
Introduction
Gliomas are the most common type of primary brain tumors in adults, classified by the World Health Organization (WHO) based on histopathological criteria into four grades: I to IV, with glioblastoma (GBM; grade IV) representing the most frequent and aggressive form [1, 2]. The challenges of GBM treatment include involvement of multiple molecular pathways which result in rapid development of drug resistance, blood-brain barrier considerations as well as molecular heterogeneity of the tumor [3]. The standard-of-care for GBM involves optimal surgical resection followed by a combination of radiation and chemotherapy with the oral DNA alkylating agent temozolomide, which together translates into a median survival of 14.6 months. Almost all GBM patients experience recurrence, and second-line treatments provide only modest benefit for the vast majority of patients. Therefore, there is a desperate need for novel treatment options [4], [5], [6].
Histologically similar GBMs can be driven by distinct genetic events that result in varied clinical behaviors and prognoses. Therefore, effort has been focused on classifying GBMs according to molecular aberrations to better direct therapy [7], [8], [9]. For example, promoter methylation of the MGMT (O6-methylguanine-DNA methyltransferase) gene (MGMT-Me) reduces transcription and, consequently, decreased DNA repair, resulting in enhanced temozolomide sensitivity [10], [11]. Additional actionable alterations including mutations in the active site of isocitrate dehydrogenases (IDH1/2) and EGFR aberrations, e.g. gene amplification and deletion of exon 2-7 (EGFRvIII), have been shown to play an important role in oncogenesis and progression of glioma, and may carry important theranostic significance [7, 12] [13]. TCGA researchers used gene expression-based molecular classification and grouped GBMs into four subgroups of proneural, classical , mesenchymal and neural, as defined by alterations including PDGFRA/IDH1, EGFR and NF1, demonstrating differences in prognosis and responsiveness to aggressive therapies [7]. A comprehensive understanding of the frequencies of these and other important biomarkers in a large cohort of GBM samples from patients would aid clinical trial design and expedite the incorporation of tumor profiling into clinical practice.
Our study aims to investigate biomarker data collected from the molecular profiles obtained in a CLIA-certified laboratory on 1035 clinical GBM tumors to assist in prognostic and therapeutic decisions. Up to 76 biomarkers were selected based on an association with therapeutic responses in clinical studies on various cancer types and tested using multiple technologies. Biomarker results and associated therapies are presented for the full cohort and for subgroups defined by biomarker characteristics such as IDH1 and TP53 mutations. Previous reports have revealed molecular changes during progression of lower grade gliomas to GBM, driving tumor growth and treatment resistance [14]; however such changes during progression of high-grade gliomas have not been systematically reported. Paired tumors available in the database were therefore analyzed for potential biomarker changes over time. Further, we explored the associations of biomarker status with patient survival.
Results
Patient and tumor characteristics
A total of 1454 consecutive adult gliomas samples that received tumor profiling from July of 2009 to June of 2015 were identified, from which 177 grade III tumors, 115 grade II tumors 37 grade IV tumors with gliosarcoma features as well as 90 tumors with insufficient tumor grade annotation were excluded. The remaining 1035 GBM tumors were used for biomarker analysis (Figure 1). The average age of the GBM patients were 57.1 years old (interquartile range: 49-66), 413 (40%) were female. From the 177 grade III tumor cohort, 107 tumors of grade III astrocytoma with no indication of oligodendroglial component were used for comparison.
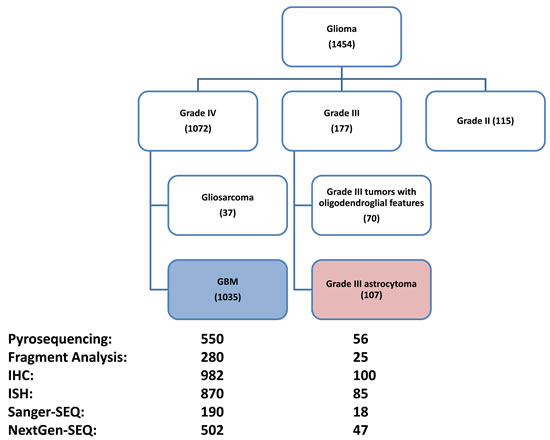
Figure 1: Flow chart showing patient composition included in this study. Numbers in parentheses represent the N number of each subgroup. Data analysis was done on the GBM cohort (N=1035); a comparison with grade III astrocytoma (N=107) was also performed. Numbers next to test platforms represent number of tumors with results from each platform. The tests done and test platform used on each tumor are variable, the details of which are shown in Table 1.
Distribution frequencies of biomarkers revealed by molecular profiling
A total of 76 biomarkers tested by immunohistochemistry, in-situ hybridization, pyrosequencing, fragment analysis, Sanger sequencing as well as next-generation sequencing were analyzed in the GBM cohort. As different biomarker tests were performed for each tumor, the total N for each biomarker tested varied from 58 to 932 (Tables 1, 2)
Overall, MGMT promoter methylation was seen in 43% of GBM tumors, and EGFRvIII was seen in 19%. In-situ hybridization revealed EGFR amplification occurring in 56%, while 1p19q co-deletion and cMET amplification occurring in less than 2%. Among 22 IHCs, TUBB3 and EGFR overexpression were seen in over 80% of GBM tumors, while intact PTEN expression was seen in 74%. Notably, PD-1 expression on tumor-infiltrating lymphocytes was seen in 46% of GBM tumors using the cutoff of 1/high power field, and PD-L1 expression on cancer cells was seen in 19%, using the cutoff of 5% ([16]). ALK IHC was done on a very small subset of tumors (n=58) and 24% showed overexpression (Table 1).
