
INTRODUCTION
Preoperative chemotherapy is an important part of the management of patients with colorectal liver metastases (CLM). Response evaluation is usually assessed on a radiological basis using the RECIST criteria [1]. Response is determined as the total change in sum of diameters of all pre-defined target lesions following chemotherapy. The RECIST criteria do not take account individual lesion response. Recently, Van Kessel et al. showed a radiological heterogeneity in approximately 35% of patients with CLM, suggesting underlying pathological heterogeneity [2]. The pathological response (PR) has been validated as a morphologic variable to assess response to chemotherapy. To date, the PR is assessed by four methods based on the presence of viable cancer cells, which however differed in patients with multiple CLM [3–6]. CLM are expected to demonstrate a similar behaviour since the organ microenvironment and tumour cell chemosensitivity were thought similar. However, no study has addressed the intermetastatic behaviour on a pathological level.
Information not only on KRAS but also in NRAS mutational status is now an essential prerequisite for the selection of a targeted therapy based on anti-EGFR therapies. Moreover, mutations in KRAS and other genes such as BRAF and PIK3CA are also associated with worse survival [7]. Variations in genetic alterations between primary colorectal cancer and metastatic lesions of different sites in the same patient have been extensively reported. No study has investigated the incidence and the impact of intermetastatic heterogeneity on a genetic level within CLM.
This study aimed to assess the occurrence of pathological heterogeneity in patients with multiple CLM and its predictive factors, and the relationship between genetic and pathological heterogeneity.
RESULTS
Patient characteristics
The study included 157 patients with multiple CLM. Of them, 87 patients (55%) received oxaliplatin-based chemotherapy, and 70 patients (45%) received irinotecan-based chemotherapy. Sixty patients (38%) received bevacizumab, and 26 patients (16.5%) received cetuximab. The median number of chemotherapy cycles was six (range, 3–34). The mean and median numbers of CLM were 4.68 (+ 3.6) and 4 (range, 2 to 32), respectively. The mean and median maximum tumour size were 3.3 cm (+ 2.2 cm) and 3 cm (range: 0.1–11 cm), respectively. One hundred and six patients (67%) had positive resection margins on pathological examination of the specimen (R1 resection).
Pathological heterogeneity and associated factors
Seven patients had a complete response. The remaining 150 patients were classified as follows: according to the method by Blazer et al. [4], 83 patients had a major and 67 patients a minor tumour response. According to the method by Sebagh et al. [6], 55 patients had < 6 cm-residual tumour and 95 patients had > 6 cm-residual tumour. There was no difference in the mean PR between the 86 patients treated by chemotherapy plus biotherapy and the 71 patients treated by chemotherapy alone (38.9% versus 41.9% according to the method by Blazer).
The mean and median difference in the PR between the most heterogeneous CLM were 30.4% (+ SD = 29.6%) and 25% (range: 0–100%), respectively. Pathological homogeneity was observed in 126 patients (80.2%) including the 7 patients with complete response. Pathological heterogeneity of > 50% was observed in 31 patients (19.7%).
Pathological heterogeneity of > 50% was significantly associated with none of the clinical and pathological variables (Table 1). The mean difference in the PR was significantly higher in patients with > 3 CLM (37.1% vs 13.8%, p < 0.0001) and in patients undergoing preoperative portal vein embolization (PVE) (37.9% vs 27.2%, p = 0.05), tended to be higher in patients with a number of preoperative chemotherapy cycles > 6 (p = 0.07) and was not impacted by the global PR, nor the addition of targeted therapies under univariate analysis (Table 2). The multivariate analysis identified a number of CLM > 3 and the use of preoperative PVE as independent factors (p = 0.002 and 0.04, respectively).
