LNCaP C. PC-3 D. and C4-2 E. as indicated at a final concentration of 10 nM in media in 6-well plates. Cells were harvested 3 days later for qPCR assays. After normalized with S18 gene expression levels, fold induction against the mock control transfection (set of 1) was calculated and graphed. Data were shown as the average from three independent experiments and the error bar represents the SEM. The asterisk indicates a significant difference compared to the control (p < 0.05, student’s t-test).
We then evaluated the saRNAs on gene expression at the protein level. Based on the qPCR data, we chose five saRNAs, including the non-effective saV2-1, less effective saV2-4 and saV2-7, plus the most effective saV2-5 and saV2-9. Western blot data showed a consistent upregulation of CRMP4a protein levels after transfection of the saRNAs in a similar order as seen in qPCR assays (Figure 3A-3B). In detail, saV2-5 and saV2-9 exerted a stronger effect on CRMP4a protein expression compared to other saRNAs. They also exerted a dramatic dose-dependent enhancing effect on CRMP4a protein expression (Figure 3C-3D). These data indicate that saV2-5 and saV2-9 saRNAs are potent enhancers in upregulating DPYSL3 gene expression.
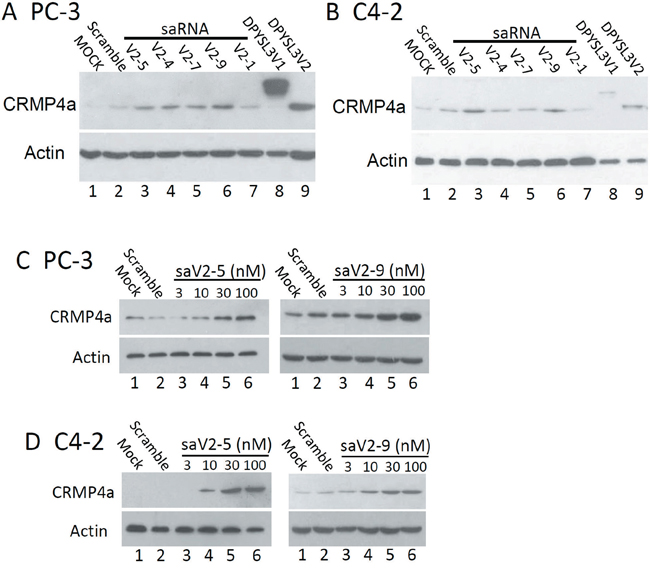
Figure 3: Promoter-targeting saRNAs enhance CRMP4a expression in prostate cancer cells. Selective saRNAs were transfected into PC-3 or C4-2 cells at a concentration of 10 nM A and B. or as indicated in each panel C and D. for 3 days, and cell lysates were subjected to western blot assays for CRMP4a expression. The scramble control saRNAs were used at 10 nM. Cellular protein lysates from PC-3 or C4-2 cells over-expressing exogenous CRMP4a or CRMP4b were used as positive control. Actin blot served as protein loading control.
To further examine if saRNA-enhanced DPYSL3 gene expression is a promoter-specific activity, we created a luciferase reporter construct driven by a 1250-bp segment (-1275/-25 from the transcription start site) of DPYSL3v2 promoter, as illustrated in Figure 4A. The reporters were packaged as lentiviral particles. Transfection of the saRNAs in PC-3 cells infected with the lentiviral luciferase reporter (DPYSL3v2p-LUC) significantly enhanced the reporter activity (Figure 4B), of which the saV2-9 saRNA displayed the most potent effect compared to others (Figure 4C). To confirm the sequence specificity of the saRNAs, we generated two mutant reporters by deleting the targeting segments of saV2-5 (Δv2-5p-LUC) and saV2-9 (Δv2-9l-LUC) saRNAs on the promoter individually (Figure 4A). As expected, deleting the corresponding segment on the promoter abolished the saRNA-induced reporter activity (Figure 4D). These data demonstrate that saRNA-mediated gene activation is indeed a promoter-specific event of transcriptional activation.
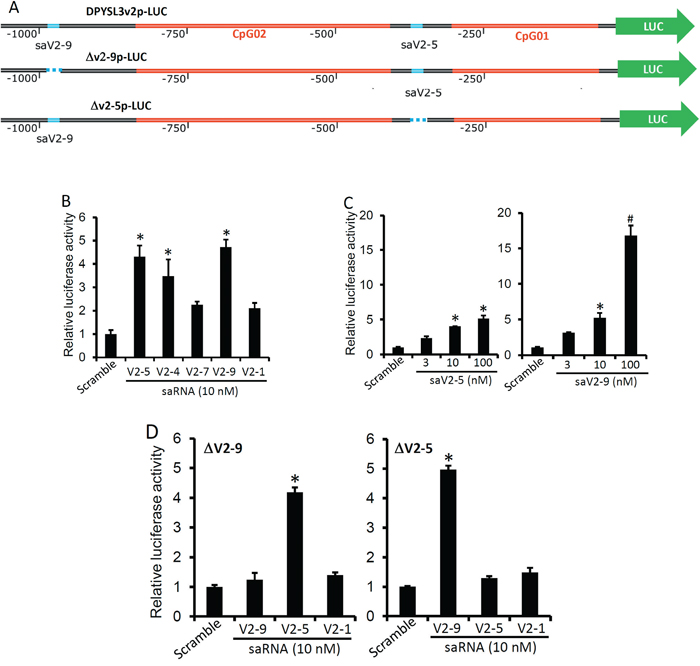
Figure 4: Promoter-specific effect of the saRNAs on DPYSL3 expression. A. Scheme of DPYSL3v2 promoter-driven luciferase reporters. Δv2-9 or Δv2-5 indicates a fragment deletion of the saRNA-targeted site. B-D PC-3 cells plated in 6-well plates were infected with lentiviral particle containing the parental reporter (B & C) or individual mutant reporter (D) and then transfected with the saRNAs at a dose of 10 nM B. or as indicated in the panel C and D.. The scramble saRNA was used at 10 nM. Luciferase activity was measured one day later and the fold induction was calculated against the scramble control. Data were shown as the average and the error bar represents the SEM. The asterisk indicates a significant difference compared to the control (p < 0.05, student’s t-test).
