
INTRODUCTION
In the past, cancer immunotherapy mainly focused on attacking tumor cells. Cancer immunotherapy is a promising treatment strategy against solid tumors; however, it cannot completely eradicate them. The biological complexity of the tumor microenvironment (TME) seems to be an obstacle for cancer immunotherapy, suggesting that using a strategy in which only tumor cells are targeted is inadequate to overwhelm the aggressively growing tumor. Therefore, a synergistic TME-targeted strategy is required for the development of more potent cancer immunotherapy [1]. Modern immunotherapy has shifted to an approach that also targets the TME. This approach is founded on the “seed and soil hypothesis” which has illuminated that the complex interplay between TME components plays an important role in tumor metastasis [2]. Among tumor stromal cell types, cancer-associated fibroblasts (CAFs) are the dominant cellular component in the TME, and they play critical roles in promoting tumor progression. Given the close interaction between tumor cells and CAFs in the TME, CAF-targeted strategies would be promising for developing integrated cancer immunotherapy [3]. Fibroblast activation protein α (FAP α) is overexpressed by CAFs and is the predominant component of the stroma in most types of cancer, while it is not detectable in normal adult human tissues [4, 5]. Studies have confirmed that FAP α plays multiple roles in neoangiogenesis, invasion, and metastasis; thus, FAP α has been explored as a target for cancer therapy. Combined immunotherapy treatment with T cells that target cancer cells and an additional agent that targets FAP α-expressing cells for destruction could increase the success of solid tumor elimination [6, 7].
In order to determine the role of FAP α-expressing stromal cells in immune suppression within the TME, a transgenic mouse model has been created with established Lewis lung carcinomas. Only 2% of the tumor cells in this model express FAP α; however, when all FAP α expressing cells (stromal and cancerous) are destroyed, the tumors begin to die rapidly. Therefore, FAP α expressing cells are a non-redundant, immunosuppressive component of the TME [8]. Furthermore, it was reported that a FAP α vaccine combined with curcumin stimulates FAP α antibody production and CD8+ T cell-mediated killing of FAP α-expressing stromal cells and prolongs the survival of mice implanted with melanoma [9]. All of these results suggest that FAP α is an adaptive tumor-associated antigen useful for tumor immunotherapy. In this article, we will review the role of FAP α in tumor development, discuss FAP α as a potential target, and examine its immunotherapeutic benefits.
THE BIOLOGICAL CHARACTERISTICS OF FIBROBLAST ACTIVATION PROTEIN α
Extracellular matrix (ECM) breakdown, detachment of neoplastic cells from the primary site, and their subsequent invasion into the lymphatic vessels, capillaries, and the surrounding normal tissues, is the result of a complex interplay of numerous proteolytic enzymes, including serine proteases. Overexpression of serine proteases in carcinomas is correlated with enhanced tumorigenicity and adverse prognosis [10-12]. Therapeutic strategies targeting FAP α, a membrane-bound serine protease of the prolyl oligopeptidase family that is expressed on CAFs within the tumor stroma, offer another tumor treatment option [13].
FAP α was originally identified by a group in pursuit of a selective marker for activated fibroblasts. A monoclonal antibody named F19 was produced to define the FAP α-positive cell, which strongly labeled cultured fibroblasts, fibroblasts in fetal mesenchymal tissues, the reactive stromal fibroblasts of epithelial tumors, and tumor cells of sarcomas [5, 14]. The name FAP α was given to the F19 antigen because of its unique expression profile [15]. Subsequently, another group identified a 170-KDa membrane-bound protease expressed by invadopodia, the protrusions of invasive melanoma cells, which was called “seprase”[16, 17]. Molecular cloning suggested that FAP α and seprase were the same cell surface serine protease [18-20].
FAP α is a type II integral membrane serine protease that belongs to the dipeptidyl peptidase (DPP) subfamily, which has the ability to cleave the bond between proline and any other amino acids. This enzymatic activity has been shown to have an impact on a wide variety of bioactive signaling molecules [21]. There is 50% sequence homology between DPP-IV and FAP α, and 70% homology in the catalytic domain. Both peptides have the same domain structure and belong to the family of post-prolyl peptidases [22, 23]. Human FAP α, expressed in activated stromal fibroblasts and remodeling tissue, is a type II cell-surface-bound transmembrane glycoprotein with Mr 95,000, it consists of 760 amino acids and is composed of a short 6 amino acid cytoplasmic domain, an 18 amino acid trans-membrane domain, and a large extracellular domain of 736 amino acids. The critical structure of the catalytic triad is formed by serine (Ser624), aspartate (Asp702), and histidine (His734) [24] (Figure 1). Ser624 is essential for enzymatic activity, for when this serine is changed into alanine, the proteolytic activity of FAP α no longer exists [25]. According to the crystal structure, FAP α exists as a homodimer which, when activated, must assemble into a heterodimer [26]. FAP heterodimers which composed by FAP α and FAP β participate in the migration of fibroblasts to collagen substrates [27], probably because together they can effectively degrade the substrate and regulate tumor cell growth, differentiation, adhesion, metastasis.
FAP α has both dipeptidyl peptidase and collagenase activity, and can degrade gelatin and type I collagen. However, the biological significance of FAP α cleavage of gelatin and type I collagen is still unknown. Nonetheless, FAP α is required for the generation of biologically active fragments of denatured collagen [28]. Recently, Neuropeptide Y (NPY), B-type natriuretic peptide, substance P, and peptide YY were found to be natural substrates for FAP α dipeptidyl peptidase activity. These proteins are also substrates for DPPIV; however, FAP α is distinguished from DPPIV by its effects on the half-life of substrates and its endopeptidase activity [29, 30].