Table 1: Univariate analysis of factors associated with pathological heterogeneity of more than 50%
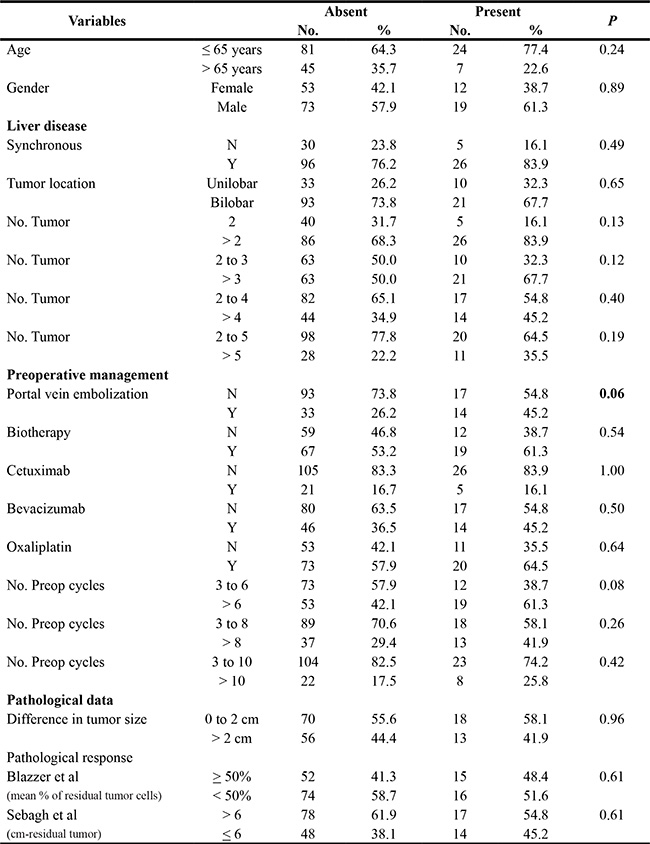
Table 2: Univariate and multivariate analysis for the mean difference in the pathological response (between the CLM with the highest and the lowest response)
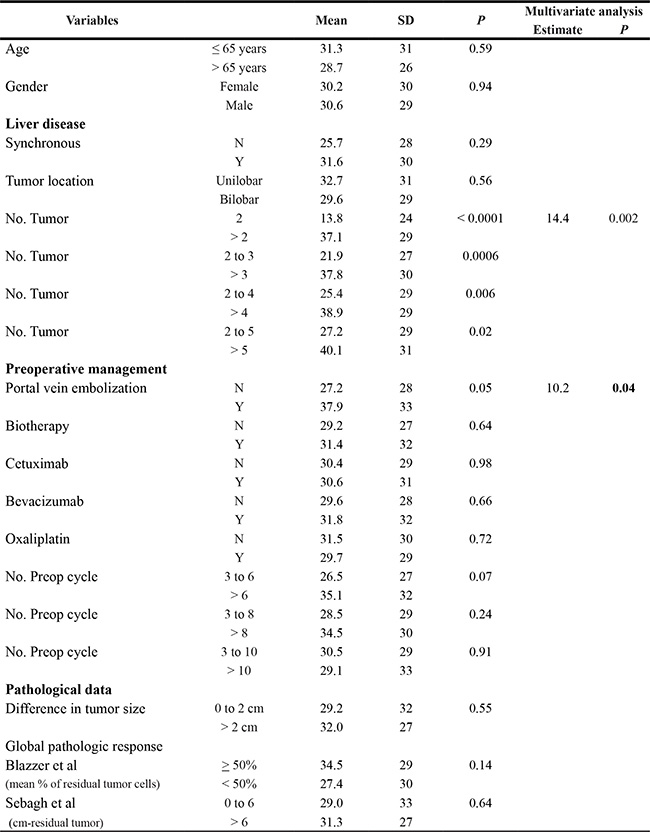
SD, Standard deviation; Intercept = 23.10, R2 = 0.1
Gene mutation status (Table 3)
Table 3: Gene somatic profile in the overall population
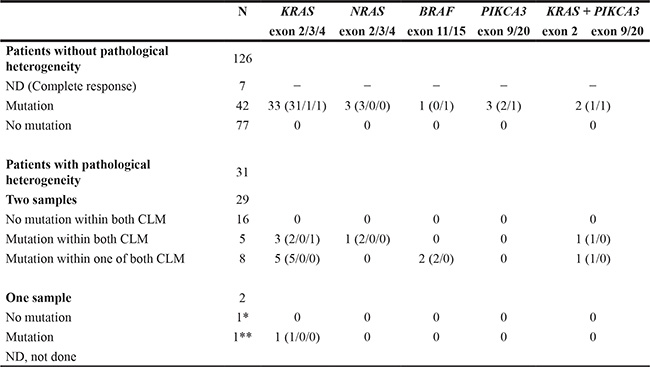
*complete remission of the second CLM.