DPYSL3 saRNAs suppress cancer cell migration and invasion
To assess the potential of DPYSL3v2 saRNAs as anti-metastasis agents, we then tested their effect on tumor cell migration and invasion in vitro. Cell migration was assessed with a wound healing assay. As shown in Figure 5A-5B, transfection of saV2-9 molecules significantly suppressed cell migration in both PC-3 and C4-2 cells compared to the scramble and the negative saV2-1 saRNAs, while the saV2-5 also significantly suppressed cancer cell migration at a less extent.
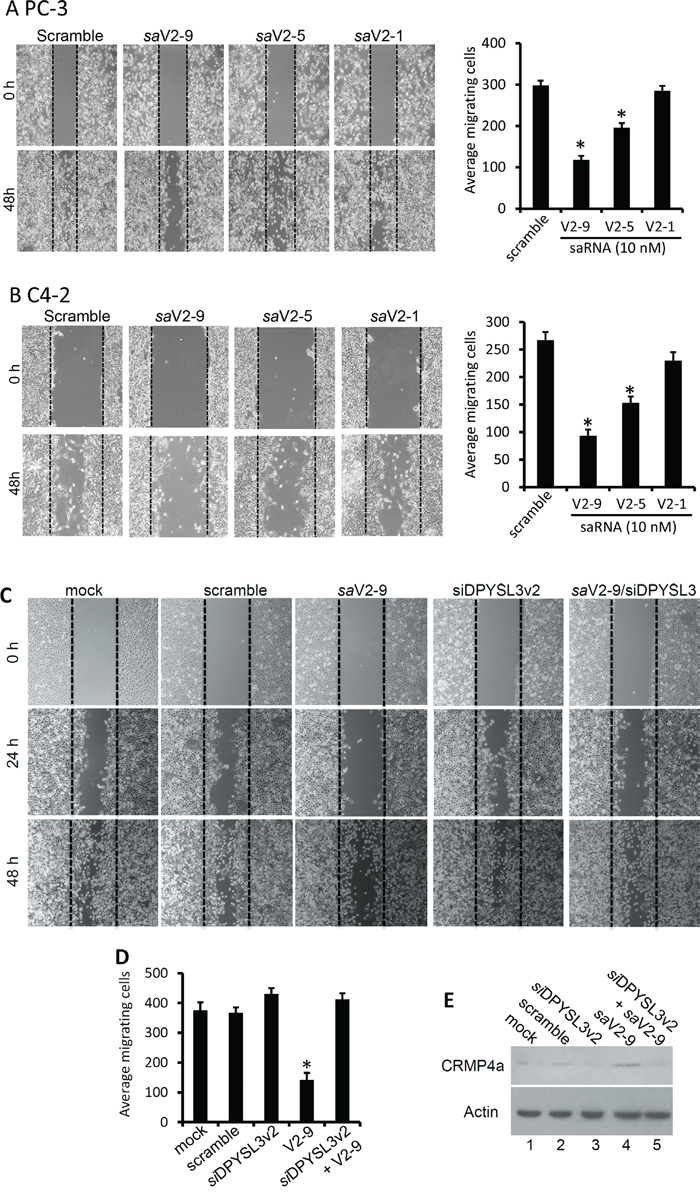
Figure 5: DPYSL3v2-targeted saRNAs suppress cell migration. A and B. Cells as indicated were seeded in 24-well plates and transfected with the saRNAs as indicated (10 nM). A wound scratch was made with a sterile 200-μl pipet tip and cells migrated into the wound space were counted at day 3 and the representing microscopic fields were recorded (A-B). The quantitative data of migrated cells from 3 microscopic fields were summarized in the graph panels next to the image panel. C and D. Similar wound healing assay was conducted except a siRNA against DPYSL3v2 was transfected with or without the saV2-9 saRNAs. Microscopic images in panel C were taken from each treatment and the quantitative data from 3 microscopic fields were graphed in panel D. All small RNAs were used at a concentration of 10 nM in the media. E. CRMP4a protein levels in the experiments of panel C were evaluated by western blot assay. Actin blot served as protein loading control.
To verify if saRNA-mediated DPYSL3 gene upregulation played a critical role in suppressing cancer cell migration, we co-transfected the saV2-9 saRNA together with a small interfering RNA (siRNA) of DPYSL3 gene in PC-3 cells. Comparing to the saV2-9 alone that significantly reduced cell migration, co-transfection of DPYSL3 siRNAs abolished saV2-9-induced suppressing effect on cell migration (Figure 5C-5D). Notably, DPYSL3 siRNA only had a slight enhancing effect on cell migration, possibly due to a lower level of the endogenous gene expressed in PC-3 cells (Figure 5E). These data suggest that DPYSL3v2 gene upregulation is a critical event in saV2-9-mediated suppression on cell migration.
To assess the anti-invasion effect of the saRNAs, we utilized the Boyden-Chamber assay. As shown in Figure 6A, saV2-9 transfection significantly reduced the numbers of migrated PC-3 cells compared to the scramble saRNA, in contrast, silencing DPYSL3v2 expression with siRNA approach largely increased PC-3 migration into low chamber. Consistently, co-transfection of the saV2-9 with DPYSL3 siRNA abolished the saV2-9-mediated suppression but increased cell migration. To rule out the possibility that these anti-migration and anti-invasion effect was due to reduced overall cell growth, we tested the effect of saRNAs on cell growth in a sulforhodamine B (SRB) assay [30]. As shown in Figure 6B-6C, the saRNAs used in the assays, either active or inactive on DPYSL3 gene expression, had no significant effect on cell growth compared to the scramble saRNA in both PC-3 and C4-2 cells. Taken together, these data demonstrate that the saV2-9 molecule is capable in suppressing cell migration by potently up-regulating DPYSL3v2 gene expression in prostate cancer cells.