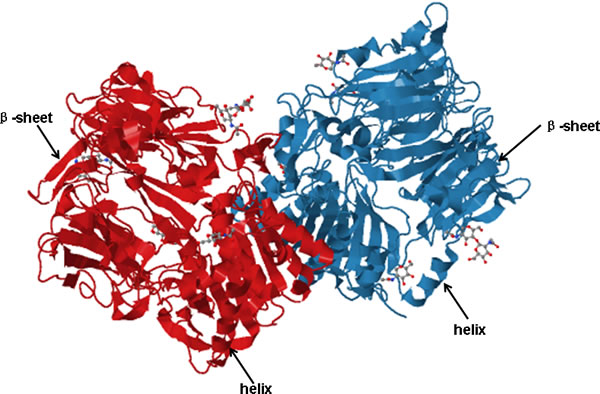
Figure 1: Cartoon architecture of the FAP α homodimer. The critical structure of the catalytic triad is formed by serine (Ser624), aspartate (Asp702), and histidine (His734), and Ser624 is essential for enzymatic activity. The figure was generated using JSmol (PDB ID 1Z68). The blue and red represents two same subunit of FAP α which contains helixes and β-sheets.
THE ASSOCIATION BETWEEN FIBROBLAST ACTIVATION PROTEIN α AND HUMAN CARCINOMA
While different kinds of tumors have general features in common, they also have their own characteristics related to tumor location, size, stage, degree of cell malignancy, involvement of lymph nodes, and metastasis. In this section, we will examine the role of FAP α in different carcinomas. FAP α is expressed by more than 90% of human epithelial tumors, and there has been much interest in exploring FAP α as a therapeutic target in breast cancer. In vivo studies have demonstrated that increased FAP α expression is associated with increased tumor growth rate and promotes neovascularization [31]. Another study using shRNA to target FAP α in a mouse model carrying 4T1 breast cancer came to the same conclusions [32]. These two results demonstrate the important role of FAP α and its potential value as an effective therapeutic target.
FAP α is overexpressed by CAFs in 85-90% of primary and metastatic colorectal cancers [33]. High levels of FAP α in human colon tumors promote tumor growth, progression, metastasis, and recurrence [34]. Moreover, the level of FAP α in rectal carcinomas, which have received preoperative chemo- or radiotherapy, is a negative prognostic factor [35]. Not only the level of FAP α, but also the location of FAP α, is related to poor prognosis of colon cancer patients [33]. All of these findings provide rationale for the development of FAP α-directed therapy.
A series of findings about the expression and role of FAP α in pancreatic carcinoma has suggested that FAP α-targeted immunotherapy may be a new treatment for pancreatic cancer patients. FAP α-induced reorganization of the ECM in TME promotes the invasiveness of pancreatic cancer cells [36]. There is also growing evidence that high FAP α expression in pancreatic cancer is related to poor clinical outcome and its location is associated with its clinical results [37]. In pancreatic carcinoma, FAP α is not only expressed in stromal fibroblast cells, but also in carcinoma cells, in contrast to previous studies which had shown FAP α to be selectively expressed in malignant cells of bone and soft tissue sarcomas. In addition, similar to previous findings, high expression of FAP α in fibroblasts and carcinoma cells is associated with poor clinical outcomes. Therefore, FAP α is a link between the TME and pancreatic cancer cells, which indicates that blocking the activity of FAP α directly or depleting the FAP α-expressing cells may obtain the expected anti-tumor effects [38]. Although the exact function of FAP α in the development of the different diseases remains unclear, it is believed to participate in the progression and metastasis of cancer, angiogenesis, and the suppression of the antitumor response of the immune system [4]. In sum, these findings support the hypothesis that FAP α is a novel target for tumor therapy.
The relationship between FIBROBLAST ACTIVATION PROTEIN α and immune suppression in The TUMOR MicROENVIRONMENT
The complex interactions between the stroma and tumor, along with the regulatory signaling molecules in the TME, contribute to oncogenesis and tumor progression. The process of tumor invasion and metastasis is accompanied by angiogenesis and ECM degradation [39]. In most epithelial cancers greater than 1-2mm3 in size, tumor progression is critically dependent on the supporting TME [40]. Previous studies in murine models have shown that vaccination against tumor vasculature in tumor stroma, results in tumor repression without significant adverse effects, suggesting that TME-targeted immunotherapy is likely to bring a benefit to cancer patients [41-43].
However, tumor immune tolerance is a major impediment in cancer immunotherapy. For example, tumor vaccines proven to have therapeutic effects in vitro have the ability to activate the host immune system. Even the use of tumor-specific antibodies and activation of antitumor immune cells does not alter the overall capabilities of these agents [44]. Therefore, researchers began to take a fresh look at the relationship between the tumor and the TME, and determined that the failure of these vaccines is probably due to the existence of special cells in the TME that are immune-suppressive. “Antitumor” immune cells include cytotoxic CD8+ T lymphocytes (CTLs), T helper type 1 (Th1) cells, type 1 macrophages (M1), type 1 neutrophils (N1), natural killer (NK) cells, natural killer T (NK-T) cells, eosinophils, and mature dendritic cells (DCs) [45-48], all of which are known to support the clearance of tumor cells. An effective antitumor immune response can be divided into three steps: First, there is full activation of T lymphocytes by mature DCs in the tumor-draining lymph node; then, cancer-specific effector T cells leave the blood vessels and enter the tumor site; and finally, tumor-infiltrating lymphocytes (TIL) eventually cause tumor regression [49].
In contrast to normal tissues, the vast majority of immune cells in the TME have lost their function. Furthermore, in cancer patients the composition of immune cells undergoes a change wherein the inhibitory subgroups, such as regulatory immune cells, myeloid-derived suppressor cells (MDSCs), and M2 macrophages are the dominate components [50]. In addition, the TME helps the tumor cells to escape from the attack of effector cells by recruiting inhibitory cells, therefore escaping the body’s immune surveillance, moreover, the origin of MDSCs in the TME is converted by a specific mature subset of NK T-cells [51]. Furthermore, the abnormal distribution, migration barrier, and anergy of T lymphocytes and other immune cells are important reasons for T lymphocyte-mediated antitumor activity failure [52-54]. Thus, immunosuppression in the TME leads to inefficient or ineffective cancer treatments, and immunotherapy strategies aimed at activating T cells are currently under investigation in preclinical and clinical studies.