Table 1: Biomarker frequency in GBM tumors tested by pyrosequencing, fragment analysis, in-situ hybridization and immunohistochemistry
Pyro Sequencing |
Positive (N) |
Total (N) |
Percent |
Pyro SEQ-MGMT |
235 |
550 |
43% |
Fragment Analysis |
Positive (N) |
Total (N) |
Percent |
FA-EGFRvIII |
53 |
280 |
19% |
In-situ hybridization |
Positive (N) |
Total (N) |
Percent |
FISH-EGFR |
163 |
289 |
56% |
FISH-1p19q |
3 |
171 |
1.8% |
ISH-cMET |
9 |
500 |
1.8% |
ISH-HER2/Neu |
0 |
609 |
0 |
Immunohistochemistry |
Positive (N) |
Total (N) |
Percent |
TUBB3 |
401 |
485 |
82.7% |
EGFR |
206 |
254 |
81.1% |
PTEN |
691 |
932 |
74.1% |
TOP2A |
410 |
786 |
52.2% |
TS |
421 |
819 |
51.4% |
TOPO1 |
435 |
847 |
51.4% |
PD-1 |
112 |
243 |
46.1% |
ERCC1 |
166 |
431 |
38.5% |
RRM1 |
298 |
803 |
37.1% |
TLE3 |
202 |
601 |
33.6% |
PDGFR |
35 |
129 |
27.1% |
ALK |
14 |
58 |
24.1% |
PD-L1 |
47 |
242 |
19.4% |
SPARC |
100 |
733 |
13.6% |
PGP |
59 |
780 |
7.6% |
PR |
52 |
814 |
6.4% |
AR |
51 |
807 |
6.3% |
MGMT |
25 |
434 |
5.8% |
cMET |
10 |
633 |
1.6% |
c-kit |
3 |
298 |
1.0% |
ER |
1 |
818 |
0.1% |
Her2/Neu |
0 |
906 |
0 |
List of abbreviations: TUBB3: Class III beta-tubulin; PTEN: phosphatase and tensin homolog; TOPO1: Topoisomerase I; EGFR: epidermal growth factor receptor; TS: Thymidylate synthase; TOP2A: topoisomerase II alpha; RRM1: ribonucleotide reductase subunit M1; ERCC1: excision repair cross-complementation group 1; TLE3: transducing-like enhancer of split 3; PDGFR: platelet-derived growth factor receptor alpha; SPARC: secreted protein acidic and rich in cysteine; PR: progesterone receptor; PGP: P-glycoprotein; MGMT: O-6-methylguanine-DNA methyltransferase; AR: Androgen Receptor; cMET: MET or hepatocyte growth factor receptor; ER: estrogen receptor; Her2/neu: human epidermal growth factor receptor 2; PD-1: programmed death 1; PD-L1: programmed cell death ligand 1; ALK: anaplastic lymphoma kinase; cKIT: CD117 or stem cell factor receptor.
Mutation analysis revealed that 39 of 48 genes tested carried mutations, with frequencies ranging from 0.2% to 34% (Table 2) as calculated from 186-663 samples per gene tested. The highest rates were seen in TP53 (34%), PTEN (16%), EGFR (point mutations and small insertions-deletions) (10%), IDH1 (9%), PIK3CA (8%), BRCA2 (7%) and BRCA1 (5%). The remaining 32 genes showed mutation rates below 5%. Specific protein changes observed in each gene can be found in Supplementary materials.
Of the 500 GBM tumors profiled with NGS, 67% had at least one mutation (Figure 2). Co-mutations of 2 genes or more were observed in 31% of patients, 10% had co-mutations of 3 or more genes, and 3 patients showed mutations in 7 or more genes. The genes mutated in these three highly mutated GBM cases are shown in Figure 1. The only common mutated gene among the three cases is TP53. In the complete GBM cohort, TP53-mutated cases were significantly more likely to carry additional mutations in other genes: 67% (111of 166) while only 51% (167 of 327) of TP53-wild type cases had additional mutations (RR=1.31 [1.13-1.52], p=0.0011).
Table 2: Mutation rates of 48 genes tested by sequencing
Sequencing |
Positive (N) |
Total (N) |
Percent |
Sequencing |
Positive (N) |
Total (N) |
Percent |
TP53 |
167 |
494 |
33.8% |
SMO |
3 |
404 |
0.7% |
PTEN |
75 |
460 |
16.3% |
HNF1A |
3 |
433 |
0.7% |
EGFR |
50 |
515 |
9.7% |
ERBB4 |
3 |
496 |
0.6% |
IDH1 |
44 |
500 |
8.8% |
KDR |
3 |
496 |
0.6% |
PIK3CA |
49 |
596 |
8.2% |
GNA11 |
2 |
426 |
0.5% |
BRCA2 |
12 |
186 |
6.5% |
FLT3 |
2 |
492 |
0.4% |
BRCA1 |
9 |
186 |
4.8% |
FGFR2 |
2 |
493 |
0.4% |
APC |
21 |
499 |
4.2% |
NOTCH1 |
2 |
494 |
0.4% |
ATM |
20 |
490 |
4.1% |
CSF1R |
2 |
496 |
0.4% |
PTPN11 |
13 |
494 |
2.6% |
AKT1 |
2 |
498 |
0.4% |
JAK3 |
12 |
494 |
2.4% |
IDH2 |
1 |
309 |
0.3% |
BRAF |
16 |
663 |
2.4% |
SMARCB1 |
1 |
497 |
0.2% |
KRAS |
13 |
617 |
2.1% |
MLH1 |
1 |
498 |
0.2% |
RB1 |
10 |
494 |
2.0% |
CTNNB1 |
1 |
499 |
0.2% |
cMET |
10 |
498 |
2.0% |
GNAS |
1 |
500 |
0.2% |
c-KIT |
11 |
566 |
1.9% |
ALK |
0 |
498 |
0% |
PDGFRA |
7 |
491 |
1.4% |
CDH1 |
0 |
498 |
0% |
ABL1 |
6 |
466 |
1.3% |
ERBB2 |
0 |
479 |
0% |
NRAS |
6 |
551 |
1.1% |
FGFR1 |
0 |
500 |
0% |
STK11 |
5 |
472 |
1.1% |
GNAQ |
0 |
352 |
0% |
VHL |
4 |
422 |
0.9% |
HRAS |
0 |
404 |
0% |
RET |
4 |
478 |
0.8% |
JAK2 |
0 |
500 |
0% |
FBXW7 |
4 |
495 |
0.8% |
MPL |
0 |
487 |
0% |
SMAD4 |
4 |
496 |
0.8% |
NPM1 |
0 |
496 |
0% |
While PIK3CA, BRAF, EGFR, cKIT, KRAS and NRAS data are a combination of NextGen and Sanger sequencing, and IDH2 mutation data is from Sanger sequencing, all other mutations were collected using NextGen.