**non amplified DNA of the second CLM.
In the overall population, a 30.6% rate of KRAS mutations, 2% rate of BRAF mutations, 2.7% rate of NRAS mutations and 4.7% rate of PIK3CA mutations were observed. In total, 37.3% of patients showed at least one mutation. The frequency of gene mutation was not significantly different between the patients with and without pathological heterogeneity (14/31 versus 42/119).
In the patients without pathological heterogeneity (N = 126), the mutational status was necessarily unavailable in the 7 patients with complete response. of the remaining 119 patients, 77 patients did not exhibit mutations and 42 patients (35.2%) had one or more mutations: 35 patients had KRAS mutations, 3 NRAS mutations, 1 BRAF mutation and 5 PIK3CA mutations, associated with KRAS mutations in 2.
In the patients with pathological heterogeneity (N = 31), genotyping was available from the two most heterogeneous CLM in 29 patients. In the remaining 2 patients, the mutational status was available in only one CLM: In the first patient having 2 CLM, one CLM was not tested because of a complete response and the second CLM did not exhibit mutation. In the second patient, one CLM exhibited a KRAS mutation and the DNA from the second CLM was unamplified.
In the 29 patients with available genotyping from the two most heterogeneous CLM, 13 patients had at least one mutation within at least one CLM: eight patients had KRAS mutations, one had NRAS mutation, two had BRAF mutation and two had concurrent PIK3CA and KRAS mutations. Table 4 showed that both tested CLM were mutated in five patients. The same mutational status was present within each CLM in 5/5 patients (100%). Both CLM were wild-type in 16 patients. One mutated CLM and one wild-type CLM were present in eight patients, giving a genetic heterogeneity of 27.6% of the patients with pathological heterogeneity. In 6 of these 8 patients, the mutated CLM was the less florid.
Table 4: Mutation status of the most heterogeneous CLM in the 31 patients with pathological heterogeneity
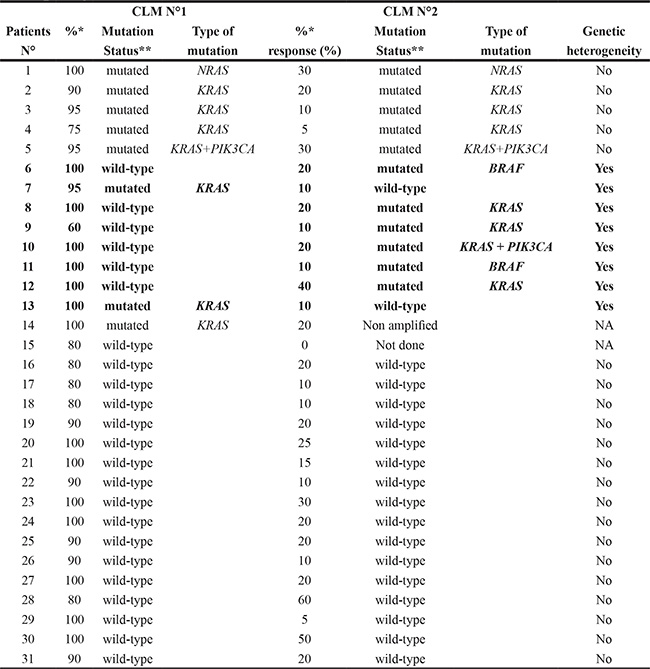
*Percentage of remaining tumour cells
**Mutation status included the relevant genes usually tested in primary colorectal tumor and CLM (i. e., KRAS, NRAS, BRAF and PIK3CA).
Survival (Figure 1)
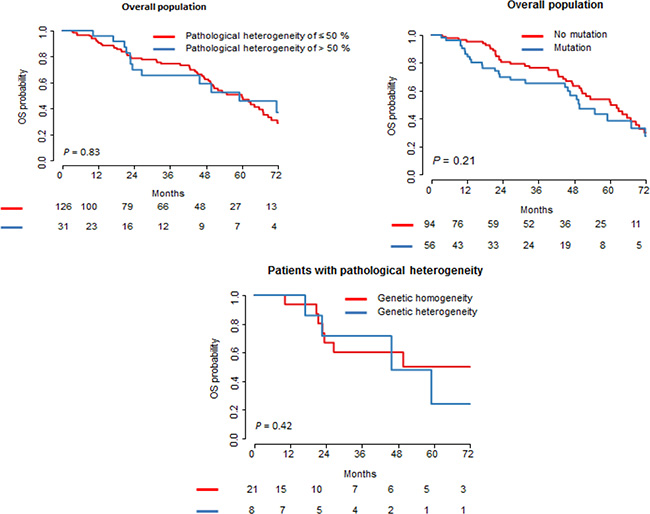
Figure 1: Survival curves according to the PR and genetic status. There was no statistical difference in the overall survival between patients with and without pathological heterogeneity, and between patients with and without mutation. In the patients with pathological heterogeneity, there was also no difference in the overall survival between patients with and without genetic heterogeneity.