FAP α is an immune-suppressive component in the TME. The first experiments that assessed the immunologic effects of perturbing the FAP α+ stromal cells used a direct approach of conditionally depleting this cell type from a mouse bearing immunogenic Lewis lung carcinoma cells expressing ovalbumin (LL2/OVA). These studies demonstrated that administering diphtheria toxin to these mice depletes approximately 80% of the tumoral FAP α+ cells, which comprised only approximately 2% of all tumoral cells, and caused rapid, adaptive immune-dependent reduction in tumor volume. Furthermore, when FAP α expression was inhibited, tumor shrinkage was seen accompanied by increased expression of IFN-γ and TNF-α, indicating the existence of immune suppression that was restricted to the FAP α+ cells [8]. However, the mechanism of FAP α action on immunosuppression has not been clarified in detail. In subcutaneous tumors established with immunogenic LL2/OVA, the FAP α+ population is comprised of CD45+ and CD45- cells, and the tumoral FAP α+/CD45+ population was identified as a minor sub-population of F4/80hi/CCR2+/CD206+ M2 macrophages [55]. Using bone marrow chimeric mice in which the primate diphtheria toxin receptor (DTR) is restricted either to the FAP α+/CD45+ or to the FAP α+/CD45- subset, it was demonstrated by conditionally depleting each subset that both independently contribute to the immune suppressive properties of the TME. The immune inhibitory enzyme, heme oxygenase-1 (HO-1), is the basis for the function of the FAP α+/CD45+ subset. FAP α+/CD45+ cells are the major tumoral source of HO-1, and an inhibitor of HO-1, Sn-mesoporphyrin, causes the same extent of immune-dependent arrest of LL2/OVA tumor growth as does the depletion of these cells. Since this observation of immune suppression by the FAP α+/CD45+ stromal cell has been replicated in a transplanted model of pancreatic ductal adenocarcinoma, tumoral immune suppression is likely mediated by macrophages expressing FAP α and HO-1.
In subsequent studies, immune suppression by the FAP α+ CAFs was mediated by CXCL12, the chemokine that binds to cancer cells and excludes T cells by a mechanism that depends on signaling by the CXCL12 receptor, CXCR4 [56]. T cells are absent from regions of the tumor containing cancer cells, which are coated with the chemokine, CXCL12, and the FAP α+ CAFs are the principal source of CXCL12 in the tumor. Administering AMD3100, a CXCL12 receptor chemokine (C-X-C motif) receptor 4 inhibitor, induces rapid T-cell accumulation and acts synergistically with α-PD-L1 to greatly diminish cancer cells, which are identified by their loss of heterozygosity of the TRP53 gene. The residual tumor is composed only of premalignant epithelial cells and inflammatory cells. Thus, a single protein, CXCL12, secreted from a single stromal cell type, the FAP α+ CAFs, explains the overriding immunosuppression by the FAP α+ cell in a model of human pancreatic ductal adenocarcinoma. CXCL12 is the reason for immune suppression, while abrogation of FAP α positive cells permits immune inhibition of tumor growth and enhances the efficacy of constructed immunotherapeutic antibodies [57]. Furthermore, a newly developed vaccine that co-targets tumor cells and FAP α, a consistent marker of CAFs, have shown greater antitumor activity with the enhanced induction of infiltration of CD8+ T cells in B16 melanoma models [58]. Therefore, the more we can understand about the role of FAP α in the suppression of the antitumor response of the immune system will lead to novel immunotherapy targets for clinical benefit.
The FAP α-targeted immunotherapy strategy
As previously mentioned, FAP α is a transmembrane serine protease that is highly expressed on CAFs present in >90% of human epithelial tumors, and plays a significant role in tumor progression and metastasis [14]. Targeting FAP α genetically with vaccines, with antibodies, or with pharmacological agents, impairs tumor progression in several preclinical cancer models [59-61]. Therefore, FAP α is considered to be an adaptive tumor-associated antigen for tumor immunotherapy. In this section we will focus on the clinical and preclinical attempts at employing FAP α-targeted treatment strategies and its prospects in tumor therapy.
As reviewed above, FAP α belongs to the serine proteinase family, has both collagenase and dipeptidase activities which can degrade gelatin, collagen, and other substrates of dipeptidase, and promotes tumor growth, migration, invasion, metastasis and ECM degradation. Therefore, it was hypothesized that selectively blocking the enzymatic activity of FAP α may be a method of targeting it in tumor development. Val-boroPro, also called Talabostat, was the first inhibitor of the proteolytic activity of FAP α used in phase II clinical trials successively performed in patients with metastatic colon cancer, non-small cell lung cancer, and melanoma. Val-boroPro was administrated alone or combined with other non-specific anti-tumor drugs, but minimal clinical response was observed with the addition of Val-boroPro [62-64]. There is still no consistent conclusion as to why the drug did not work, but there are reports suggesting that the enzymatic activity of FAP α has little to do with increased tumor growth [28], and there are also contradictory results suggesting that serine proteases can function as tumor suppressors [21, 65]. Some scientists hold the view that it was the form that Val-boroPro exists in vivo that matters [4], and that more studies are required to get the real picture.
The disappointing clinical outcome of Val-boroPro does not exclude the potential role of FAP α’s proteolytic activity in tumor invasion and metastasis, or that an inhibitor antibody may be a potent therapeutic target. An inhibitory scFv antibody, named E3 was identified, which competitively inhibits FAP α function [66]. This scFv antibody with high affinity and enhanced inhibitory effects on FAP α enzyme activity, seems very likely to be exploited as a tool for the treatment of FAP α driven tumors. Studies to evaluate the effects of scFv antibody deserve more exploration and further characterization to confirm previous findings. In one study, human scFvs were transformed into bivalent minibodies of completely human origin, which worked far better than murine or humanized antibody derivatives. Thus, the successful use of mini-antibodies in immunohistology for a variety of carcinomas is encouraging for in vivo diagnostic and tumor-targeting studies [67]. Combinatorial strategies addressing the two key issues of cancer immunotherapy (ie. targeting the tumor cells and modulating the T-cell response), the production of a bio-specific single chain antibody (scFv) directed against FAP α and CD3 (T cell receptor component), and the subsequent construction of a bio-specific antibody combined with co-stimulatory antibody-ligand fusion proteins, show the potential for initiating and regulating immune response at the TME in addition to modulating tumor progress [68, 69]. Advanced combinatorial strategies could result in unprecedented clinical outcomes with great beneficial effects.