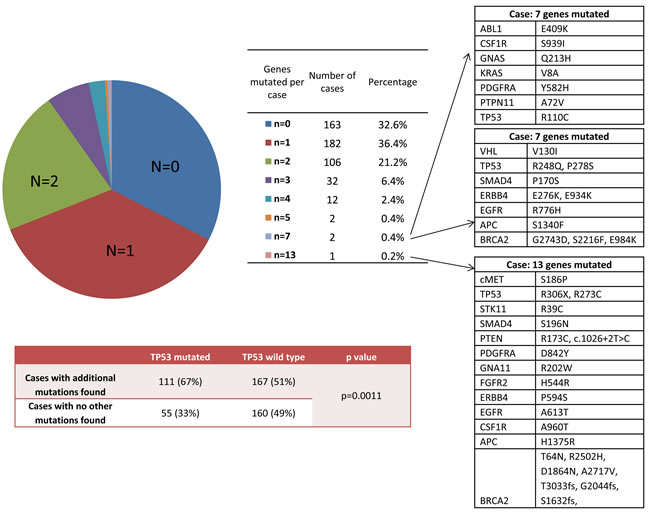
Figure 2: Frequencies of multiple mutations per case (N: number of simultaneous mutations found per case). The 3 cases with the highest number of simultaneous mutations are listed with the specific mutations found. The frequency of TP53 mutation associated with additional mutations is also shown.
In order to identify molecular features specific to GBM (WHO grade IV astrocytoma), the GBM cohort was compared to 107 grade III astrocytoma tumors that don’t show any indication of oligodendroglial component. As shown in Figure 3, EGFR aberrations including gene amplification and EGFRvIII mutation, PTEN mutation, as well as TOP2A, RRM1 and TS overexpression were significantly more prevalent in GBM than grade III astrocytomas. In contrast, MGMT promoter methylation, TP53 and IDH1 mutations were more frequent in grade III astrocytomas.
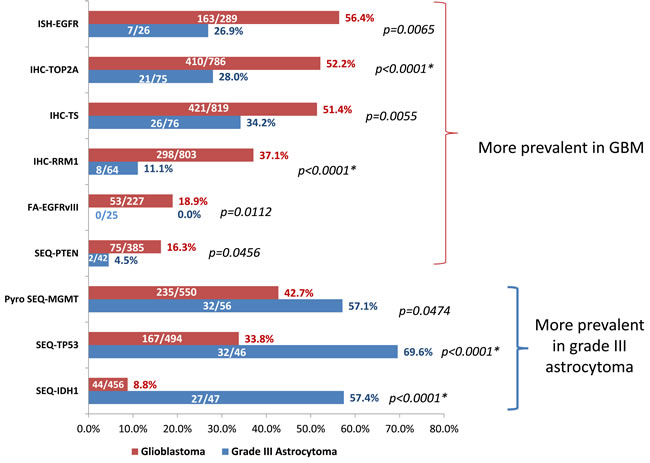
Figure 3: A: Differential biomarker features tested by promoter methylation, fragment analysis, in-situ hybridization and IHC in GBM and grade III astrocytomas. Shown are biomarkers that are statistically different in GBM and grade III astrocytomas by two-tailed Fisher-Exact test. Asterisks indicate comparisons that remain statically significant after correcting for multiple comparisons by Bonferroni correction. Numbers on the bar indicates positive N/total N for each biomarker tested.
Tumor profiles are differentiated by IDH1 mutation
IDH1 mutation identifies GBMs that are developed from lower grade gliomas, i.e., secondary GBM, and are associated with prolonged patient survival [17]. In our GBM cohort, IDH1 mutation was highly associated with MGMT methylation and TP53 mutation. In contrast, IDH1 mutation and EGFRvIII were mutually exclusive, in that all 52 EGFRvIII mutations were found in IDH1 wild type tumors. (Table 3) The relative relationships of IDH1, TP53, MGMT methylation and EGFRvIII are further illustrated in the Venn diagram shown in Figure 4.
Table 3: Differential biomarker characteristics in IDH1-mutated and IDH1-wild type GBM
|
All GBM tumors |
|||
|
IDH1 MT N/Total (%) |
IDH1 WT N/Total (%) |
RR [95% CI] |
p value |
MGMT Methylation |
30/41 (73%) |
469/423 (40%) |
3.63 (1.87-7.07) |
<0.0001* |
TP53 mutation |
40/44 (91%) |
126/449 (28%) |
19.7 (7.17-54.1) |
<0.0001* |
EGFR vIII |
0/23 (0%) |
52/242 (21%) |
0 (n/a) |
0.0105 |
PTEN mutation |
1/40 (2.5%) |
74/420 (18%) |
0.13 (0.02-0.94) |
0.0117 |
(Asterisks indicate comparisons that remain statically significant after correction for multiple comparisons.)
Analysis of paired GBM tumor samples reveals biomarker changes over time
Metachronous paired GBM tumors were available on 17 patients (Figure 5). The mean interval between sample collection times was 499 (91-1605) days. The paired tumors were comprised of primary and recurrent disease (N=6) as well as paired recurrent tumors (N=5), while for 6 pairs it’s unclear if the first specimen was from the primary tumor or a recurrence. 94% of the pairs (16 of 17) had at least one biomarker change; patient q only had paired data on 4 biomarkers, and did not show a biomarker change.