The median follow-up period was 52.8 months (range: 1–109 months) CI [48.9–68.7 months]. The cumulative 5-year overall survival rate was 48%. There was no statistical difference in the overall survival between patients with and without pathological heterogeneity, and between patients with and without mutation. In the patients with pathological heterogeneity, there was also no difference in the overall survival between patients with and without genetic heterogeneity.
DISCUSSION
This study demonstrated a pathological heterogeneity of > 50% between the CLM in response to therapy in 19.7% of patients. The mean difference in the PR between CLM was independently associated with the use of preoperative PVE and a number of CLM > 3. A genetic heterogeneity was observed in 27.6% of the patients with pathological heterogeneity. The pathological and genetic heterogeneity did not impact the overall survival.
To date, four methods for assessing the PR to preoperative chemotherapy have been validated [3–6]. These methods based on the presence of viable cancer cells however differed in patients with multiple CLM. Rubbia-brandt et al. defined five tumour regression grades (TRG) [3]; Blazer et al. accounted for the percentage of viable cancer cells [4]. Maru et al. defined the tumour cell viability by the maximum tumour thickness at the tumour-normal interface [5]. Recently, we proposed a new method which accounted for the number of nodules, their size and their respective percentage of viable tumour cells [6]. The means of calculating the PR by the three latter methods implied that the response to chemotherapy could vary from one CLM to another. However, the pathological heterogeneity between CLM was not explicitly studied. For Rubbia-brandt et al., the morphology of CLM within the same patient was similar, with TRG being equal between nodules or within one grade range in most cases (90%). The variability of TRG between nodules in range of two grades was observed in 10% [3]. This could justify that this method accounted for one CLM (i.e., the most florid one) in contrast to the other methods. In this study, the rate of pathological heterogeneity >50% between CLM was of 19.7% (31/157), near 2-fold higher than that reported by Rubbia-brandt et al. (10%, 12/111). As for CLM, only a few prior studies focused on the comparison of PR in primary colorectal tumours, regional lymph nodes and CLM. In the study by Gervaz et al. [8], CLM exhibited a better PR than primary tumours and an identical poor response for primary tumours and lymph nodes. Two studies on patients with T3/T4 rectal cancer downstaged by neaoadjuvant chemoradiation to pT0 reported a 17% rate of positive mesorectal lymph nodes [9, 10].
Our result demonstrated that a greater heterogeneity in PR was associated with a number of CLM > 3 and the use of preoperative PVE. Because PVE is mainly used in patients with a great number of CLM, which is a significant factor of heterogeneity, a potential confounding impact of PVE could be opposed. However, under the multivariate analysis, the use of PVE and a CLM number > 3 remained independent factors for pathological heterogeneity. CLM are thought to mainly depend on arterial supply, which serves as a rational for hepatic arterial infusion strategy. The suppression of portal flow following PVE may enhance arterial flow [11], and then improve the efficacy of post PVE/ligation chemotherapy via a better drug delivery and higher concentrations of cytotoxic drugs. Thus, pathological heterogeneity would result from difference in CLM locations affected heterogeneously by embolization. In contrast, pathological heterogeneity was not statistically associated with the number of preoperative chemotherapy cycles. These results argue that the histological differences in response to chemotherapy are probably related more to the microenvironment than to tumour cell chemosensitivity.