Early work on FAP α-targeting monoclonal antibodies investigated the toxicity, imaging, and bio-distribution of a 131-labeled monoclonal antibody (131I-mAbF19) against FAP α in patients with hepatic metastases from colorectal carcinoma. The 131I-mAbF19 was administered by intravenous injection and no toxicity was observed [70]. Further work has defined the population pharmacokinetics of 131I-mAbF19 with the data from two phase I studies in cancer patients [71]. These studies indicated potential diagnostic and therapeutic applications of humanized mAbF19. Then, sibrotuzumab, an antibody directed against humanized F19, was produced and used in a Phase I dose-escalation study, which showed the safety of this antibody in patients who suffer from advanced or metastatic FAP α-positive cancers [6]. Interestingly, there was a relationship between body weight and this antibody [72]. Unconjugated sibrotuzumab (BIBH 1) was investigated in an early phase II trial with metastatic colorectal cancer patients, and, although it was well tolerated and safe, the trial was suspended because of its minimal clinical response [73]. Despite the disappointing results, the study of more efficient FAP α antibodies continues. Novel human-mouse cross-reactive antibodies, ESC11 and ESC14 labeled with radionuclide (177) Lu, were recently engineered and characterized [74]. Accumulation of these two antibodies is specific to tumor tissue, particularly the (177) Lu-labeled ESC11, suggesting these antibodies could be potential tumor growth retardants in a melanoma xenograft model. (177)Lu-ESC11 has advantage over (177) Lu-ESC14 and (177) Lu-vF19 in prolonging survival time. However, more preclinical and clinical experiments are needed to explore the diagnostic and therapeutic effects of these potent antibody-drug conjugates in patients with FAP α-expressing tumors.
Vaccines targeting FAP α provide another therapeutic strategy that takes advantage of the restricted distribution of FAP α in tumor sites. Scientists have constructed a DNA vaccine directed against FAP α [75], and immune tolerance against the FAP α self-antigen can be inhibited through delivery of FAP α cDNA as a subcutaneous DNA vaccine. In prophylactic experiments, the CD8+ T-cell-mediated antitumor immune response induced by pFAP α vaccination inhibited tumor growth, significantly suppressed growth of pulmonary metastases and prolonged the life spans of vaccinated mice, consistent with a previous study [60]. Similar results were also observed for adaptive immunity induced by adoptive transfer of T cells from pFAP α-immunized mice. Non-specific immune responses were unlikely, because the cytotoxic effects mediated by CD8+ T cells in vitro were restricted to target cells overexpressing the FAP α antigen, consistent with other studies [59, 60]. More importantly, an in vitro screening method was used to determine whether dendritic cells transfected with mRNA encoding products of FAP α are capable of stimulating cytotoxic CD8+ (CTL) responses from human peripheral blood mononuclear cells. It was demonstrated that CTL responses could be consistently generated against FAP α. To enhance the immunogenicity of the mRNA-translated FAP α product, a lysosomal targeting signal derived from lysosome-associated membrane protein-1 (LAMP-1) was fused to the COOH terminus of FAP α to redirect the translated product into the class II presentation pathway. Dendritic cells transfected with mRNA encoding the FAP α-LAMP fusion product stimulated enhanced CD4+ and CD8+ T-cell responses [76].
Furthermore, in our research, we have developed a new tumor vaccine, FAP α τ-MT, which was produced by conjugating 1-MT to a FAP α. The in vitro results confirmed that 1-MT could be dissociated from the FAP α τ-MT vaccine and inhibit intracellular IDO activity [77]. In a FAP α-positive tumor model, the FAP ατ-MT vaccine elicited an antitumor response that was similar to systemic treatment with the FAP α τ vaccine plus 1-MT. Most importantly, administration of the FAP α τ-MT vaccine did not lead to pregnancy failure in mice carrying allogeneic fetuses. These findings that FAP α τ-MT breaks tumor immune tolerance as a local IDO inhibitor, suggesting that conjugation of 1-MT to a tumor antigen peptide is a potentially effective clinical cancer immunotherapy. In subsequent studies, we used the main catalytic domain of dipeptidyl peptidase of murine FAP α as a vaccine that contains abundant T-cell epitopes and B-cell epitopes, combined with curcumin lavage that inhibits the expression of IDO to relieve tumor immune tolerance, to treat mice implanted with melanoma cells. We demonstrated that FAP α vaccine combined with curcumin lavage inhibits tumor growth and prolongs the survival of mice implanted with melanoma cells. The combination of a FAP α vaccine and curcumin stimulated FAP α antibody production and CD8+T cell-mediated killing of FAP α-expressing stromal cells without adverse reactive effects [9]. These results suggest that FAP α, a product preferentially expressed by CAFs, would be a more effective antigen to target in the setting of cancer immunotherapy.
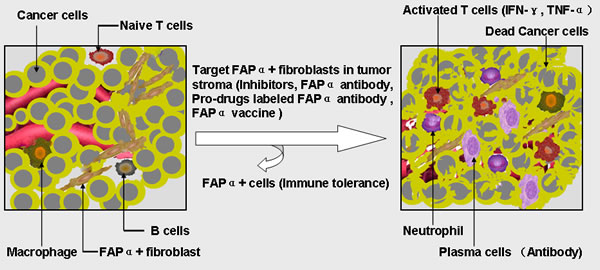
Figure 2: The diagram demonstrating the role of FAP α in immune suppression and the application of FAP α-targeted immunotherapy strategy.
CONCLUSIONS
Cancer cells are embedded in stroma, the connective tissue framework of solid tumors. Stromal cells and cancer cells depend on each other for mutual paracrine stimulation, and stromal fibroblasts are probably required for cancer cells to survive and grow [78, 79]. Recently, tumor stromal cells have been suggested as a target for tumor immunotherapy because tumor stromal cells, unlike tumor cells, are diploid, genetically stable, and open to immunological attack. Thus, immunizing against stromal fibroblasts in tumors may unmask an immune response to cancer [80-82].
FAP α is a tumor-associated antigen which is a serine protease involved in extracellular matrix remodeling and highly expressed on reactive stromal fibroblasts in >90% of human epithelial carcinomas, but is not detectable in normal adult human tissues [83, 84]. Stromal cells expressing FAP α may suppress the immune response to tumors as a consequence of producing massive amounts of stromal cell-derived factor-1 (SDF-1/CXCL12). SDF-1 attracts regulatory T cells (CD4+ subtype) into the tumor [85]. Furthermore, because of the multiple roles played by FAP α in neoangiogenesis, invasion and metastasis, it is being explored as a target for cancer therapy.