Paired MGMT methylation data was available on 12 patients. While 8 tumor pairs had the same methylation status, 3 pairs changed from methylated to unmethylated (Patients d, g and k) and one pair changed from methylation equivocal to methylated (Patient e). (7% methylated to 54%). EGFR gene amplification status changed in 2 out of 13 pairs, with one patient acquiring amplification and one losing amplification (patients a and m, respectively). EGFRvIII changed from absent to present in 1 of 6 pairs tested.
Sequencing data was available on 8 pairs (Patients a-h). While 4 pairs carried the same mutational profiles, 4 pairs acquired new mutations: 2 acquired EGFR mutations (Patient a with EGFR D770_N771insN, Patient d with EGFR T790M), one (Patient b) acquired an IDH2 mutation (P167L) and one (Patient e) acquired three mutations between paired samples collected 4.4 years apart. The first profile of patient e had wild type cKIT, PTPN11 and PIK3CA and only 7% MGMT promoter methylation (equivocal). The second profile showed mutations in cKIT (E583K), PTPN11 (A72T) and PIK3CA (D434N) and 54% MGMT methylation. The same truncating PTEN mutation (R11X) was seen in both profiles. Mutation status of TP53 was unavailable in the first profile and TP53 P177L was seen on the second.
In addition, significant changes were observed for IHC markers. Interestingly, AR (androgen receptor) and PR (progesterone receptor) showed decreased expression in 3 and 4 patients, respectively while no increase were seen. Pgp increased in 1 out of 13 pairs while EGFR and MGMT expression did not change in the 8 and 14 patients with paired data available. No directional patterns were seen in the other markers tested.
Table 4: Patient age and selected biomarkers were statistically significantly associated with survival of GBM patients (N = 310)
Important prognostic factors |
Hazard Ratio |
95% Confidence Interval |
P Value |
Age (>70 yrs vs. <=70 yrs) |
1.75 |
1.31-2.33 |
0.00013 |
Grade IV patients <=70 years old (N = 251) |
|||
SEQ. MGMT.Me (methylated vs. unmethylated) |
0.44 |
0.22-0.87 |
0.02 |
IHC. PR (positive vs. negative) |
0.61 |
0.4-0.94 |
0.02 |
IHC. TOPO1 (positive vs. negative) |
1.34 |
0.99-1.8 |
0.05 |
Grade IV patients >70 years old (N = 59) |
|||
IHC. PGP (positive vs. negative) |
0.31 |
0.12-0.81 |
0.02 |
IHC. PR (positive vs. negative) |
0.23 |
0.07-0.8 |
0.02 |
IHC. TS (positive vs. negative) |
2.61 |
1.11-6.16 |
0.03 |
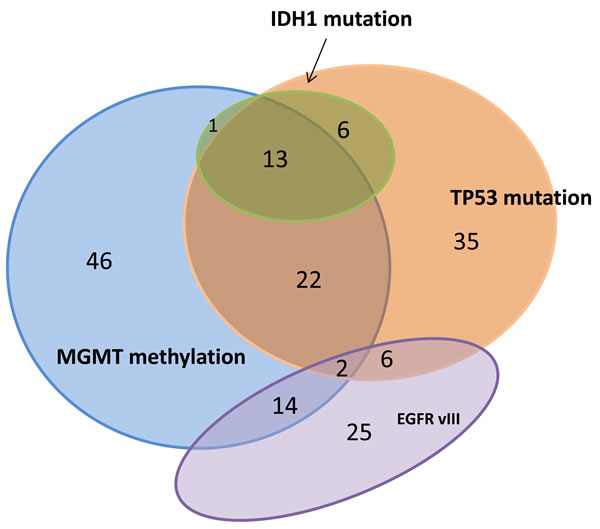
Figure 4: Venn diagram made from 238 GBM cases with IDH1, TP53, MGMT methylation and EGFRvIII evaluated. 170 cases showed at least one aberration.
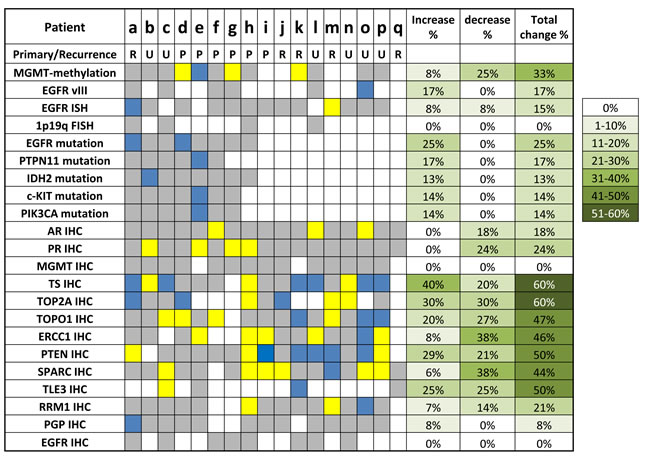
Figure 5: Comparison of biomarker profiles on metachronous GBM pairs (N=17). Primary/Recurrence: R, paired recurrent tumors; P: paired primary and recurrent tumors; U: unknown. Yellow: biomarkers that decreased over time, which included loss of protein overexpression by IHC; loss of gene amplification by ISH and loss of gene mutation by sequencing; loss of MGMT promoter methylation by pyrosequencing. Blue: biomarkers that increased over time, which included acquisition of protein expression by IHC, acquisition of gene amplification by ISH; acquisition of gene mutation by sequencing; acquisition of MGMT promoter methylation by pyrosequencing. Gray: no biomarker change over time.
Tumor grade, patient age and biomarker status were associated with survival
Patient death data was extracted from SSDI by a research intermediate and death data was available for 310 GBM patients. Patient age ranged from 21 to 89 (mean 60) years and mean survival was 543 days. Patients were categorized into the elderly (>70 years old) and young (<=70 years old) based on NCCN treatment stratification. As expected, patients > 70 years old (N=63) had a significantly shorter survival than those <=70 years old (N=247) (HR=1.75, p=0.00013).
