
INTRODUCTION
Reactive oxygen species (ROS) are an integral part of life and are a byproduct of respiration and exposure to the environment [1, 2]. When in excess, ROS can damage DNA, proteins and lipids and promote mutations that may contribute to onset of a wide spectrum of human diseases [3-5] particularly diseases that are associated with aging [6-8] such as neurodegeneration [9-12] and cancer [13-15]. To maintain redox and prevent aberrant ROS accumulation molecular-genetic mechanisms have evolved that sense and respond to oxidative stress. These genetic programs involve expression of proteins that directly detoxify ROS [16], or that are part of metabolic programs that generate anti-oxidants such as glutathione [16-24].
While these mechanisms are important to prevent disease onset, it is now becoming evident that the same mechanisms are being exploited by cancer cells in order to survive and thrive under oxidative stress insults and to armor themselves against therapeutic interventions that rely on oxidative stress as their mechanism of action [25-29]. Here we will review two transcription factors that promote transcriptional-metabolic anti-oxidative stress programs, NRF2 and p53. Both nrf2 and p53 knockout mice show enhanced susceptibility to induced or spontaneous tumors, and are therefore tumor suppressors, but have now been implicated as cancer promoting in some pathological contexts. We will discuss evidence for crosstalk between these transcription factors and possible therapeutic strategies arising from these observations.
The NRF2 anti-oxidative stress transcriptional program plays an important role in tumor prevention
The NRF2-KEAP1 molecular system is a sensor of oxidative stress [30]. Under ambient conditions, NRF2 interacts with KEAP1, an adaptor molecules which directs its targets to the CUL3 E3 ligase [31] for ubiquitylation and subsequent degradation by the proteasome [32, 33]. This mechanism results in low basal NRF2 levels and activity. Under oxidative stress, specific KEAP1 cysteines become oxidized resulting in disruption of the KEAP1-NRF2 complex, leading to a release of NRF2 from KEAP1 inhibition and promoting its stabilization [34, 35]. Newly synthesized NRF2 then translocate to the nucleus [36, 37] to transcribe genes encoding a battery of anti-oxidant proteins [38] such as Heme Oxygenase (HMOX1) [39], NAD(P)H dehydrogenase (quinine; NQO1) [40] and key enzymes in the glutathione biosynthesis/recycling pathway such as γ-glutamylcysteine ligase (GCL) [7, 41, 42], collectively known as phase II detoxifying enzymes [43].
ROS can promote tumor initiation by damaging DNA that will lead to mutations and by augmenting signaling pathways that promote cell growth and proliferation [44]. It has long been observed that anti-oxidants and some natural compounds can be beneficial in preventing cancer initiation [45]. It was hypothesized that the protective activity of these compounds may be linked to their ability to increase expression of endogenous anti-oxidants, more specifically, the phase II detoxifying enzymes [46]. This principal was demonstrated using genetic approach where the chemo-protectant oltipraz induced phase II detoxifying enzymes and reduced cancer incidents in Nrf2 WT but not KO mice treated with a chemical carcinogen [47]. These experiments also established NRF2 as an important factor in promoting the activity of cancer preventing compounds. Similarly, other natural compounds and genetic models were shown to rely on NRF2 for their protective activity leading to the notion that activating NRF2 is an attractive strategy to prevent cancer and reduce oxidative damage [48-56].
The p53 anti-oxidative stress transcriptional program plays a role in preventing ROS induced DNA damage and cancer initiation
p53 is both a positive and negative regulator of ROS [57]. p53 protein levels in the cells are tightly regulated [58-60]. In resting conditions, p53 protein is maintained at low levels by MDM2 mediated proteasomal degradation [61-66] and at this low level of expression reduces ROS levels by inducing the expression of anti-oxidative stress proteins such as SESN1, SESN2 and GPX1 [67-70]. Using a p53 KO model, it was then demonstrated that lack of expression of these anti-oxidative stress proteins is associated with increased cellular ROS which leads to increases in DNA oxidation and in the mutation rate thus promoting tumorigenesis in p53 KO mice. Later, it was also shown that p53 regulates GLS2 expression promoting glutathione generation by increasing glutaminolysis [71, 72], a metabolic process that promotes the conversion of glutamine to glutamate that is often active in cancer cells [73-77]. These findings raise the possibility that the tumor suppressor activity of p53 is related to its role in maintaining cellular redox by regulating cellular metabolism [78, 79] (Fig. 1).