In the context of immunotherapy involving T cells targeting cancer cells, an agent targeting FAP α-expressing cells might increase therapeutic efficacy against both solid tumors and metastatic cells [86-89]. Eliminating FAP α+ stromal fibroblasts activating cancer-specific T cells should inhibit growth of small spontaneous tumors and thus may help eliminate clinically undetectable cancer cells that have already metastasized before excision of the primary tumor [90]. Thus, combined immunotherapy treatment consisting of T cells that target cancer cells and an agent targeting FAP α-expressing cells for destruction could increase the success of eliminating solid tumors and metastatic cells. Because FAP α is a robust target for immunotherapy, preclinical and clinical studies targeting FAP α have been initiated adopting methods such as small inhibitor molecules, FAP α-activated prodrugs, monoantibodies, DNA vaccines, peptide vaccines, and modified vaccines, etc. (Figure 2). In the future, there will be more and more combinatorial immuno(chemo) therapeutic regimens and other promising methods targeting FAP α.
Acknowledgments and funding
Grant support: This work was funded by the National Natural Science Foundation of China (No. 81502104, No. 81201331 ), Hunan Natural Science Foundation (No. 13JJ4078, No. 11JJ4076) and the Bureau of Hunan Provincial Science and Technology( No. 2012sk2007, No. 2011SK3172).
Conflicts of interest
No potential conflicts of interest were disclosed.
References
1. Zi F, He J, He D, Li Y, Yang L and Cai Z. Fibroblast activation protein a alpha in tumor microenvironment: recent progression and implications (review). Molecular medicine reports. 2015; 11:3203-3211.
2. Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nature reviews Cancer. 2003; 3:453-458.
3. Ohshio Y, Teramoto K, Hanaoka J, Tezuka N, Itoh Y, Asai T, Daigo Y and Ogasawara K. Cancer-associated fibroblast-targeted strategy enhances antitumor immune responses in dendritic cell-based vaccine. Cancer science. 2015; 106:134-142.
4. Kelly T, Huang Y, Simms AE and Mazur A. Fibroblast activation protein a-alpha: a key modulator of the microenvironment in multiple pathologies. International review of cell and molecular biology. 2012; 297:83-116.
5. Rettig WJ, Garin-Chesa P, Beresford HR, Oettgen HF, Melamed MR and Old LJ. Cell-surface glycoproteins of human sarcomas: differential expression in normal and malignant tissues and cultured cells. Proceedings of the National Academy of Sciences of the United States of America. 1988; 85:3110-3114.
6. Scott AM, Wiseman G, Welt S, Adjei A, Lee FT, Hopkins W, Divgi CR, Hanson LH, Mitchell P, Gansen DN, Larson SM, Ingle JN, Hoffman EW, et al. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein a-positive cancer. Clinical cancer research. 2003; 9:1639-1647.
7. Jackson RC. Contributions of protein structure-based drug design to cancer chemotherapy. Seminars in oncology. 1997; 24:164-172.
8. Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, Gopinathan A, Tuveson DA and Fearon DT. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein a-alpha. Science. 2010; 330:827-830.
9. Jiang GM, Xie WY, Wang HS, Du J, Wu BP, Xu W, Liu HF, Xiao P, Liu ZG, Li HY, Liu SQ, Yin WJ, Zhang QG, et al. Curcumin combined with FAP aalphac vaccine elicits effective antitumor response by targeting indolamine-2,3-dioxygenase and inhibiting EMT induced by TNF-alpha in melanoma. Oncotarget. 2015; 6:25932-25942. doi: 10.18632/oncotarget.4577.
10. Okada K, Chen WT, Iwasa S, Jin X, Yamane T, Ooi A and Mitsumata M. Seprase, a membrane-type serine protease, has different expression patterns in intestinal- and diffuse-type gastric cancer. Oncology. 2003; 65:363-370.
11. Iwasa S, Okada K, Chen WT, Jin X, Yamane T, Ooi A and Mitsumata M. ‘Increased expression of seprase, a membrane-type serine protease, is associated with lymph node metastasis in human colorectal cancer’. Cancer letters. 2005; 227:229-236.
12. Mori Y, Kono K, Matsumoto Y, Fujii H, Yamane T, Mitsumata M and Chen WT. The expression of a type II transmembrane serine protease (Seprase) in human gastric carcinoma. Oncology. 2004; 67(5-6):411-419.
13. Lin X, Huang C, Kong L, He Y, Zhou D and Li J. Fibroblast activation protein a, a potential diagnostic and therapeutic target for cancer. Human pathology. 2014; 45:1553.
14. Garin-Chesa P, Old LJ and Rettig WJ. Cell surface glycoprotein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proceedings of the National Academy of Sciences of the United States of America. 1990; 87:7235-7239.
15. Rettig WJ, Garin-Chesa P, Healey JH, Su SL, Ozer HL, Schwab M, Albino AP and Old LJ. Regulation and heteromeric structure of the fibroblast activation protein a in normal and transformed cells of mesenchymal and neuroectodermal origin. Cancer research. 1993; 53:3327-3335.
16. Aoyama A and Chen WT. A 170-kDa membrane-bound protease is associated with the expression of invasiveness by human malignant melanoma cells. Proceedings of the National Academy of Sciences of the United States of America. 1990; 87:8296-8300.
17. Monsky WL, Lin CY, Aoyama A, Kelly T, Akiyama SK, Mueller SC and Chen WT. A potential marker protease of invasiveness, seprase, is localized on invadopodia of human malignant melanoma cells. Cancer research. 1994; 54:5702-5710.
18. Goldstein LA, Ghersi G, Pineiro-Sanchez ML, Salamone M, Yeh Y, Flessate D and Chen WT. Molecular cloning of seprase: a serine integral membrane protease from human melanoma. Biochimica et biophysica acta. 1997; 1361:11-19.