Within GBM patients younger than 70 years old, MGMT-Me and positive PR expression were significantly associated with longer survival, and TOPO1 expression was associated with shorter survival. The other biomarkers with data available were not associated with survival. In patients who were older than 70 years old, PR remained associated with a longer survival but MGMT-Me and TOPO1 were not. Instead, PGP expression was associated with longer survival and TS expression with shorter survival. (Supplementary Figures 1 and 2)
Discussion
Despite tremendous progresses in the molecular characterization of GBM, options for effective treatments are still limited. Clinical trials of targeted therapies and chemotherapies in unselected patient cohorts have shown limited benefit with the exception of temozolomide. There is clearly an unmet need to determine if existing therapies or investigational agents in clinical trials could benefit this population. The use of tumor profiling to guide treatment has generated promising results in various cancers, especially in refractory disease [18] [19] [20] [21]. Even though prospective randomized trials have not been published for all of the biomarker-drug associations utilized in these studies, the improved outcomes observed in patients treated with tumor profiling-guided therapies suggests the potential effectiveness of such an approach. The current study describes a large cohort of GBM tumor samples analyzed with the goal of providing the best treatment options for individual patients (Supplementary Table 3). This approach allowed for identification of potential therapeutic opportunities, both those that are part of standard-of-care and those that are not routinely considered. For example, 43% tumors showed MGMT-Me, suggesting benefit from temozolomide [22] [23] [24]. The presence of EGFRvIII suggests potential utility of EGFRvIII-targeted therapies [25] [26] [27]. Further, agents including gemcitabine and fluoropyrimidines are suggested for a portion of patients based on low RRM1 [28] and TS [29], respectively. While published efficacies of these agents in unselected GBM patients are variable, using predictive markers to select patients who are more likely to respond may increase response rates [30] [31]. Of special interest, tumor expression of PD-L1 and tumor-infiltrating lymphocyte expression of PD-1 are seen in 27% and 48% of tumors, suggesting gliomas as a promising potential tumor type for immune modulatory agents [32]; identification of BRCA1/2 mutations makes potential usage of PARP inhibitors of particular interest [33]; the known high rate of EGFR aberration in GBM was also shown by multiple platforms including IHC, ISH, fragment analysis (for EGFRvIII) and NextGen sequencing (for point mutations and small in-dels), confirming EGFR as an important therapeutic target in GBM [3, 34]. Further investigation into the molecular subgroups of GBM showed that TP53 mutation is indicative of mutations on additional genes and that IDH1-mutated GBM tumors lack EGFRvIII mutations and are more likely to carry TP53 mutation and MGMT promoter methylation. These characteristics of molecular features observed in a large cohort of clinical GBM samples confirm the important role of these genetic events in the genesis of glial tumors and thus the molecular heterogeneity of GBM [9] [35] [36].
We describe here that 94% of metachronous tumor pairs show biomarker changes, potentially resulting from tumor progression and/or treatment-driven selection. Changes in MGMT promoter methylation status have been previously reported with conflicting results [37], [38, 39]. Our observation that 4 of 12 patients experience changes and that loss of methylation is more frequent than acquisition supports the notion that during glioma progression, frequent MGMT promoter methylation changes occur, and therefore patients’ responsiveness to temozolomide potentially may also change. Acquired EGFR exon 20 mutations were seen in two patients, one with a T790M mutation and one with an exon 20 insertion (D770_N771insN), both of which are well-studied in NSCLC as an acquired [40] and de novo resistance mechanism to EGFR tyrosine kinase inhibitors [41], respectively. While EGFR is one of the most important oncogenic drivers in glioma, the clinical efficacy of EGFR-targeted therapy has been disappointing [42]. This observation of acquired intracellular domain EGFR mutations may serve as one of the mechanisms accounting for the low efficacy of EGFR-targeted therapies in glioma. Interestingly, one patient showed acquisition of MGMT methylation and mutations across multiple genes including cKIT, PIK3CA and PTPN11. This patient also carried TP53 and PTEN mutations, which are known to cause genomic instability [43] [44] . These results demonstrate that patients at high risk for genetic instability, as identified by an initial NGS profile, should be profiled at recurrence to identify any new, targetable aberrations. The paired tumor analyses carry some caveats: since only one profile was performed on each particular sample, tumor heterogeneity could not be fully addressed. To mitigate this issue, multiple H&E slides were cut and the most representative area of the sample was circled for testing. Additional caveat of the analysis lies in the unavailability of treatment information. Nevertheless, the high probability of biomarker changes support profiling prior to treatment initiation.
Using SSDI as the source of patient survival, our data shows that the mean overall survival of the 310 GBM patients was 500 days (16.7months), comparable to reported results from the RTOG0525 [45] and RTOG0825 [46] trials and in the TCGA database [8], and therefore, representative of the general glioma population. Our analysis confirms patient age as an important prognostic factor, and that within subgroups defined by age, biomarkers are also closely associated with survival. MGMT-methylation is predictive of longer survival in grade IV patients <=70 years old, but is not in patients > 70 years old. Even though patient treatments were not included in our data, since 2007, temozolomide has become the standard-of-care for newly diagnosed, younger GBM patients, and is increasingly used in older patients, though not as commonly [47]. Therefore, our data is consistent with, but does not prove, the predictive value of MGMT-methylation for temozolomide responsiveness. The prognostic effect of IDH1 mutation was not seen likely due to lack of NGS results before early 2013. Additional markers shown to associate with survival in GBM include PR expression, which has been described as a favorable prognosticator in meningiomas [48], but the effect in GBM [49] [50] remains unknown. Recent preclinical study has suggested progesterone with synergistic effect when combined with temozolomide [51]. Our novel finding in GBM warrants further research to elucidate any implications on therapy and survival. TOPO1 overexpression is indicative of poor prognosis in young GBM patients. Based on the potential association of TOPO1 with irinotecan response [52], it is plausible to design prospective randomized trials validating the TOPO1-irinotecan association in glioma. The poor prognostic effect of TS expression has been reported in other cancer types including NSCLC, and is reported in glioma here for the first time [53]. It’s important to note that our survival analysis is limited by only having this data for a small proportion of the patients, and that information are patients’ treatments, responses, performance and extent of resection is lacking. Public databases including SSDI have been shown to be reliable sources for patient survival and have aided in cancer research; however, with the limitations stated above, the survival analysis shown here remains exploratory and needs to be validated in an independent cohort or a randomized trial.