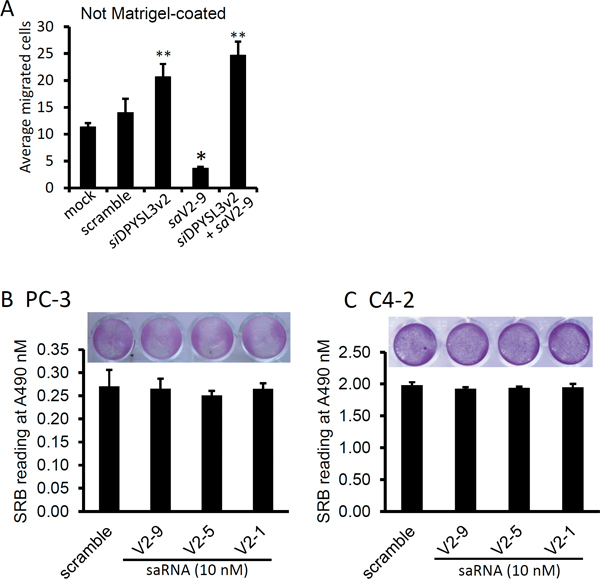
Figure 6: DPYSL3v2-targeted saRNAs suppress cell invasion. A. After transfection with the saRNAs or siRNA at 10 nM concentration as indicated, PC-3 cells were loaded onto the upper chamber. Three days later the migrated cells were stained with Hoechst 33342 (0.5 μg/ml) and counted (Mean ± SEM). The asterisks indicate a significant difference compared to the mock control (* p < 0.01, ** p < 0.05, student’s t-test). B and C. PC-3 or C4-2 cells were plated in 24-well plates and transfected with the saRNAs at indicated for 3 days. Cell growth was evaluated with SBR assay as described in the text. Representing microscopic images were inserted in the panels.
DPYSL3 saRNAs suppress tumor metastasis in orthotopic xenograft model
To validate the saV2-9 as a metastasis inhibitor in vivo, we conducted a proof-of-concept experiment in nude mice bearing orthotopic prostate xenografts derived from C4-2 cells. First, to achieve a prostate cancer cell-specific delivery of the saRNA molecule, we conjugated the small hairpin-structured saV2-9 RNA molecule with the prostate cancer-specific RNA aptamer A10-3.2 (Figure 7A). A10-3.2 is a specific ligand for prostate-specific membrane antigen (PSMA) [31, 32]. PSMA-dependent cellular intake was confirmed with a fluorescent-labeled APT-saV2-9 conjugate in PSMA-positive C4-2 but not in PSMA-negative PC-3 cells (Figure 7B). Western blot assays demonstrated that this A10-3.2-conjugated saV2-9 drastically enhanced CRMP4a protein expression in PSMA-positive C4-2 and LNCaP cells (Figure 7C).
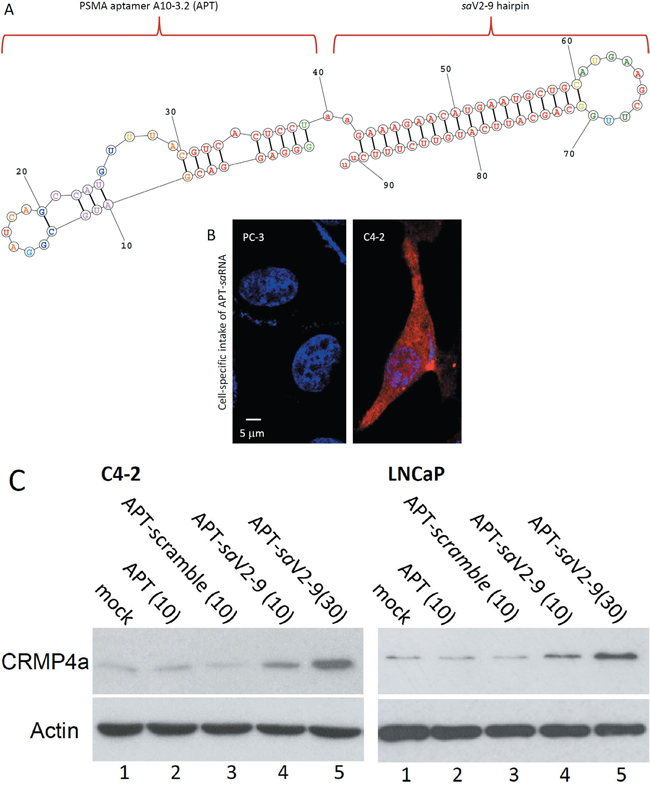
Figure 7: PSMA aptamer-conjugated saV2-9 enhances CRMP4a expression. A. Scheme of the RNA conjugate of PSMA aptamer A10-3.2 with saV2-9 hairpin. Predicted RNA folding was performed online at http://rna.urmc.rochester.edu/RNAstructureWeb/. B. PC-3 or C4-2 cells were seeded in a chambered glass slide and the A10-3.2-V2-9 conjugates that were 5′-labeled with a fluorescent dye TYE™ 563 were added into cell culture at 10 nM for 30 min. After nuclear staining with Hoechst 33342, cells were evaluated under a confocal fluorescent microscope for cellular intake of the conjugate molecules. C. C4-2 and LNCaP cells were seeded in 6-well plates and the RNA conjugates were added into cell culture for 3 days as indicated (concentration at nM). Cells were harvested for western blot assays with anti-CRMP4 antibodies. Actin blot served as protein loading control. Data represent two separate experiments.
Next, we evaluated the anti-metastasis potential of the A10-3.2-saV2-9 conjugates in mice bearing orthotopic C4-2 xenografts. A scramble RNA molecule conjugated with the A10-3.2 aptamer was used as the negative control. C4-2 cells were stably infected with lentiviral CMV-LUC reporter to express luciferase for in vivo imaging. Once the orthotopic tumor was established as monitored by living animal imaging (IVIS system) at about 4 weeks after tumor cell inoculation in dorsal lobe of the prostate as described [14], animals were treated daily by intraperitoneal injection of the conjugates at 1.0 nM per treatment for 10 consecutive days. At the end of treatment, animals were sacrificed and xenograft tumors, major organs and reginal lymph nodes were harvested for further analyses. Histological evaluation of major organs was conducted to determine regional and distal metastasis. As summarized in Table 3, there were no significant differences in animal body weight and the wet weight of orthotopic xenografts between the scramble control and saV2-9 groups. Animals received the scramble saRNA treatment all displayed metastatic lessons at regional lymph nodes and lung. One of the animals even had a big liver mass as a metastatic tumor (Figure 8A). However, animals received the saV2-9 treatment only had one incidence of reginal (lymph node) but not distal (lung) metastasis as evaluated histologically on H&E sections (Figure 8A). No sign of tumor metastasis in kidney.