Information not only on KRAS but also in NRAS mutational status is now an essential prerequisite for the selection of a targeted therapy based on anti-EGFR therapies. Moreover, mutations in other genes such as BRAF and PIK3CA are also associated with resistance to anti-EGFR therapies and/or worse survival [7]. A lot of studies have focused on the comparison of mutational status in primary colorectal tumours, regional lymph nodes and metastasis. In term of frequency, an association between RAS mutations and more aggressive tumour biology was reported: A higher incidence of gene mutation was shown in patients with primary tumours of stage III/IV compared with stage I/II [12], in patients with metachronous CLM detected after chemotherapy for the primary colorectal cancer [13–15], as well as in patients with metastases at particular sites such as lung and brain [16–18]. Because of our centre referral pattern, the gene profile was assessed in CLM and not in primary colorectal cancer. We observed a 30.6% rate of KRAS mutations, similar to that reported [16, 19], 2% rate of BRAF mutation, 2.7% rate of NRAS mutations and 4.7% rate of PIK3CA mutations and concomitant mutations of PIK3CA and KRAS occurred in 2.7% of patients. In total, 37.3% of patients showed at least one mutation.
Usually only one single sample is analysed per patient and no general recommendation exist as to which tumour sample should preferentially be tested [20]. Some studies demonstrated a high concordance between primary tumours and metastases [21–23]. Thus, the genomic profiling is usually tested in primary tumour biopsies: Material from metastatic sites is not routinely collected, generally of poorer quality and contains fewer tumour cells as a result of tumour necrosis or chemotherapy-induced changes. Other studies demonstrated discordance and thus suggested that metastatic tissues should be tested [12, 18, 24, 25]. Lymph nodes metastases were not reliable tissue specimens to define the KRAS mutation [12].
To our best knowledge, this study is the first to address the intermetastatic heterogeneity not only on a pathological but also genetic level within CLM in a given patient. We showed that genetic heterogeneity (one mutated CLM/one wild-type CLM) was observed in 27.6% (8/29) of the patients with pathological heterogeneity and genetic homogeneity corresponding to either any mutation (16/29, 55%) or mutations (5/29, 17.2%) within both CLM in 72.4%. In both mutated CLM, there was concordance of mutation type in 100% (5/5). Our results should lead to test additional CLM in case of wild-type results in the particular subpopulation of patients with pathological heterogeneity. Indeed, the genotyping of multiple CLM is unusual in routine practice because a such strategy is time and money consuming. This explains the lack of genotyping of all paired CLM in the 157 patients which is a limitation of our study. We also found that the mutated CLM was the less florid one in 6/8 patients (75%). This result should help pathologists in the selection of the block for the genotyping. Nevertheless, the development of liquid biopsy based on genetic testing from circulating cell free tumour DNA containing mutation in plasma has been reported as a non-invasive, specific and highly sensitive approach for monitoring disease load [26–30]. Liquid biopsies should advantageously replace tumour-section analysis although caution must be taken with respect to the short half life of the cell free DNA and its decrease during cytotoxic chemotherapy. The perspectives include the assessment of radicality of primary and secondary resections, early detection of recurrence after liver metastasectomy and monitoring of response/resistance to chemotherapy. Before surgery, it should also be a tool to solve the potential existence of a genetic heterogeneity between the primary tumour and the metastases, and between metastases since cell free DNA can be released from each CLM or all parts of the tumour.
Numerous classical factors including carcinoembryonic antigen, maximal tumor size or tumor number…, are associated with recurrence and poor survival while some recent studies have questioned their prognostic value. There is increasing evidence that the PR to preoperative chemotherapy is a major determinant of outcome after resection [3–6], independently of others prognostics factors. This study showed that pathological heterogeneity was not associated with PR. This could explain that the pathological heterogeneity did not impact the overall survival.
In conclusion, we showed an intermetastatic pathological heterogeneity in 19.7% of patients, which was independently associated with the use of PVE and a number of CLM > 3. We showed intermetastatic heterogeneity in gene expression in 27.6% of patients of the patients with pathological heterogeneity and homogeneity in type of gene mutation in 100% of patients. Existing intermetastatic heterogeneity could have clinical implications and refine guide management for tissue sampling.
PATIENTS AND METHODS
Study population
We included patients who were previously identified and characterized [6]. Briefly, among the 425 patients who underwent elective liver resection for CLM at Paul Brousse Hospital (Villejuif, France) between 2004 and 2011, patients with operative mortality and those with incomplete surgery (R2 resection) have been excluded. In total, 223 patients with either R0 or R1 resection who received at least 3 cycles of preoperative chemotherapy with no more than two lines in association and in whom the tissue material was available for pathologic review have been eligible for the previous study. For the purpose of the current study concerning the intermetastatic heterogeneity, the 66 patients with a single CLM were excluded from the prior study.