In conclusion, we have summarized biomarker data from 1035 GBM tumors submitted for tumor profiling for theranostic purposes. While standard chemotherapy options are limited for GBM, our data is of importance for both clinical consideration and for clinical trial design. We have identified distinct biomarker profiles defined by WHO grades and molecular characteristics including TP53 and IDH1 mutations. The demonstration of biomarker changes within the same patient over time suggests the necessity of profiling before treatment is instituted. Our results provide a biomarker database for therapy consideration and clinical trial design. Prospective trials are underway to confirm the clinical merit of this approach.
Materials and Methods
Biomarker evaluation was performed on consecutive glioma samples submitted to a CLIA-certified laboratory (Caris Life Sciences, Phoenix, AZ) between 2009 and June of 2015. A retrospective analysis was performed to identify biomarker characteristics of the complete cohort and subgroups. Relative risks with 95% confidence intervals were calculated for univariate comparisons, and associated p-values were calculated using the Fisher Exact test. A two-tailed p-value < 0.05 was considered statistically significant and Bonferroni correction was used to correct for multiple comparisons. A logistic regression model was used for multivariate analysis.
This retrospective analysis utilized previously collected, de-identified data and was deemed exempt from IRB oversight; consent requirements were waived by Western Regional Review Board, the IRB of record for Caris Life Sciences.
Multiplatform tumor profiling
Immunohistochemistry (IHC) was performed on formalin-fixed paraffin-embedded (FFPE) tumor samples using automated staining techniques. IHC results were evaluated independently by board-certified pathologists. Results were categorized into positive or negative by defined thresholds specific to each marker based on published clinical literature that associates biomarker status with patient responses to therapeutic agents. The primary antibody clones and thresholds used can be found in Supplementary Material. Fluorescent in situ hybridization (FISH) was performed to detect EGFR gene amplification and 1p19q co-deletion. Chromogenic in situ hybridization (CISH) or FISH were both used for Her2/neu and cMET gene amplification. Probes and cutoffs can be found in Supplementary Material.
Next-generation sequencing (NGS) was performed on genomic DNA isolated from FFPE tumor tissue using the Illumina MiSeq platform. Specific regions of 47 genes were amplified using the customized Illumina TruSeq Amplicon Cancer Hotspot panel [15]. All variants reported are detected with >99% confidence based on the mutation frequency present and the amplicon coverage. Average depth of coverage is larger than x1500x. Sanger sequencing included selected regions of BRAF, KRAS, c-KIT, EGFR, NRAS, IDH2 and PIK3CA and was performed using M13-linked PCR primers designed to flank and amplify targeted sequences. MGMT methylation testing was performed on extracted DNA by pyrosequencer-based analysis of 5 CpG sites (CpGs 74-78). Samples with ≥7% and <9% methylation were considered to be equivocal results. Fragment analysis of EGFRvIII was performed on RNA extracted from FFPE samples. Two sets of FAM-linked primers were used to PCR-amplify both the wild type and mutant EGFR alleles, and PCR products were visualized using an ABI 3500xl. Signals generated from the wild-type allele were used as an amplification control and samples were considered positive if EGFRvIII was detected at a level that was 5x higher than the average background signal.
Data extraction from Social Security Death Index (SSDI)
Patient dates-of-death were extracted from public databases, including the Social Security Death Index, by a research intermediary who ensured that protected health information was removed from datasets delivered to the researchers. The research intermediary estimated patient survival times by calculating the difference between date of death and date of diagnosis. To estimate the effects of biomarkers on survival, a Cox proportional-hazards model was used to calculate the hazard ratio (HR).
Conflicts of Interest
Joanne Xiu, David Spetzler, Todd Maney, Anotale Ghazalpour, Ryan Bender, Zoran Gatalica and Sandeep Reddy are employees of Caris Life Sciences. Other authors have no conflict of interest to declare.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
References
1. Dolecek TA, Propp JM, Stroup NE and Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-oncology. 2012; 14 Suppl 5:v1-49.
2. Siegel R, Ma J, Zou Z and Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014; 64(1):9-29.
3. Furnari FB, Cloughesy TF, Cavenee WK and Mischel PS. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat Rev Cancer. 2015; 15(5):302-310.
4. Kesari S. Understanding glioblastoma tumor biology: the potential to improve current diagnosis and treatments. Seminars in oncology. 2011; 38 Suppl 4:S2-10.
5. Agnihotri S, Burrell KE, Wolf A, Jalali S, Hawkins C, Rutka JT and Zadeh G. Glioblastoma, a brief review of history, molecular genetics, animal models and novel therapeutic strategies. Archivum immunologiae et therapiae experimentalis. 2013; 61(1):25-41.
6. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine. 2005; 352(10):987-996.
7. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer cell. 2010; 17(1):98-110.
8. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, et al. The somatic genomic landscape of glioblastoma. Cell. 2013; 155(2):462-477.
9. Crespo I, Vital AL, Gonzalez-Tablas M, Patino Mdel C, Otero A, Lopes MC, de Oliveira C, Domingues P, Orfao A and Tabernero MD. Molecular and Genomic Alterations in Glioblastoma Multiforme. The American journal of pathology. 2015; 185(7):1820-1833.
10. Malmstrom A, Gronberg BH, Marosi C, Stupp R, Frappaz D, Schultz H, Abacioglu U, Tavelin B, Lhermitte B, Hegi ME, Rosell J and Henriksson R. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. The Lancet Oncology. 2012; 13(9):916-926.