19. Pineiro-Sanchez ML, Goldstein LA, Dodt J, Howard L, Yeh Y, Tran H, Argraves WS and Chen WT. Identification of the 170-kDa melanoma membrane-bound gelatinase (seprase) as a serine integral membrane protease. The Journal of biological chemistry. 1997; 272:7595-7601.
20. Scanlan MJ, Raj BK, Calvo B, Garin-Chesa P, Sanz-Moncasi MP, Healey JH, Old LJ and Rettig WJ. Molecular cloning of fibroblast activation protein a alpha, a member of the serine protease family selectively expressed in stromal fibroblasts of epithelial cancers. Proceedings of the National Academy of Sciences of the United States of America. 1994; 91:5657-5661.
21. Kelly T. Fibroblast activation protein a-alpha and dipeptidyl peptidase IV (CD26): cell-surface proteases that activate cell signaling and are potential targets for cancer therapy. Drug resistance updates : reviews and commentaries in antimicrobial and anticancer chemotherapy. 2005; 8(1-2):51-58.
22. Chen WT, Kelly T and Ghersi G. DPPIV, seprase, and related serine peptidases in multiple cellular functions. Current topics in developmental biology. 2003; 54:207-232.
23. Zhang M, Xu L, Wang X, Sun B and Ding J. Expression levels of seprase/FAP aalpha and DPPIV/CD26 in epithelial ovarian carcinoma. Oncology letters. 2015; 10:34-42.
24. Aertgeerts K, Levin I, Shi L, Snell GP, Jennings A, Prasad GS, Zhang Y, Kraus ML, Salakian S, Sridhar V, Wijnands R and Tennant MG. Structural and kinetic analysis of the substrate specificity of human fibroblast activation protein a alpha. The Journal of biological chemistry. 2005; 280:19441-19444.
25. Park JE, Lenter MC, Zimmermann RN, Garin-Chesa P, Old LJ and Rettig WJ. Fibroblast activation protein a, a dual specificity serine protease expressed in reactive human tumor stromal fibroblasts. The Journal of biological chemistry. 1999; 274:36505-36512.
26. O’Brien P and O’Connor BF. Seprase: an overview of an important matrix serine protease. Biochimica et biophysica acta. 2008; 1784:1130-1145.
27. Ghersi G, Dong H, Goldstein LA, Yeh Y, Hakkinen L, Larjava HS and Chen WT. Regulation of fibroblast migration on collagenous matrix by a cell surface peptidase complex. The Journal of biological chemistry. 2002; 277:29231-29241.
28. Huang Y, Simms AE, Mazur A, Wang S, Leon NR, Jones B, Aziz N and Kelly T. Fibroblast activation protein a-alpha promotes tumor growth and invasion of breast cancer cells through non-enzymatic functions. Clinical & experimental metastasis. 2011; 28:567-579.
29. Keane FM, Nadvi NA, Yao TW and Gorrell MD. Neuropeptide Y, B-type natriuretic peptide, substance P and peptide YY are novel substrates of fibroblast activation protein a-alpha. The FEBS journal. 2011; 278:1316-1332.
30. Levy MT, McCaughan GW, Abbott CA, Park JE, Cunningham AM, Muller E, Rettig WJ and Gorrell MD. Fibroblast activation protein a: a cell surface dipeptidyl peptidase and gelatinase expressed by stellate cells at the tissue remodelling interface in human cirrhosis. Hepatology. 1999; 29:1768-1778.
31. Paulsson J and Micke P. Prognostic relevance of cancer-associated fibroblasts in human cancer. Seminars in cancer biology. 2014; 25:61-68.
32. Cai F, Li Z, Wang C, Xian S, Xu G, Peng F, Wei Y and Lu Y. Short hairpin RNA targeting of fibroblast activation protein a inhibits tumor growth and improves the tumor microenvironment in a mouse model. BMB reports. 2013; 46:252-257.
33. Wikberg ML, Edin S, Lundberg IV, Van Guelpen B, Dahlin AM, Rutegard J, Stenling R, Oberg A and Palmqvist R. High intratumoral expression of fibroblast activation protein a (FAP a) in colon cancer is associated with poorer patient prognosis. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2013; 34:1013-1020.
34. Henry LR, Lee HO, Lee JS, Klein-Szanto A, Watts P, Ross EA, Chen WT and Cheng JD. Clinical implications of fibroblast activation protein a in patients with colon cancer. Clinical cancer research. 2007; 13:1736-1741.
35. Saigusa S, Toiyama Y, Tanaka K, Yokoe T, Okugawa Y, Fujikawa H, Matsusita K, Kawamura M, Inoue Y, Miki C and Kusunoki M. Cancer-associated fibroblasts correlate with poor prognosis in rectal cancer after chemoradiotherapy. International journal of oncology. 2011; 38:655-663.
36. Lee HO, Mullins SR, Franco-Barraza J, Valianou M, Cukierman E and Cheng JD. FAP a-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells. BMC cancer. 2011; 11:245.
37. Cohen SJ, Alpaugh RK, Palazzo I, Meropol NJ, Rogatko A, Xu Z, Hoffman JP, Weiner LM and Cheng JD. Fibroblast activation protein a and its relationship to clinical outcome in pancreatic adenocarcinoma. Pancreas. 2008; 37:154-158.
38. Shi M, Yu DH, Chen Y, Zhao CY, Zhang J, Liu QH, Ni CR and Zhu MH. Expression of fibroblast activation protein a in human pancreatic adenocarcinoma and its clinicopathological significance. World journal of gastroenterology. 2012; 18:840-846.
39. Yan Y, Wang LF and Wang RF. Role of cancer-associated fibroblasts in invasion and metastasis of gastric cancer. World journal of gastroenterology. 2015; 21:9717-9726.
40. Bizzarri M and Cucina A. Tumor and the microenvironment: a chance to reframe the paradigm of carcinogenesis? BioMed research international. 2014; 2014:934038.
41. Nielsen MB and Marincola FM. Melanoma vaccines: the paradox of T cell activation without clinical response. Cancer chemotherapy and pharmacology. 2000; 46 Suppl:S62-66.
42. Church SE and Galon J. Tumor Microenvironment and Immunotherapy: The Whole Picture Is Better Than a Glimpse. Immunity. 2015; 43:631-633.