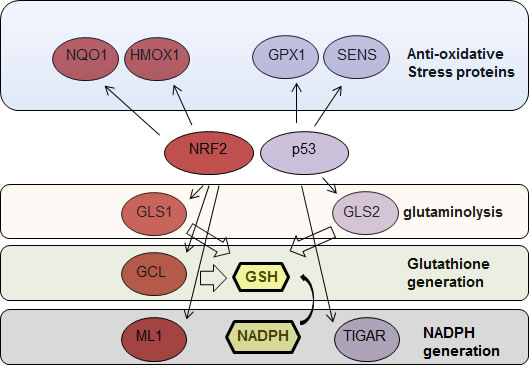
Figure 1: NRF2 and p53 regulate the expression of proteins involved in protection ageist oxidative stress. NRF2 and p53 target genes (red and blue) that are contributing to protection against oxidative stress directly or by promoting glutathione synthesis by facilitating glutaminolysis, through direct synthesis or by facilitating NADPH production. NRF2, nuclear factor (erythroid-derived 2)-like 2; HMOX1, Heme Oxygenase 1; GCL, γ-glutamylcysteine ligase; GPX, glutathione peroxidase; NQO1, NAD(P)H quinine dehydrogenase; SENS, sestrins; GLS 1/2, glutaminase 1/2; GLH, reduced glutathione; NAPDH, Nicotinamide adenine dinucleotide phosphate.
An important role for p53-mediated metabolic regulation to its tumor suppressor activity
p53 coordinates a large number of integrated transcriptional programs that result in divers biological outputs [80-84]. Deciphering the contribution of a specific aspect of p53 function to its tumor suppressor activity is one of the important questions in the p53 field [85-87]. p53 is modified by a large number of different posttranslational modifications that play an important role in the regulation of the specific cellular program that will be activated by p53 [88-90]. Manipulation of these modifications is an attractive strategy when attempting to dissect the roles of specific p53 transcriptional programs in its biological functions and pharmacological reactivation of p53 activity in cancer is an active research field [91-98]. Among other modifications, p53 is acetylated on three lysines [99-101]. Giu and coworkers studied the role of lysine acetylation to the execution of discreet p53 cellular functions related to its tumor suppressor phenotype namely, apoptosis, cell cycle arrest, senescence and the anti-oxidative stress metabolic program [102-107]. Using p53 KO mice and cells that re-express p53 mutants, in which all three acetylated lysines were replaced by arginine (p53-3KR), they showed that, like the p53 KO mice, mice and cells expressing p53-3KR were defective in the execution of apoptosis, cell cycle arrest and senescence. Surprisingly, p53-3KR mice did not succumb to cancer, as did the p53 KO mice indicating that p53-3KR retained it tumor suppressor activity. Further examination of the p53-3KR mutants revealed that they did retain the WT function in executing a transcriptional metabolic program that resulted in reducing glucose uptake, reducing glycolysis and reducing ROS generation that were associated with induction of the p53 anti-oxidative stress targets GLS2 [72] and TIGAR [108]. These findings underscore the importance of ROS regulation in the tumor suppressor activity of p53.
Cancer cells and oncogenes hijack NRF2 for anti-oxidative stress protection
Despite the established role of NRF2 in cancer prevention [55] recent genetic evidence obtained from human cancers points to possible pro-cancer activities of NRF2. In particular it was found that there are several cancer related genetic events that prevent the degradation of NRF2 through the KEAP1-CUL3 pathway that leads to elevated NRF2 activity [33, 109]. These include somatic mutations in NRF2 that disrupt its interaction with KEAP1 [110, 111] and somatic mutations in KEAP1 that disrupt its interaction with NRF2 [112-115]. It was also found that high NRF2 expression or nuclear localization and low KEAP1 expression were associated with poor prognosis [116]. These findings, and cell based experiments, point to a model that argues that NRF2 can be exploited by cancer cells in order to curb oxidative stress and perhaps to enhance their chemo-resistance [109, 111, 116-122]
Aberrant proliferation is one of the hallmarks of cancer and oncogenes will often activate growth-promoting pathways. In a provocative paper DeNocola and collaborators showed that oncogenic K-Ras(G12D), when expressed at physiological levels, reduces ROS by increasing NRF2 that in turn promotes the expression of the anti-oxidative stress response [123]. The increase in NRF2 was promoted by the activation of the RAF pathway leading to an increase in Jun activation that enhanced NRF2 gene expression. The authors demonstrated that NRF2 was important for K-Ras(G12V) tumorigenic functions using a murine model of mutant K-Ras driven lung and pancreatic cancer. Nrf2 KO mice showed reduced tumor occurrence, reduced proliferation in the tumors arising in these mice and increased overall survival [123]. These findings led to the concept of a new pro-cancer activity of NRF2 in which it supports oncogene-mediated oncogenesis in addition to its proposed chemoprotective role. In support of this premise, detailed global analysis of NRF2 target genes, under both resting and NRF2 induced conditions, revealed that a substantial proportion of NRF2 targets are cell cycle promoting genes [124].