11. Wick W, Platten M, Meisner C, Felsberg J, Tabatabai G, Simon M, Nikkhah G, Papsdorf K, Steinbach JP, Sabel M, Combs SE, Vesper J, Braun C, Meixensberger J, Ketter R, Mayer-Steinacker R, et al. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. The Lancet Oncology. 2012; 13(7):707-715.
12. Olar A and Aldape KD. Using the molecular classification of glioblastoma to inform personalized treatment. The Journal of pathology. 2014; 232(2):165-177.
13. Gan HK, Cvrljevic AN and Johns TG. The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. The FEBS journal. 2013; 280(21):5350-5370.
14. Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K, Asthana S, Jalbert LE, Nelson SJ, Bollen AW, Gustafson WC, Charron E, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science (New York, NY). 2014; 343(6167):189-193.
15. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. The New England journal of medicine. 2012; 366(26):2443-2454.
16. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, et al. IDH1 and IDH2 mutations in gliomas. The New England journal of medicine. 2009; 360(8):765-773.
17. Von Hoff DD, Stephenson JJ, Jr., Rosen P, Loesch DM, Borad MJ, Anthony S, Jameson G, Brown S, Cantafio N, Richards DA, Fitch TR, Wasserman E, Fernandez C, Green S, Sutherland W, Bittner M, et al. Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. Journal of clinical oncology. 2010; 28(33):4877-4883.
18. Tsimberidou AM, Iskander NG, Hong DS, Wheler JJ, Falchook GS, Fu S, Piha-Paul S, Naing A, Janku F, Luthra R, Ye Y, Wen S, Berry D and Kurzrock R. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clinical cancer research. 2012; 18(22):6373-6383.
19. Jameson GS, Petricoin EF, Sachdev J, Liotta LA, Loesch DM, Anthony SP, Chadha MK, Wulfkuhle JD, Gallagher RI, Reeder KA, Pierobon M, Fulk MR, Cantafio NA, Dunetz B, Mikrut WD, Von Hoff DD, et al. A pilot study utilizing multi-omic molecular profiling to find potential targets and select individualized treatments for patients with previously treated metastatic breast cancer. Breast cancer research and treatment. 2014; 147(3):579-588.
20. Tsimberidou AM, Wen S, Hong DS, Wheler JJ, Falchook GS, Fu S, Piha-Paul S, Naing A, Janku F, Aldape K, Ye Y, Kurzrock R and Berry D. Personalized medicine for patients with advanced cancer in the phase I program at MD anderson: validation and landmark analyses. Clinical cancer research. 2014; 20(18):4827-4836.
21. Febbo PG, Ladanyi M, Aldape KD, De Marzo AM, Hammond ME, Hayes DF, Iafrate AJ, Kelley RK, Marcucci G, Ogino S, Pao W, Sgroi DC and Birkeland ML. NCCN Task Force report: Evaluating the clinical utility of tumor markers in oncology. Journal of the National Comprehensive Cancer Network. 2011; 9 Suppl 5:S1-32; quiz S33.
22. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC and Stupp R. MGMT gene silencing and benefit from temozolomide in glioblastoma. The New England journal of medicine. 2005; 352(10):997-1003.
23. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. The Lancet Oncology. 2009; 10(5):459-466.
24. Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, Friedman HS, Gilbert MR, Herndon JE, McLendon RE, Mitchell DA, Reardon DA, Sawaya R, Schmittling R, Shi W, Vredenburgh JJ, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro-oncology. 2011; 13(3):324-333.
25. Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, Gilbert MR, Herndon JE, 2nd, McLendon RE, Mitchell DA, Reardon DA, Sawaya R, Schmittling RJ, Shi W, Vredenburgh JJ and Bigner DD. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. Journal of clinical oncology. 2010; 28(31):4722-4729.
26. Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD, Mrugala MM, Jensen R, Baehring JM, Sloan A, Archer GE, Bigner DD, Cruickshank S, Green JA, Keler T, Davis TA, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro-oncology. 2015; 17(6):854-861.
27. Gong W, Zhang X, Wu J, Chen L, Li L, Sun J, Lv Y, Wei X, Du Y, Jin H and Dong J. RRM1 expression and clinical outcome of gemcitabine-containing chemotherapy for advanced non-small-cell lung cancer: a meta-analysis. Lung cancer. 2012; 75(3):374-380.
28. Qiu LX, Tang QY, Bai JL, Qian XP, Li RT, Liu BR and Zheng MH. Predictive value of thymidylate synthase expression in advanced colorectal cancer patients receiving fluoropyrimidine-based chemotherapy: evidence from 24 studies. International journal of cancer. 2008; 123(10):2384-2389.
29. Metro G, Fabi A, Mirri MA, Vidiri A, Pace A, Carosi M, Russillo M, Maschio M, Giannarelli D, Pellegrini D, Pompili A, Cognetti F and Carapella CM. Phase II study of fixed dose rate gemcitabine as radiosensitizer for newly diagnosed glioblastoma multiforme. Cancer chemotherapy and pharmacology. 2010; 65(2):391-397.
30. Grunda JM, Fiveash J, Palmer CA, Cantor A, Fathallah-Shaykh HM, Nabors LB and Johnson MR. Rationally designed pharmacogenomic treatment using concurrent capecitabine and radiotherapy for glioblastoma; gene expression profiles associated with outcome. Clinical cancer research. 2010; 16(10):2890-2898.
31. Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wohrer A, Dieckmann K, Filipits M, Brandstetter A, Weller M, Kurscheid S, Hegi ME, Zielinski CC, Marosi C, Hainfellner JA, Preusser M, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro-oncology. 2015; 17(8):1064-1075.
32. Barazzuol L, Jena R, Burnet NG, Meira LB, Jeynes JC, Kirkby KJ and Kirkby NF. Evaluation of poly (ADP-ribose) polymerase inhibitor ABT-888 combined with radiotherapy and temozolomide in glioblastoma. Radiation oncology (London, England). 2013; 8:65.