Pathological evaluation
In each liver resection specimens, all CLM have been sampled entirely for CLM < 2 cm and extensively from the centre to the periphery (as recommended, one sample per cm along the greatest dimension) for bigger lesions, respectively. Formalin-fixed paraffin embedded tissue blocks were cut at 4 μm thickness and stained with haematoxylin and eosin. All stained sections have been reviewed by a single pathologist blinded to clinical information. In each CLM, the percentage of area with remaining viable tumour cells in relation to the total area of the CLM was evaluated. Pathological heterogeneity was arbitrarily defined as a difference of > 50% in the percentage of viable tumour cells between the CLM with the lower and higher response in a given patient. In each patient, the PR has been assessed according to the method by Blazer and al [4] and Sebagh et al. [6].
Tumour DNA preparation and gene mutation profiling
In our Pathology Department, pathologists usually choose a paraffin-embedded block from the most florid CLM. Following the assessment of the percentage of tumour cells in relation with the sample area (including non tumoral liver and stroma of the tumour), the block was subsequently cut at 30μm and microdissected if containing less than 10% of tumoural cells. In patients with pathological heterogeneity, DNA extraction was performed within two samples from the most heterogeneous CLM on a pathological level. DNA extraction was performed using the QIAmp DNA Mini kit (Qiagen, Courtaboeuf, France) according to the manufacturer instructions.
All the patients were retrospectively and completely tested for the relevant genes usually tested in primary colorectal tumor and CLM (i.e., KRAS, NRAS, BRAF and PIK3CA). Somatic gene mutations were detected using the MassARRAY iPLEX platform (Sequenom-Agena Bioscience, San Diego, US), which involves a three-step process consisting of the initial PCR reaction, inactivation of unincorporated nucleotides by shrimp alkaline phosphatase and a single-base primer extension. Then, the products are nano-dispensed onto a matrix-loaded silicon chip (SpectroChipII, Sequenom-Agena Bioscience, San Diego, US) and finally, the mutations are detected by MALDI-TOF (matrix-assisted laser desorption-ionization-time of flight) mass spectrometry. The experimental sensitivity of the assay was estimated to be below 5% for each gene mutation.
Statistical analysis
Continuous data were expressed as a median (range) and/or mean (standard deviation) whereas categorical data were expressed as percentages. Categorical data were compared using Fisher’s exact test or chi-square test as appropriate. Univariate and multivariate analysis were used to examine the relationship between various clinical and histological factors and 1) pathological heterogeneity (as present or absent), and 2) mean difference in the PR between the CLM with the lower and the higher response. Variables with a P value 0.15 under univariate analysis were included for multivariate analysis. The cumulative survival rate was calculated by the Kaplan-Meier and compared by log-rank test. Statistical significance was indicated by a P value < 0.05. Calculations were performed using both R (2.14.1).
Abbreviations
CLM, colorectal metastases; PR, pathological response; TRG, tumor regression grade; PVE, portal vein embolization.
GRANT SUPPORT
No financial support to carry out this research.
CONFLICTS OF INTEREST
Disclose any potential conflicts of interest.
REFERENCES
1. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). European Journal of Cancer. 2009; 45:228–247.
2. Van Kessel CS, Samim M, Koopman M, van den Bosch MAAJ, Borel Rinkes IHM, Punt CJA, van Hillegersberg R. Radiological heterogeneity in response to chemotherapy is associated with poor survival in patients with colorectal liver metastases. European Journal of Cancer. 2013; 49:2486–2493.
3. Rubbia-Brandt L, Giostra E, Brezault C, Roth AD, Andres A, Audard V, Sartoretti P, Dousset B, Majno PE, Soubrane O, Chaussade S, Mentha G, Terris B. Importance of histological tumor response assessment in predicting the outcome in patients with colorectal liver metastases treated with neo-adjuvant chemotherapy followed by liver surgery. Annals of Oncology. 2007; 18:299–304.
4. Blazer DG 3rd, Kishi Y, Maru DM, Kopetz S, Chun YS, Overman MJ, Fogelman D, Eng C, Chang DZ, Wang H, Zorzi D, Ribero D, Ellis LM, et al. Pathologic response to preoperative chemotherapy: a new outcome end point after resection of hepatic colorectal metastases. Journal of Clinical Oncology. 2008; 26:5344–5351.