43. Tsang YW, Huang CC, Yang KL, Chi MS, Chiang HC, Wang YS, Andocs G, Szasz A, Li WT and Chi KH. Improving immunological tumor microenvironment using electro-hyperthermia followed by dendritic cell immunotherapy. BMC cancer. 2015; 15:708.
44. Vasievich EA and Huang L. The suppressive tumor microenvironment: a challenge in cancer immunotherapy. Molecular pharmaceutics. 2011; 8:635-641.
45. de Visser KE, Eichten A and Coussens LM. Paradoxical roles of the immune system during cancer development. Nature reviews Cancer. 2006; 6:24-37.
46. Mantovani A, Allavena P, Sica A and Balkwill F. Cancer-related inflammation. Nature. 2008; 454:436-444.
47. Verbeke H, Struyf S, Laureys G and Van Damme J. The expression and role of CXC chemokines in colorectal cancer. Cytokine & growth factor reviews. 2011; 22(5-6):345-358.
48. Burkholder B, Huang RY, Burgess R, Luo S, Jones VS, Zhang W, Lv ZQ, Gao CY, Wang BL, Zhang YM and Huang RP. Tumor-induced perturbations of cytokines and immune cell networks. Biochimica et biophysica acta. 2014; 1845:182-201.
49. Peranzoni E, Rivas-Caicedo A, Bougherara H, Salmon H and Donnadieu E. Positive and negative influence of the matrix architecture on antitumor immune surveillance. Cellular and molecular life sciences. 2013; 70:4431-4448.
50. Kerkar SP and Restifo NP. Cellular constituents of immune escape within the tumor microenvironment. Cancer research. 2012; 72:3125-3130.
51. Park YJ, Song B, Kim YS, Kim EK, Lee JM, Lee GE, Kim JO, Kim YJ, Chang WS and Kang CY. Tumor microenvironmental conversion of natural killer cells into myeloid-derived suppressor cells. Cancer research. 2013; 73:5669-5681.
52. Fisher DT, Chen Q, Appenheimer MM, Skitzki J, Wang WC, Odunsi K and Evans SS. Hurdles to lymphocyte trafficking in the tumor microenvironment: implications for effective immunotherapy. Immunological investigations. 2006; 35(3-4):251-277.
53. Vazquez-Cintron EJ, Monu NR and Frey AB. Tumor-induced disruption of proximal TCR-mediated signal transduction in tumor-infiltrating CD8+ lymphocytes inactivates antitumor effector phase. Journal of immunology. 2010; 185:7133-7140.
54. Wang SF, Fouquet S, Chapon M, Salmon H, Regnier F, Labroquere K, Badoual C, Damotte D, Validire P, Maubec E, Delongchamps NB, Cazes A, Gibault L, et al. Early T cell signalling is reversibly altered in PD-1+ T lymphocytes infiltrating human tumors. PloS one. 2011; 6:e17621.
55. Arnold JN, Magiera L, Kraman M and Fearon DT. Tumoral immune suppression by macrophages expressing fibroblast activation protein a-alpha and heme oxygenase-1. Cancer immunology research. 2014; 2:121-126.
56. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, Teichmann SA, Janowitz T, Jodrell DI, Tuveson DA and Fearon DT. Targeting CXCL12 from FAP a-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proceedings of the National Academy of Sciences of the United States of America. 2013; 110:20212-20217.
57. Fearon DT. The carcinoma-associated fibroblast expressing fibroblast activation protein a and escape from immune surveillance. Cancer immunology research. 2014; 2:187-193.
58. Gottschalk S, Yu F, Ji M, Kakarla S and Song XT. A vaccine that co-targets tumor cells and cancer associated fibroblasts results in enhanced antitumor activity by inducing antigen spreading. PloS one. 2013; 8:e82658.
59. Lee J, Fassnacht M, Nair S, Boczkowski D and Gilboa E. Tumor immunotherapy targeting fibroblast activation protein a, a product expressed in tumor-associated fibroblasts. Cancer research. 2005; 65:11156-11163.
60. Loeffler M, Kruger JA, Niethammer AG and Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. The Journal of clinical investigation. 2006; 116:1955-1962.
61. Teichgraber V, Monasterio C, Chaitanya K, Boger R, Gordon K, Dieterle T, Jager D and Bauer S. Specific inhibition of fibroblast activation protein a (FAP a)-alpha prevents tumor progression in vitro. Advances in medical sciences. 2015; 60:264-272.
62. Narra K, Mullins SR, Lee HO, Strzemkowski-Brun B, Magalong K, Christiansen VJ, McKee PA, Egleston B, Cohen SJ, Weiner LM, Meropol NJ and Cheng JD. Phase II trial of single agent Val-boroPro (Talabostat) inhibiting Fibroblast activation protein a in patients with metastatic colorectal cancer. Cancer biology & therapy. 2007; 6:1691-1699.
63. Eager RM, Cunningham CC, Senzer N, Richards DA, Raju RN, Jones B, Uprichard M and Nemunaitis J. Phase II trial of talabostat and docetaxel in advanced non-small cell lung cancer. Clinical oncology. 2009; 21:464-472.
64. Eager RM, Cunningham CC, Senzer NN, Stephenson J, Jr., Anthony SP, O’Day SJ, Frenette G, Pavlick AC, Jones B, Uprichard M and Nemunaitis J. Phase II assessment of talabostat and cisplatin in second-line stage IV melanoma. BMC cancer. 2009; 9:263.
65. Ramirez-Montagut T, Blachere NE, Sviderskaya EV, Bennett DC, Rettig WJ, Garin-Chesa P and Houghton AN. FAP aalpha, a surface peptidase expressed during wound healing, is a tumor suppressor. Oncogene. 2004; 23:5435-5446.
66. Zhang J, Valianou M, Simmons H, Robinson MK, Lee HO, Mullins SR, Marasco WA, Adams GP, Weiner LM and Cheng JD. Identification of inhibitory scFv antibodies targeting fibroblast activation protein a utilizing phage display functional screens. FASEB journal. 2013; 27:581-589.
67. Schmidt A, Muller D, Mersmann M, Wuest T, Gerlach E, Garin-Chesa P, Rettig WJ, Pfizenmaier K and Moosmayer D. Generation of human high-affinity antibodies specific for the fibroblast activation protein a by guided selection. European journal of biochemistry / FEBS. 2001; 268:1730-1738.