33. Gan H. A phase 1 study evaluating ABT-414 in combination with temozolomide (TMZ) for subjects with recurrent or unresectable glioblastoma (GBM). Journal of clinical oncology. 2015; 32(5s):suppl; abstr 2021.
34. Wiestler B, Capper D, Sill M, Jones DT, Hovestadt V, Sturm D, Koelsche C, Bertoni A, Schweizer L, Korshunov A, Weiss EK, Schliesser MG, Radbruch A, Herold-Mende C, Roth P, Unterberg A, et al. Integrated DNA methylation and copy-number profiling identify three clinically and biologically relevant groups of anaplastic glioma. Acta neuropathologica. 2014; 128(4):561-571.
35. Suzuki H, Aoki K, Chiba K, Sato Y, Shiozawa Y, Shiraishi Y, Shimamura T, Niida A, Motomura K, Ohka F, Yamamoto T, Tanahashi K, Ranjit M, Wakabayashi T, Yoshizato T, Kataoka K, et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nature genetics. 2015; 47(5):458-468.
36. Park CK, Kim JE, Kim JY, Song SW, Kim JW, Choi SH, Kim TM, Lee SH, Kim IH and Park SH. The Changes in MGMT Promoter Methylation Status in Initial and Recurrent Glioblastomas. Translational oncology. 2012; 5(5):393-397.
37. Brandes AA, Franceschi E, Tosoni A, Bartolini S, Bacci A, Agati R, Ghimenton C, Turazzi S, Talacchi A, Skrap M, Marucci G, Volpin L, Morandi L, Pizzolitto S, Gardiman M, Andreoli A, et al. O(6)-methylguanine DNA-methyltransferase methylation status can change between first surgery for newly diagnosed glioblastoma and second surgery for recurrence: clinical implications. Neuro-oncology. 2010; 12(3):283-288.
38. Tonn J. MGMT promoter methylation status and DNA mismatch repair genes in paired primary and recurrent glioblastoma: A translational study of the German Glioma Network. Journal of clinical oncology. 2010; 28(2076).
39. Su KY, Chen HY, Li KC, Kuo ML, Yang JC, Chan WK, Ho BC, Chang GC, Shih JY, Yu SL and Yang PC. Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. Journal of clinical oncology. 2012; 30(4):433-440.
40. Yasuda H, Kobayashi S and Costa DB. EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. The Lancet Oncology. 2012; 13(1):e23-e31.
41. Raizer JJ, Abrey LE, Lassman AB, Chang SM, Lamborn KR, Kuhn JG, Yung WK, Gilbert MR, Aldape KA, Wen PY, Fine HA, Mehta M, Deangelis LM, Lieberman F, Cloughesy TF, Robins HI, et al. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro-oncology. 2010; 12(1):95-103.
42. Hanel W and Moll UM. Links between mutant p53 and genomic instability. Journal of cellular biochemistry. 2012; 113(2):433-439.
43. Sun Z, Huang C, He J, Lamb KL, Kang X, Gu T, Shen WH and Yin Y. PTEN C-terminal deletion causes genomic instability and tumor development. Cell reports. 2014; 6(5):844-854.
44. Gilbert MR, Wang M, Aldape KD, Stupp R, Hegi ME, Jaeckle KA, Armstrong TS, Wefel JS, Won M, Blumenthal DT, Mahajan A, Schultz CJ, Erridge S, Baumert B, Hopkins KI, Tzuk-Shina T, et al. Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. Journal of clinical oncology. 2013; 31(32):4085-4091.
45. Gilbert MR DJ. RTOG 0825: Phase III double-blind placebo-controlled trial evaluating bevacizumab (Bev) in patients (Pts) with newly diagnosed glioblastoma (GBM). J Clin Oncol 31, 2013b (suppl; abstr 1).
46. Network. NCC. NCCN Clinical Practice Guidelines in Oncology. Central Nervous System Cancers. National Comprehensive Cancer Network. Version 1.2014;.
47. Hsu DW, Efird JT and Hedley-Whyte ET. Progesterone and estrogen receptors in meningiomas: prognostic considerations. Journal of neurosurgery. 1997; 86(1):113-120.
48. Kabat GC, Etgen AM and Rohan TE. Do steroid hormones play a role in the etiology of glioma? Cancer epidemiology, biomarkers & prevention. 2010; 19(10):2421-2427.
49. Assimakopoulou M, Sotiropoulou-Bonikou G, Maraziotis T and Varakis J. Does sex steroid receptor status have any prognostic or predictive significance in brain astrocytic tumors? Clinical neuropathology. 1998; 17(1):27-34.
50. Atif F, Patel NR, Yousuf S and Stein DG. The Synergistic Effect of Combination Progesterone and Temozolomide on Human Glioblastoma Cells. PloS one. 2015; 10(6):e0131441.
51. Braun MS, Richman SD, Quirke P, Daly C, Adlard JW, Elliott F, Barrett JH, Selby P, Meade AM, Stephens RJ, Parmar MK and Seymour MT. Predictive biomarkers of chemotherapy efficacy in colorectal cancer: results from the UK MRC FOCUS trial. Journal of clinical oncology. 2008; 26(16):2690-2698.
52. Sun JM, Ahn JS, Jung SH, Sun J, Ha SY, Han J, Park K and Ahn MJ. Pemetrexed Plus Cisplatin Versus Gemcitabine Plus Cisplatin According to Thymidylate Synthase Expression in Nonsquamous Non-Small-Cell Lung Cancer: A Biomarker-Stratified Randomized Phase II Trial. Journal of clinical oncology. 2015; 33(22):2450-2456.
53. Millis SZ, Bryant D, Basu G, Bender R, Vranic S, Gatalica Z and Vogelzang NJ. Molecular profiling of infiltrating urothelial carcinoma of bladder and nonbladder origin. Clinical genitourinary cancer. 2015; 13(1):e37-49.