Table 3: DPYSL3 saRNA suppresses tumor metastasis in vivo
parameters |
APT-scramble |
APT-saV2-9 |
p value |
|---|---|---|---|
body weight (gm ± SEM) |
32.3 ± 1.4 |
31.8 ± 1.7 |
n.s. |
O.T. tumor weight (gm ± SEM) |
2.56 ± 0.87 |
2.71 ± 0.57 |
n.s. |
lymph node met cases (%) |
5/5 (100%) |
1/5 (20%) |
< 0.05 |
Lung/Liver met cases (%) |
5/5 (100%) |
0/5 (0%) |
< 0.05 |
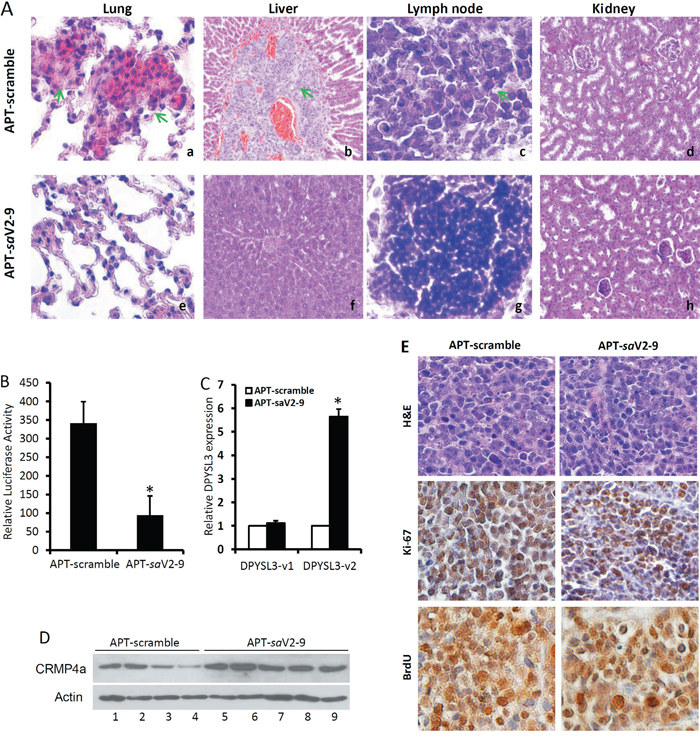
Figure 8: A10-3.2-saV2-9 conjugates suppress xenograft tumor metastasis in vivo. A. Representing H&E section images from major organs as indicated harvested from animals received the APT-saV2-9 or the scramble control. The green arrows indicate metastatic cancer cells in left lung (a), liver (b) and lymph node (c) sections from animals received the control saRNA. No metastatic cancer cells were seen in kidney sections or other organs. B. The whole right lobes of lung harvested from animals treated with the scramble or saV2-9 conjugates were homogenized and protein extracts were collected for luciferase activity measurement. The readings were normalized with the corresponding protein concentrations expressed as relative LUC reading/mg lung proteins. Data presented are MEAN ± SEM and the asterisk indicates a significant difference compared to the scramble control (P < 0.05, Student’s t-test). C. Total RNAs were extracted from xenograft tissues and subjected to qPCR analysis for DPYSl3 gene expression. After normalized with KRT18 gene expression levels, relative fold induction against the value in the samples from the scramble control group was calculated and presented as MEAN ± SEM. The asterisk indicates a significant difference compared to the scramble control (p < 0.05, student’s t-test). D. Total proteins were extracted from xenograft tumors and subjected to western blot assays for CRMP4a protein expression. Actin blot served as protein loading control. E. Xenograft tumors obtained from animals treated with the scramble or saV2-9 conjugates were processed for H&E staining, Ki-67 and BrdU immunohistochemical staining. Representing microscopic images were taken from a 200x magnification.
Because it is not easy to identify all micro-metastasis foci with conventional histological method, we performed a luciferase measurement of lung tissue protein extracts to determine total metastatic C4-2-LUC cells, as described recently [33]. As shown in Figure 8B, the luciferase activity in whole right lobe of lung tissues from saV2-9 treated animals was significantly lower than that in the control group, indicating a dramatic suppression of lung metastasis.
We also examined DPYSL3 gene expression in xenograft tissues. As expected, DPYSL3v2 (not DPYSL3v1) gene expression at the mRNA (Figure 8C) and CRMP4a protein (Figure 8D) levels were largely elevated in orthotopic xenograft tissues received saV2-9 treatment compared to that from the control group. Consistent with cell growth data (Figure 6B-6C), cell proliferation markers (Ki-67 and BrdU labeling) remained similar in orthotopic xenograft tissues from these two treatments (Figure 8E). These data strongly suggest that the saRNA approach is a potent way to enhance endogenous gene expression in vivo and that prostate cancer cell-specific delivery of DPYSL3v2 promoter-targeting saRNA is feasible in suppressing tumor metastasis in prostate cancer model.
DISCUSSION
As a proof-of-concept project, this study demonstrated that the saRNA approach is a potent method to enhance the expression of tumor metastasis suppressor DPYSL3 gene, which depends on the sequence specificity of the target gene promoter. Most importantly, this study provided the feasibility of in vivo application of saV2-9 RNA to suppress tumor metastasis.
The association of reduced CRMP4a (DPYSL3v2 gene) protein level with prostate cancer metastasis was first established in screening metastasis-associated proteins using proteomic approach [14]. Further investigation confirmed DPYSL3 gene/CRMP4 protein as a tumor metastasis suppressor because CRMP4a overexpression led to suppressed tumor cell motility in vitro and metastasis in vivo of prostate cancer cells [14]. In this study, we provided further evidences that DPYSL3 gene expression (variant 2 only) is significantly lower in primary prostate cancers than that in benign tissues and that CRMP4a protein levels are largely reduced in prostate cancers compared to the surrounding benign tissues. In addition, a potential alteration on CRMP4a protein modification is evidenced in malignant tissues compared to benign tissues, depending on further analysis. In contrast, DPYSL3v1 gene expression in prostate tissues is very low at the mRNA level and its coding protein is missing or undetectable.