5. Maru DM, Kopetz S, Boonsirikamchai P, Agarwal A, Chun YS, Wang H, Abdalla EK, Kaur H, Charnsangavej C, Vauthey J-N, Loyer EM. Tumor thickness at the tumor-normal interface: a novel pathologic indicator of chemotherapy response in hepatic colorectal metastases. The American Journal of Surgical Pathology. 2010; 34:1287–1294.
6. Sebagh M, Allard M-A, Cunha AS, Ruiz A, Araujo R, Lemoine A, Paule B, Delvart V, Cherqui D, Vibert E, Adam R. A proposed new method for assessing the pathological response to chemotherapy in resected colorectal liver metastases. British Journal of Cancer. 2014; 111:470–476.
7. De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. The Lancet Oncology. 2010; 11:753–762.
8. Gervaz P, Rubbia-Brandt L, Andres A, Majno P, Roth A, Morel P, Mentha G. Neoadjuvant chemotherapy in patients with stage IV colorectal cancer: a comparison of histological response in liver metastases, primary tumors, and regional lymph nodes. Annals of Surgical Oncology. 2010; 17:2714–2719.
9. Hughes R, Glynne-Jones R, Grainger J, Richman P, Makris A, Harrison M, Ashford R, Harrison RA, Livingstone JI, McDonald PJ, Meyrick Thomas J, Mitchell IC, Northover JMA, et al. Can pathological complete response in the primary tumour following pre-operative pelvic chemoradiotherapy for T3-T4 rectal cancer predict for sterilisation of pelvic lymph nodes, a low risk of local recurrence and the appropriateness of local excision? International Journal of Colorectal Disease. 2006; 21:11–17.
10. Bedrosian I, Rodriguez-Bigas MA, Feig B, Hunt KK, Ellis L, Curley SA, Vauthey JN, Delclos M, Crane C, Janjan N, Skibber JM. Predicting the node-negative mesorectum after preoperative chemoradiation for locally advanced rectal carcinoma. Journal of Gastrointestinal Surgery. 2004; 8:56–62; discussion 62–63.
11. Nagino M, Nimura Y, Kamiya J, Kanai M, Hayakawa N, Yamamoto H. Immediate increase in arterial blood flow in embolized hepatic segments after portal vein embolization: CT demonstration. AJR American Journal of Roentgenology. 1998; 171:1037–1039.
12. Baldus SE, Schaefer K-L, Engers R, Hartleb D, Stoecklein NH, Gabbert HE. Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clinical Cancer Research. 2010; 16:790–799.
13. Vauthey J-N, Zimmitti G, Kopetz SE, Shindoh J, Chen SS, Andreou A, Curley SA, Aloia TA, Maru DM. RAS mutation status predicts survival and patterns of recurrence in patients undergoing hepatectomy for colorectal liver metastases. Annals of Surgery. 2013; 258:619–26; discussion 626–627.
14. Losi L, Baisse B, Bouzourene H, Benhattar J. Evolution of intratumoral genetic heterogeneity during colorectal cancer progression. Carcinogenesis. 2005; 26:916–922.
15. Messick CA, Sanchez J, Dejulius KL, Church JM, Kalady MF. Genetic and molecular diversity of colon cancer hepatic metastases. Surgery. 2009; 146:227–231.
16. Tie J, Lipton L, Desai J, Gibbs P, Jorissen RN, Christie M, Drummond KJ, Thomson BNJ, Usatoff V, Evans PM, Pick AW, Knight S, Carne PWG, et al. KRAS mutation is associated with lung metastasis in patients with curatively resected colorectal cancer. Clinical Cancer Research. 2011; 17:1122–1130.
17. Kim M-J, Lee HS, Kim JH, Kim YJ, Kwon JH, Lee J-O, Bang S-M, Park KU, Kim D-W, Kang S-B, Kim J-S, Lee JS, Lee K-W. Different metastatic pattern according to the KRAS mutational status and site-specific discordance of KRAS status in patients with colorectal cancer. BMC Cancer. 2012; 12:347.
18. Cejas P, López-Gómez M, Aguayo C, Madero R, de Castro Carpeño J, Belda-Iniesta C, Barriuso J, Moreno García V, Larrauri J, López R, Casado E, Gonzalez-Barón M, Feliu J. KRAS mutations in primary colorectal cancer tumors and related metastases: a potential role in prediction of lung metastasis. PloS One. 2009; 4:e8199.