68. Wuest T, Moosmayer D and Pfizenmaier K. Construction of a bispecific single chain antibody for recruitment of cytotoxic T cells to the tumour stroma associated antigen fibroblast activation protein a. Journal of biotechnology. 2001; 92:159-168.
69. Hornig N, Kermer V, Frey K, Diebolder P, Kontermann RE and Muller D. Combination of a bispecific antibody and costimulatory antibody-ligand fusion proteins for targeted cancer immunotherapy. Journal of immunotherapy. 2012; 35:418-429.
70. Welt S, Divgi CR, Scott AM, Garin-Chesa P, Finn RD, Graham M, Carswell EA, Cohen A, Larson SM, et al. Antibody targeting in metastatic colon cancer: a phase I study of monoclonal antibody F19 against a cell-surface protein of reactive tumor stromal fibroblasts. Journal of clinical oncology. 1994; 12:1193-1203.
71. Tanswell P, Garin-Chesa P, Rettig WJ, Welt S, Divgi CR, Casper ES, Finn RD, Larson SM, Old LJ and Scott AM. Population pharmacokinetics of antifibroblast activation protein a monoclonal antibody F19 in cancer patients. British journal of clinical pharmacology. 2001; 51:177-180.
72. Kloft C, Graefe EU, Tanswell P, Scott AM, Hofheinz R, Amelsberg A and Karlsson MO. Population pharmacokinetics of sibrotuzumab, a novel therapeutic monoclonal antibody, in cancer patients. Investigational new drugs. 2004; 22:39-52.
73. Hofheinz RD, al-Batran SE, Hartmann F, Hartung G, Jager D, Renner C, Tanswell P, Kunz U, Amelsberg A, Kuthan H and Stehle G. Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie. 2003; 26:44-48.
74. Fischer E, Chaitanya K, Wuest T, Wadle A, Scott AM, van den Broek M, Schibli R, Bauer S and Renner C. Radioimmunotherapy of fibroblast activation protein a positive tumors by rapidly internalizing antibodies. Clinical cancer research. 2012; 18:6208-6218.
75. Wen Y, Wang CT, Ma TT, Li ZY, Zhou LN, Mu B, Leng F, Shi HS, Li YO and Wei YQ. Immunotherapy targeting fibroblast activation protein a inhibits tumor growth and increases survival in a murine colon cancer model. Cancer science. 2010; 101:2325-2332.
76. Fassnacht M, Lee J, Milazzo C, Boczkowski D, Su Z, Nair S and Gilboa E. Induction of CD4(+) and CD8(+) T-cell responses to the human stromal antigen, fibroblast activation protein a: implication for cancer immunotherapy. Clinical cancer research. 2005; 11:5566-5571.
77. Yi YM, Zhang G, Zeng J, Huang SC, Li LL, Fang R, Jiang GM, Bu XZ, Cai SH and Du J. A new tumor vaccine: FAP atau-MT elicits effective antitumor response by targeting indolamine2,3-dioxygenase in antigen presenting cells. Cancer biology & therapy. 2011; 11:866-873.
78. Erez N, Truitt M, Olson P, Arron ST and Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer cell. 2010; 17:135-147.
79. Lacina L, Plzak J, Kodet O, Szabo P, Chovanec M, Dvorankova B, Smetana K Jr. Cancer Microenvironment: What Can We Learn from the Stem Cell Niche. International Journal of Molecular Sciences. 2015; 16: 24094-24110.
80. Reisfeld RA. The tumor microenvironment: a target for combination therapy of breast cancer. Critical reviews in oncogenesis. 2013; 18(1-2):115-133..
81. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, Antzis M, Cotner CE, Johnson LA, Durham AC, Solomides CC, June CH, Pure E and Albelda SM. Targeting fibroblast activation protein a in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer immunology research. 2014; 2:154-166.
82. Turley SJ, Cremasco V, Astarita JL. Immunological hallmarks of stromal cells in the tumour microenvironment. Nature reviews immunology. 2015; 15: 669-682.
83. El Khoury J, Kurban M, Kibbi AG and Abbas O. Fibroblast-activation protein a: valuable marker of cutaneous epithelial malignancy. Archives of dermatological research. 2014; 306:359-365.
84. Liu F, Qi L, Liu B, Liu J, Zhang H, Che D, Cao J, Shen J, Geng J, Bi Y, Ye L, Pan B and Yu Y. Fibroblast activation protein a overexpression and clinical implications in solid tumors: a meta-analysis. PloS one. 2015; 10:e0116683.
85. Kryczek I, Wei S, Keller E, Liu R and Zou W. Stroma-derived factor (SDF-1/CXCL12) and human tumor pathogenesis. American journal of physiology Cell physiology. 2007; 292:C987-995.
86. Baird SK, Rigopoulos A, Cao D, Allan L, Renner C, Scott FE and Scott AM. Integral membrane protease fibroblast activation protein a sensitizes fibrosarcoma to chemotherapy and alters cell death mechanisms. Apoptosis. 2015; 20:1483-1498.
87. Fang J, Xiao L, Joo KI, Liu Y, Zhang C, Liu S, Conti PS, Li Z and Wang P. A potent immunotoxin targeting fibroblast activation protein a for treatment of breast cancer in mice. International journal of cancer. 2015. [Epub ahead of print]
88. Chen M, Xiang R, Wen Y, Xu G, Wang C, Luo S, Yin T, Wei X, Shao B, Liu N, Guo F, Li M, Zhang S, Li M, Ren K, Wang Y, Wei Y. A whole-cell tumor vaccine modified to express fibroblast activation protein a induces antitumor immunity against both tumor cells and cancer-associated fibroblasts. Scientific Reports. 2015; 5: 14421.
89. Baird SK, Rigopoulos A, Cao D, Allan L, Renner C, Scott FE, Scott AM. Integral membrane protease fibroblast activation protein a sensitizes fibrosarcoma to chemotherapy and alters cell death mechanisms. Apoptosis. 2015; 20: 1483-1498.
90. Spiotto MT, Rowley DA and Schreiber H. Bystander elimination of antigen loss variants in established tumors. Nature medicine. 2004; 10:294-298.