Prevention of metastasis has been a major effort to reduce cancer mortality [34]. In attempt to develop a novel anti-metastasis therapy for prostate cancer, we utilized the newly established saRNA technique to up-regulate the expression of tumor metastasis suppressor DPYSL3 gene. Our data revealed that only DPYSL3v2 but not DPYSL3v1 gene is responding to the saRNA-mediated gene activation. In a total of 14 DPYSL3v2 promoter-targeted saRNAs, 4 of them showed a significant up-regulation of DPYSL3v2 gene expression at variable levels in multiple prostate cancer cell lines. The targeting sites of the most potent saRNAs on the promoter region are within 1kb range up-stream of the transcription start site and off the CpG island, consistent to published design criteria [17, 20]. These saRNAs induced a significant suppression of cell motility in multiple assays. Most significantly, delivery of the hairpin-structured saRNAs to prostate cancer cells via a PSMA aptamer largely suppressed tumor metastasis in an orthotopic xenograft model of prostate cancer. These findings are supported by a recent report using a TALE-dependent approach [15], in which promoter-specific demethylation caused a significant up-regulation of DPYSL3 gene/CRMP4 protein expression, leading to reduced cancer cell migration and tumor metastasis. Taken together, it is conceivable that targeting DPYSL3 gene promoter with our saRNA approach or the TALE-dependent demethylation is feasible to enhance endogenous DPYSL3v2 gene expression, resulting in increased CRMP4a protein levels in vivo and subsequent suppression of tumor metastasis. However, our saRNA approach possesses much more drug-like properties, including easy synthesis and delivery, and potential tissue-specific targeting. These saRNA molecules might be used as adjunctive therapy to conventional cytotoxic drugs in prostate cancer management, although the mechanism of CRMP4a-mediated suppression of tumor metastasis requires further investigation.
Successful delivery of therapeutic agents efficiently and specifically to the target tissue is desirable for treatment of human cancers [35]. Prostate specific membrane antigen (PSMA) is a well-known tumor antigen [36]. It is primarily expressed on the surface of prostate cancer epithelial cells and also is highly expressed in metastatic prostate cancer cells and the microvasculature of most studied tumors. This restricted expression pattern warranted it as a promising target for the diagnosis, detection, and management of prostate cancer. As such, PSMA has currently been used for molecular imaging, cancer vaccine development and targeted drug delivery in prostate cancers [37]. Particularly, an RNA-based aptamer targeting human PSMA was developed, which interacts specifically with the PSMA extra-cellular domain and has been widely used in targeted drug delivery and molecular imaging [31]. Recent reports defined two truncated PSMA aptamers with only 39-41mer in length but retraining the PSMA binding function [19, 32]. These truncated PSMA Aptamers make the chemical synthesis much easier to carry out and less non-specific binding [38]. In this study, we created a chimeric RNA molecule that the truncated A10-3.2 aptamer is conjugated to a hairpin-structured saRNA sequence. Consistent to previous reports from our group and others [32, 37, 39–41], A10-3.2 aptamer exerted a clean PSMA-specific cellular intake of the saRNA molecules in prostate cancer cells.
In conclusion, we identified 4 saRNA molecules that are potent in enhancing DPYSL3v2 gene expression in vitro and confirmed one of them is potent in suppressing tumor metastasis in vivo. The saRNA approach by targeting tumor suppressor gene such as DPYSL3 represents a novel direction of cancer gene modulation and drug development.
MATERIALS AND METHODS
Cell lines, antibodies and reagents
Prostate cancer cell LNCaP, C4-2, 22RV1 and PC-3 lines were described in our previous publications [39, 40, 42] and maintained in a humidified atmosphere of 5% CO2, RPMI-1640 media supplemented with 10% fetal bovine serum (FBS) and antibiotics. The CMV-driven luciferase reporter stable PC-3 and C4-2 sub-lines were established using the pLenti CMV-Puro-LUC (w168-1) construct [43], a gift from Dr Eric Campeau (Addgene plasmid # 17477). All luciferase reporters were packaged in 293T cells with a lentiviral packaging psPAX2/pMD2.G system obtained from Dr Didier Trono as a gift (Addgene plasmid #12259-12260). Human embryonic kidney cell 293T line was purchased from American Type Culture Collection (ATCC, Manassas, VA) and cultured in DMEM media with 10% FBS and antibiotics. Antibodies for CRMP4 (sc-100323), Actin (sc-1616), Ki-67 (sc-23900) and DPYSL3 siRNA (sc-44487) were purchased from Santa Cruz Biotech (Santa Cruz, CA).
Small activating RNA design, Aptamer-saRNA conjugate synthesis and transfection
DPYSL3 gene and promoter information on chromosome 5 was extracted on UCSD genome browser (https://genome.ucsc.edu/) and illustrated in supplemental Figure S1. Small activating RNAs were designed based on published protocols and criteria as described previously [20]. All saRNA sense targeting sequences were listed in Table 1 with information of chromosome location and relative distance from the transcription start site. Their locations on the promoter are illustrated in supplemental Figure S2. CpG islands were predicted using an online MethPrimer program (http://www.urogene.org/methprimer/). RNA duplexes including a fluorescence TYE™ 563-labeled APT-V2-9 molecule were chemically synthesized by IDT (Coralville, IA). Transfection was conducted with RNAiMAX obtained from Life Technologies (Carlsbad, CA).
PSMA aptamer A10-3.2 conjugate with the scramble and saV2-9 saRNAs were synthesized with DuraScribe® T7 Transcription Kit from Epicentre (Madison, WI) based on the manufacturer’s protocol as well as a previous report [32]. The DNA templates and primers were listed as follow: A10-3.2-scramble saRNA: TAA TAC GAC TCA CTA TAG GGA GGA CGA TGC GGA TCA GCC ATG TTT ACG TCA CTC CTA tct act gtc act cag tag t ATGAAGC TTG a cta ctg agt gac agt aga dTdT, A10-3.2-saV2-9: TAA TAC GAC TCA CTA TAG GGA GGA CGA TGC GGA TCA GCC ATG TTT ACG TCA CTC CTA gaa aga aca tga atg ctg c ATGAAGCTTG g cag cat tca tgt tct ttc dTdT. PSMA aptamer sequence is in upper case, the saRNA is in lower case and the hairpin loop is in bold upper case. The primers for in vitro T7-based synthesis are listed as follow: Aptamer-F: TAA TAC GAC TCA CTA TAG GGA GGA CG; Aptamer-R: AAT AGG AGT GAC GTA AAC ATG GCT; A1032-V2-9R: AAG AAA GAA CAT GAA TGC TGC C; A1032-scramble-R: AAT CTA CTG TCA CTC AGT AGT CAA GCT T. In vitro transcriptions were performed with modified T7 (Y639F) polymerase and nucleotides as described [32, 44].