In order to support rapid proliferation, tumor cells rely on catabolic processes for generation of building blocks such as lipids, proteins and nucleic acids [125]. In a recent study, Mitsuishi et al, asked whether the increased levels of NRF2 observed in some cancer cells play a role in their rapid proliferation [126]. Using NRF2 knock down in A549 lung cancer cells, that harbor a KEAP1 mutation and therefore express high levels of NRF2 [114], they found that NRF2 was indeed important for proliferation of these cells. In order to identify the mechanism by which NRF2 supports proliferation they used microarray analysis to identify NRF2 target genes. In addition to well-established NRF2 targets, several new target genes involved in the pentose phosphate pathway were identified suggesting that NRF2 promotes a distinct proliferation enhancing metabolic program. Indeed, high levels of NRF2 promoted the expression of proteins that support glucose flux through pentose phosphate pathway to generate purines, the building blocks of DNA and RNA, at the expense of the glycolytic pathway. Furthermore, metabolic analysis reveled that loss of NRF2 resulted in increase in cellular levels of glutamine and glutamate suggesting that NRF2 was important for catabolizing these amino acids. The authors then used a tracer study to show that NRF2 promotes glutaminolysis, the conversion of glutamine to glutamate, and directs glutamate into two metabolic pathways. One pathway is governed by up regulation of GCL that utilizes glutamate to generate glutathione. This finding is in accord with previous reports that NRF2 is important for glutathione generation [7, 41, 42]. The second pathway is induced by up regulation of ME1, where glutamate is utilized for the generation of another anti-oxidant-reducing agent, NADPH. Therefore, NRF2 induces a metabolic program that supplies building blocks to support proliferation and anti-oxidants that can protect these cancer cells from oxidative stress (Figure 1). This particular NRF2-driven pro-proliferation metabolic program was shown to be dependent on the hyperactivation of the PI3K-AKT pathway, a pathway that is typically deregulated in cancer [127, 128] providing further support to the premise that cancer cells and their oncogenic pathways utilize NRF2 to their own advantage.
p53 in the service of cancer
The tumor suppressor activity of p53 is well documented [82, 129-132]. This activity is achieved by the employment of different pathways, spanning from gene and microRNA regulation to protein-protein interaction [130, 133-137]. However, genetic evidence obtained by analyzing patient data indicates, that in specific breast cancer sub types, WT p53 status predicts poor response to aggressive therapy [138]. One possible explanation for this observation is that WT p53 will promote cell cycle arrest and help tumor cells resists chemotherapy which targets dividing cells [139, 140]. Indeed, it was recently shown that tumors bearing WT p53 resist chemotherapy by inducing a senescence program that leads to cell cycle arrest and production of cytokines that in turn encourage the growth of drug resistant cells within the tumors [141]. These findings together with the fact that p53 promotes an anti-oxidative stress metabolic program, raise the question whether tumor cells might also use p53 in order to battle oxidative stress to gain chemo resistance, as is the case for NRF2. Indeed, p53 was shown to protect A549 cells used in the NRF2 study discussed above [126], from toxicity and radio sensitization using the metabolic inhibitor 2-Deoxy-glucose (2DG), by increasing anti-oxidative stress response and promoting oxidative phosphorylation [142, 143]. These observations, and the recent appreciation that p53 promotes a metabolic program that results in increased levels of cellular anti-oxidants [78], raise the concern that in specific pathological contexts, p53 may be exploited by cancer cells in order increase anti-oxidants to gain chemo resistance, much like NRF2.