19. Allard MA, Saffroy R, de la Maisonneuve PB, Ricca L, Bosselut N, Hamelin J, Lecorche E, Bejarano MA, Innominato P, Sebagh M, Adam R, Morère JF, Lemoine A. Colorectal liver metastases are more often super wild type. Toward treatment based on metastatic site genotyping? Targeted Oncology. 2015; 10:415–421.
20. Vignot S, Besse B, André F, Spano J-P, Soria J-C. Discrepancies between primary tumor and metastasis: a literature review on clinically established biomarkers. Critical Reviews in Oncology/Hematology. 2012; 84:301–313.
21. Knijn N, Mekenkamp LJM, Klomp M, Vink-Borger ME, Tol J, Teerenstra S, Meijer JWR, Tebar M, Riemersma S, van Krieken JHJM, Punt CJA, Nagtegaal ID. KRAS mutation analysis: a comparison between primary tumours and matched liver metastases in 305 colorectal cancer patients. British Journal of Cancer. 2011; 104:1020–1026.
22. Vakiani E, Janakiraman M, Shen R, Sinha R, Zeng Z, Shia J, Cercek A, Kemeny N, D’Angelica M, Viale A, Heguy A, Paty P, Chan TA, et al. Comparative genomic analysis of primary versus metastatic colorectal carcinomas. Journal of Clinical Oncology. 2012; 30:2956–2962.
23. Vignot S, Lefebvre C, Frampton GM, Meurice G, Yelensky R, Palmer G, Capron F, Lazar V, Hannoun L, Miller VA, Andre F, Stephens PJ, Soria J-C, et al. Comparative analysis of primary tumour and matched metastases in colorectal cancer patients: Evaluation of concordance between genomic and transcriptional profiles. European Journal of Cancer. 2015; 51:791–799.
24. Albanese I, Scibetta AG, Migliavacca M, Russo A, Bazan V, Tomasino RM, Colomba P, Tagliavia M, La Farina M. Heterogeneity within and between primary colorectal carcinomas and matched metastases as revealed by analysis of Ki-ras and p53 mutations. Biochemical and Biophysical Research Communications. 2004; 325:784–791.
25. López-Gómez M, Moreno-Rubio J, Suárez-García I, Cejas P, Madero R, Casado E, Jiménez AM, Sereno M, Gómez-Raposo C, Zambrana F, Merino M, Fernández-Luengas D, Feliu J. Gene expression differences in primary colorectal tumors and matched liver metastases: chemotherapy related or tumoral heterogeneity? Clinical & Translational Oncology. 2015; 17:322–329.
26. Buim ME, Fanelli MF, Souza VS, Romero J, Abdallah EA, Mello CA, Alves V, Ocea LM, Mingues NB, Barbosa PN, Tyng CJ, Chojniak R, Chinen LT. Detection of KRAS mutations in circulating tumor cells from patients with metastatic colorectal cancer. Cancer Biology & Therapy. 2015; 1–7.
27. Kidess E, Heirich K, Wiggin M, Vysotskaia V, Visser BC, Marziali A, Wiedenmann B, Norton JA, Lee M, Jeffrey SS, Poultsides GA. Mutation profiling of tumor DNA from plasma and tumor tissue of colorectal cancer patients with a novel, high-sensitivity multiplexed mutation detection platform. Oncotarget. 2015; 6:2549–2561. doi: 10.18632/oncotarget.3041.
28. Reinert T, Schøler LV, Thomsen R, Tobiasen H, Vang S, Nordentoft I, Lamy P, Kannerup A-S, Mortensen FV, Stribolt K, Hamilton-Dutoit S, Nielsen HJ, Laurberg S, et al. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut. 2015.
29. Thierry AR, Mouliere F, Messaoudi S El, Mollevi C, Lopez-Crapez E, Rolet F, Gillet B, Gongora C, Dechelotte P, Robert B, Del Rio M, Lamy P-J, Bibeau F, et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nature Medicine. 2014; 20:430–435.
30. Zonta E, Nizard P, Taly V. Assessment of DNA Integrity, Applications for Cancer Research. Advances in Clinical Chemistry. 2015; 70:197–246.