Plasmids, promoter cloning, luciferase reporter construction and luciferase detection
Plasmid constructs for human CRMP4a and CRMP4b expression in the pcDNA3.1-V5 backbone were described previously [45]. Stable expression sub-cell lines in PC-3, C4-2 and 293T cells were established with linearized constructs by Lipofactamine®2000 transfection (Life technologies) and single colons were selected in G418 with standard technology.
A 1250-kb fragment of DPYSL3v2 promoter sequence (-1275/-25 from the TSS) was cloned from human genome (NM_001387) and inserted into the up-stream of a Gaussia luciferase (GLuc) on a plasmid pEZX-LvPG02 by GeneCopoeia (Rockville, MD). A PCR-based three-step mutagenesis approach was utilized to generate the saRNA targeting site deletion mutants on the promoter, as illustrated in the supplementary Figure S3. The PCR primers were listed in supplemental Table S1. The mutated promoter fragments were ligated to the luciferase constructs with the unique EcoRI and BamHI sites and the final constructs were packaged into lentiviral particle using psPAX2/pMD2.G system as described above. The successful deletion was confirmed by direct sequencing (Genewiz, South Plainfield, NJ). Luciferase activity was measured with a Gaussia luciferase glow assay kit obtained from Life Technologies (Carlsbad CA).
Total RNA extraction, quantitative RT-PCR and western blot analysis
Surgical tissue specimens from prostate cancers and the procedure for total RNAs extraction from cells and tissues were described in our recent publications [28, 46]. SYBR Green-based real-time PCR (RT-PCR) assays were conducted on the Bio-Rad iQ5 system. The primer pairs for individual genes were listed as follow: DPYSL3v1 forward 5′-GGT CCC GCG GCA GAA ATA C-3′; reverse 5′-GGC ATC GAA ATC CAG CGT CT-3′; DPYSL3v2 forward 5′-CGC CAC CAT GTC CTA CCA AG-3′; reverse 5′- GAC GAT TCT GCC TCC CTT GA-3′. PCR data analysis and primers for house-keeping gene S18 and KRT18 were described in our previous publications [28, 47].
Total cellular proteins were extracted from cells and tissues with radio-immuno-precipitation buffer (RIPA, Cell Signal) supplemented with protease inhibitor cocktails. Equal amount of proteins from each lysates was subjected to SDS-PAGE gels, electrophoresed, and transferred onto PVDF membrane. The membrane was incubated with primary antibody overnight at 4°C after blocking with 10% nonfat dried milk for 1 hour and then incubated with horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotech). Actin blot was used as an internal protein loading control in all blotting membranes.
SBR cell growth, wound healing and Boyden Chamber assays
Cell growth was assessed using the sulforhodamine B assay as described [30, 48]. Wound healing assay was conducted to assess cell migration in vitro as described [49]. Briefly, sub-confluent monolayer cells in 24-well plates were scratched with a sterile 200 μl pipette tip to create a wound line in the middle of the well before transfection with saRNAs as indicated in the figures. Cell migration was monitored for 3 days.
Boyden Chamber transwells from Corning (Tewksbury, MA) were used to assess cell migration in vitro as described [50] with modifications. Briefly, 2 x 104 cells were transfected with the saRNAs as indicated in 6-well plates and transferred into the upper chambers. Migrated cells in the lower chamber were stained with fluorescent dye Hoechst-33342 (final concentration at 5 μg/ml) and counted at 3 days after transfection.
Animal experiments, tissue processing, immunohistochemistry and tissue luciferase detection
Athymic NCr-nu/nu male mice were obtained from NCI-Frederick and housed in accordance with the Institutional Animal Care and Use Committee (IACUC) procedures and guidelines. Human prostate cancer cells C4-2 expression luciferase reporter were trypsinized and resuspended in PBS. A total of 106 cells were resuspended in RPMI-1640 and injected into the dorsal lobe of mouse prostate using a 1-ml disposable syringe. Xenograft tumor development was monitored every week with an IVIS living image system as described [14]. D-Luciferin potassium salt was purchased from GoldBiotech (St. Louis, MO). Once the tumor is established, animals were randomly divided into two groups (n = 5 mice per group) and received intraperitoneal injection of either A10-3.2-scramble or A10-3.2-saV2-9 saRNAs at a dose of 1.0 nmol/injection. Treatment was performed in 10 consecutive days. The mice were euthanized one day after the last injection. One hour before sacrifice, animals were received an intraperitoneal injection of 0.5 ml BrdU solution (10 mM, Roche Diagnostics, Indianapolis, IN) for in situ proliferation assay. Major organs including lung, liver, kidney, reginal lymph nodes and prostate, and xenograft tissues were excised. Half of the tissues were fixed with 4% paraformaldehyde for immunostaining and other half of the tissues were stored at -80°C for further experimentation.
Immunohistochemistry and H&E staining were conducted as described [51]. Briefly, paraffin embedded tissue sections were deparaffinized, rehydrated, followed by antigen retrieval and endogenous peroxidase blocking. The slides were incubated with primary antibody overnight at 4°C. Immunosignals were detected with DAKO LSABt System by following the manufacturer’s manual. BrdU detection was conducted with a BrdU IHC kit (EMD Millipore, Billerica, MA). The picture was taken under a microscope (Nikon Inc., Melville, NY).
Luciferase activity in protein extracts from the whole lobe of right lung was assessed to evaluate micro-metastasis in the lung. Luciferase readings were normalized using total protein concentration.
Statistical analysis
Statistical analysis was conducted using one-way ANOVA analysis with SPSS software (SPSS, Inc., Chicago, IL). A student T-test was performed to compare two groups. Tumor metastasis rate was compared with the Wilcoxon Rank-Sum Test. A p value of 0.05 was considered to indicate a significant difference. The mean and SEM from at least three repeated experiments are shown for all of the quantitative data.
ACKNOWLEDGMENTS
We are very grateful for Dr Alyson E. Fournier (Department of Neurology and Neurosurgery, Montreal Neurological Institute, Montreal, Quebec, Canada H3A 2B4) for providing the CRMP4 constructs. This project is supported by NIH/NCI R21CA175279-01A1 to Dr Benyi Li. It is also partially supported by grants from Chinese NSF #81172427 (Dr Benyi Li) and NSF#81070348 (Dr Changbai Liu). The research activities in Dr Benyi Li’s lab were supported by KU William L. Valk Foundation, KUCC Mason’s Foundation and KUMC Lied Foundation.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Aus G, Robinson D, Rosell J, Sandblom G, Varenhorst E, South-East Region Prostate Cancer G. Survival in prostate carcinoma—outcomes from a prospective, population-based cohort of 8887 men with up to 15 years of follow-up: results from three countries in the population-based National Prostate Cancer Registry of Sweden. Cancer. 2005; 103:943–951.
2. Aaronson NK, Mattioli V, Minton O, Weis J, Johansen C, Dalton SO, Verdonck-de Leeuw IM, Stein KD, Alfano CM, Mehnert A, de Boer A, van de Poll-Franse LV. Beyond treatment - Psychosocial and behavioural issues in cancer survivorship research and practice. EJC Suppl. 2014; 12:54–64.
3. Datta K, Muders M, Zhang H, Tindall DJ. Mechanism of lymph node metastasis in prostate cancer. Future oncology. 2010; 6:823–836.
4. Rosenthal SA, Sandler HM. Treatment strategies for high-risk locally advanced prostate cancer. Nature reviews Urology. 2010; 7:31–38.
5. Schmidt EF, Strittmatter SM. The CRMP family of proteins and their role in Sema3A signaling. Adv Exp Med Biol. 2007; 600:1–11.
6. Goshima Y, Nakamura F, Strittmatter P, Strittmatter SM. Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature. 1995; 376:509–514.
7. Gaetano C, Matsuo T, Thiele CJ. Identification and characterization of a retinoic acid-regulated human homologue of the unc-33-like phosphoprotein gene (hUlip) from neuroblastoma cells. The Journal of biological chemistry. 1997; 272:12195–12201.
8. Quach TT, Mosinger B, Jr., Ricard D, Copeland NG, Gilbert DJ, Jenkins NA, Stankoff B, Honnorat J, Belin MF, Kolattukudy P. Collapsin response mediator protein-3/unc-33-like protein-4 gene: organization, chromosomal mapping and expression in the developing mouse brain. Gene. 2000; 242:175–182.
9. Fukada M, Watakabe I, Yuasa-Kawada J, Kawachi H, Kuroiwa A, Matsuda Y, Noda M. Molecular characterization of CRMP5, a novel member of the collapsin response mediator protein family. The Journal of biological chemistry. 2000; 275:37957–37965.
10. Shih JY, Yang SC, Hong TM, Yuan A, Chen JJ, Yu CJ, Chang YL, Lee YC, Peck K, Wu CW, Yang PC. Collapsin response mediator protein-1 and the invasion and metastasis of cancer cells. Journal of the National Cancer Institute. 2001; 93:1392–1400.
11. Shih JY, Lee YC, Yang SC, Hong TM, Huang CY, Yang PC. Collapsin response mediator protein-1: a novel invasion-suppressor gene. Clinical & experimental metastasis. 2003; 20:69–76.
12. Mukherjee J, DeSouza LV, Micallef J, Karim Z, Croul S, Siu KW, Guha A. Loss of collapsin response mediator Protein1, as detected by iTRAQ analysis, promotes invasion of human gliomas expressing mutant EGFRvIII. Cancer research. 2009; 69:8545–8554.
13. Wu CC, Chen HC, Chen SJ, Liu HP, Hsieh YY, Yu CJ, Tang R, Hsieh LL, Yu JS, Chang YS. Identification of collapsin response mediator protein-2 as a potential marker of colorectal carcinoma by comparative analysis of cancer cell secretomes. Proteomics. 2008; 8:316–332.
14. Gao X, Pang J, Li LY, Liu WP, Di JM, Sun QP, Fang YQ, Liu XP, Pu XY, He D, Li MT, Su ZL, Li BY. Expression profiling identifies new function of collapsin response mediator protein 4 as a metastasis-suppressor in prostate cancer. Oncogene. 2010; 29:4555–4566.
15. Li K, Pang J, Cheng H, Liu WP, Di JM, Xiao HJ, Luo Y, Zhang H, Huang WT, Chen MK, Li LY, Shao CK, Feng YH, Gao X. Manipulation of prostate cancer metastasis by locus-specific modification of the CRMP4 promoter region using chimeric TALE DNA methyltransferase and demethylase. Oncotarget. 2015; 6:10030–10044. doi:10.18632/oncotarget.3192.
16. Zhou W, Xie P, Pang M, Yang B, Fang Y, Shu T, Liu C, Wang X, Zhang L, Li S, Rong L. Upregulation of CRMP4, a new prostate cancer metastasis suppressor gene, inhibits tumor growth in a nude mouse intratibial injection model. International journal of oncology. 2015; 46:290–298.
17. Wang J, Place RF, Portnoy V, Huang V, Kang MR, Kosaka M, Ho MK, Li LC. Inducing gene expression by targeting promoter sequences using small activating RNAs. J Biol Methods. 2015; 2.
18. Li LC, Okino ST, Zhao H, Pookot D, Place RF, Urakami S, Enokida H, Dahiya R. Small dsRNAs induce transcriptional activation in human cells. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103:17337–17342.
19. Rockey WM, Hernandez FJ, Huang SY, Cao S, Howell CA, Thomas GS, Liu XY, Lapteva N, Spencer DM, McNamara JO, Zou X, Chen SJ, Giangrande PH. Rational truncation of an RNA aptamer to prostate-specific membrane antigen using computational structural modeling. Nucleic Acid Ther. 2011; 21:299–314.
20. Huang V, Qin Y, Wang J, Wang X, Place RF, Lin G, Lue TF, Li LC. RNAa is conserved in mammalian cells. PloS one. 2010; 5:e8848.
21. Mao Q, Li Y, Zheng X, Yang K, Shen H, Qin J, Bai Y, Kong D, Jia X, Xie L. Up-regulation of E-cadherin by small activating RNA inhibits cell invasion and migration in 5637 human bladder cancer cells. Biochemical and biophysical research communications. 2008; 375:566–570.
22. Chen Z, Place RF, Jia ZJ, Pookot D, Dahiya R, Li LC. Antitumor effect of dsRNA-induced p21(WAF1/CIP1) gene activation in human bladder cancer cells. Molecular cancer therapeutics. 2008; 7:698–703.
23. Kang MR, Yang G, Place RF, Charisse K, Epstein-Barash H, Manoharan M, Li LC. Intravesical delivery of small activating RNA formulated into lipid nanoparticles inhibits orthotopic bladder tumor growth. Cancer research. 2012; 72:5069–5079.
24. Wang J, Place RF, Huang V, Wang X, Noonan EJ, Magyar CE, Huang J, Li LC. Prognostic value and function of KLF4 in prostate cancer: RNAa and vector-mediated overexpression identify KLF4 as an inhibitor of tumor cell growth and migration. Cancer research. 2010; 70:10182–10191.
25. Quinn CC, Chen E, Kinjo TG, Kelly G, Bell AW, Elliott RC, McPherson PS, Hockfield S. TUC-4b, a novel TUC family variant, regulates neurite outgrowth and associates with vesicles in the growth cone. The Journal of neuroscience. 2003; 23:2815–2823.
26. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, et al. Integrative genomic profiling of human prostate cancer. Cancer cell. 2010; 18:11–22.
27. Holzbeierlein J, Lal P, LaTulippe E, Smith A, Satagopan J, Zhang L, Ryan C, Smith S, Scher H, Scardino P, Reuter V, Gerald WL. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am J Pathol. 2004; 164:217–227.
28. Zhu Q, Youn H, Tang J, Tawfik O, Dennis K, Terranova PF, Du J, Raynal P, Thrasher JB, Li B. Phosphoinositide 3-OH kinase p85alpha and p110beta are essential for androgen receptor transactivation and tumor progression in prostate cancers. Oncogene. 2008; 27:4569–4579.
29. Cole AR, Causeret F, Yadirgi G, Hastie CJ, McLauchlan H, McManus EJ, Hernandez F, Eickholt BJ, Nikolic M, Sutherland C. Distinct priming kinases contribute to differential regulation of collapsin response mediator proteins by glycogen synthase kinase-3 in vivo. The Journal of biological chemistry. 2006; 281:16591–16598.
30. Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006; 1:1112–1116.
31. Lupold SE, Hicke BJ, Lin Y, Coffey DS. Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer research. 2002; 62:4029–4033.
32. Dassie JP, Liu XY, Thomas GS, Whitaker RM, Thiel KW, Stockdale KR, Meyerholz DK, McCaffrey AP, McNamara JO, 2nd, Giangrande PH. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat Biotechnol. 2009; 27:839–849.
33. Rahim S, Minas T, Hong SH, Justvig S, Celik H, Kont YS, Han J, Kallarakal AT, Kong Y, Rudek MA, Brown ML, Kallakury B, Toretsky JA, Uren A. A small molecule inhibitor of ETV1, YK-4-279, prevents prostate cancer growth and metastasis in a mouse xenograft model. PloS one. 2014; 9:e114260.
34. Guan X. Cancer metastases: challenges and opportunities. Acta Pharm Sin B. 2015; 5:402–418.
35. Zhu G, Niu G, Chen X. Aptamer-Drug Conjugates. Bioconjug Chem. 2015; 26:2186–2197.
36. Ghosh A, Heston WD. Tumor target prostate specific membrane antigen (PSMA) and its regulation in prostate cancer. Journal of cellular biochemistry. 2004; 91:528–539.
37. Ristau BT, O’Keefe DS, Bacich DJ. The prostate-specific membrane antigen: lessons and current clinical implications from 20 years of research. Urologic oncology. 2014; 32:272–279.
38. Pan W, Clawson GA. The shorter the better: reducing fixed primer regions of oligonucleotide libraries for aptamer selection. Molecules. 2009; 14:1353–1369.
39. Chen R, Zhao Y, Huang Y, Yang Q, Zeng X, Jiang W, Liu J, Thrasher JB, Forrest ML, Li B. Nanomicellar TGX221 blocks xenograft tumor growth of prostate cancer in nude mice. The Prostate. 2015; 75:593–602.
40. Yang J, Xie SX, Huang Y, Ling M, Liu J, Ran Y, Wang Y, Thrasher JB, Berkland C, Li B. Prostate-targeted biodegradable nanoparticles loaded with androgen receptor silencing constructs eradicate xenograft tumors in mice. Nanomedicine (Lond). 2012; 7:1297–1309.
41. Zhao Y, Duan S, Zeng X, Liu C, Davies NM, Li B, Forrest ML. Prodrug strategy for PSMA-targeted delivery of TGX-221 to prostate cancer cells. Molecular pharmaceutics. 2012; 9:1705–1716.
42. Sun A, Li C, Chen R, Huang Y, Chen Q, Cui X, Liu H, Thrasher JB, Li B. GSK-3beta controls autophagy by modulating LKB1-AMPK pathway in prostate cancer cells. The Prostate. 2016;76:172-83.
43. Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, Kaufman PD. A versatile viral system for expression and depletion of proteins in mammalian cells. PloS one. 2009; 4:e6529.
44. Padilla R, Sousa R. Efficient synthesis of nucleic acids heavily modified with non-canonical ribose 2’-groups using a mutantT7 RNA polymerase (RNAP). Nucleic acids research. 1999; 27:1561–1563.
45. Alabed YZ, Pool M, Ong Tone S, Fournier AE. Identification of CRMP4 as a convergent regulator of axon outgrowth inhibition. The Journal of neuroscience. 2007; 27:1702–1711.
46. Chen R, Zeng X, Zhang R, Huang J, Kuang X, Yang J, Liu J, Tawfik O, Thrasher JB, Li B. Cav1.3 channel alpha1D protein is overexpressed and modulates androgen receptor transactivation in prostate cancers. Urologic oncology. 2014; 32:524–536.
47. Liao X, Tang S, Thrasher JB, Griebling TL, Li B. Small-interfering RNA-induced androgen receptor silencing leads to apoptotic cell death in prostate cancer. Molecular cancer therapeutics. 2005; 4:505–515.
48. Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New colorimetric cytotoxicity assay for anticancer-drug screening. Journal of the National Cancer Institute. 1990; 82:1107–1112.
49. Schleef RR, Birdwell CR. The effect of fibrin on endothelial cell migration in vitro. Tissue Cell. 1982; 14:629–636.
50. Zigmond SH and Hirsch JG. Leukocyte locomotion, chemotaxis. New methods for evaluation, and demonstration of a cell-derived chemotactic factor. The Journal of experimental medicine. 1973; 137:387–410.
51. Li B, Sun A, Youn H, Hong Y, Terranova PF, Thrasher JB, Xu P, Spencer D. Conditional Akt activation promotes androgen-independent progression of prostate cancer. Carcinogenesis. 2007; 28:572–583.