Cross talk between p53 and NRF2
It is becoming more evident that, at the functional level, p53 and NRF2 play similar roles and are both providing cells with enhanced capacity to mitigate oxidative stress. Interestingly, recent findings from the Zhang lab indicate that p21, a p53 target gene [144, 145], stabilizes NRF2 by binding to KEAP1 and interfering with its ability to promote NRF2 ubiquitylaton and proteasomal degradation [146]. On the other hand, previous findings from the Shaul lab indicate that NQO1, an NRF2 target, interacts with p53 [147] and blocks its degradation by the 20S proteasome [148], a degradation process that is independent of MDM2 and ubiquitin [59, 149] (Fig. 2). These findings support the premise of an interesting cross talk between these two transcription factors and raise the question of whether there is a positive feedback loop between NRF2 and p53 and whether cancer cells enhance their resistance to oxidative stress by utilizing this putative positive feedback loop (Fig. 2). Indeed, p53 deficient HCT116 colon carcinoma cells exhibited reduced induction of NRF2 target genes as compared with p53 proficient HCT116 cells following challenge with oxidative stress [150] suggesting that p53 may be important for NRF2 activation in cancer cells. However, this model may not be complete, as it was recently shown that MDM2 is a transcriptional target of NRF2 through which NRF2 negatively regulates p53 [151] (Fig.2). In another study it was also shown that p53 binds to promoter elements activated by NRF2 and is therefore a transcriptional repressor of NRF2 target genes [152] (Fig. 2). The apparent discrepancies between these reports suggest that the relationship between NRF2 and p53 may well be dependent on the cellular and biological context.
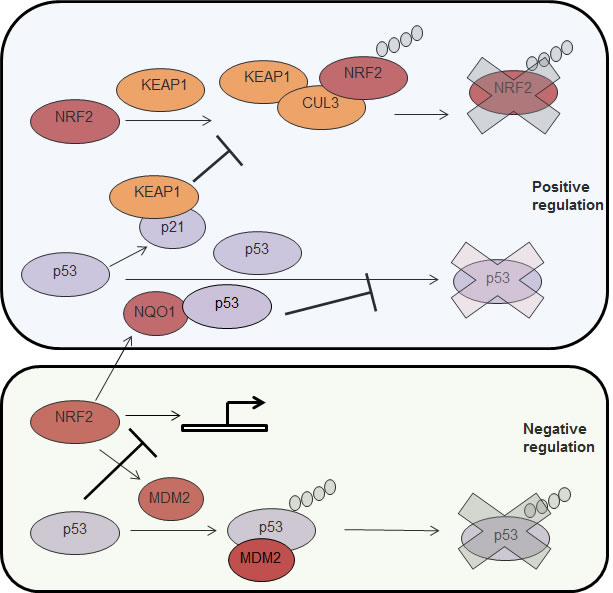
Figure 2: Positive and negative (up or down) co-regulation between p53 and NRF2. Top to bottom. KEAP1 interacts with NRF2 and forms a complex with the E3 ligase CUL3 that results in NRF2 ubiquitylation and degradation by the proteasome (depicted as a blue X). The p53 target gene, p21, interacts with KEAP1 and inhibits NRF2 ubiquitylation and degradation. p53 is degraded by the proteasome in a ubiquitin n dependent manner. The NFR2 target, NQO1, interacts with p53 and protects it from degradation. NRF2 target gene, MDM2, promotes p53 ubiquitylation and degradation by the proteasome. P53 is a transcriptional repressor of NRF2. NRF2, nuclear factor (erythroid-derived 2)-like 2NQO1, NAD(P)H quinine dehydrogenase; mdm2, mouse double minute 2.
Targeting anti-oxidative stress proteins as possible anti-cancer therapy
Stabilization or reactivation of p53 throughout the use of small molecules is a promising therapeutic strategy [96, 98, 153-157]. However, we believe that alternative approaches should be explored in order to win the war against cancer [158-163].
In light of the model that NRF2 and p53 synergize in enhancing the cellular anti-oxidative stress mechanisms, we reason that cancer cells that exhibit high NRF2 levels and harbor WT p53 will be more dependent on these pathways to sustain chemo resistance. It is therefore tempting to speculate that targeting the anti-oxidative stress modules, that are promoted by NRF2 and p53, in combination with chemotherapeutic that will increase ROS, such as Doxorubicin [164], is a rational approach for treating such cases. Some of potential drugable targets would be the glutaminolysis pathway, the glutathione generating pathway, anti-oxidative stress proteins and NRF2. As discussed above, p53 and NRF2 promote glutaminolysis that supplies the glutamate and NADPH to generate glutathione to battle oxidative stress (Fig. 1). Targeting Kidney type Glutaminase (KGA), an essential enzyme in glutaminolysis, using an inhibitor such as BPTES, could inhibit glutaminolysis [165-167]. Indeed, this compound was shown to inhibit growth of MYC transformed P493 cells in vivo by increasing ROS and reducing glutathione levels in these cells [166]. Another strategy to reduce glutathione is by targeting γ-glutaminase, a rate limiting enzyme in the generation of glutathione, using BSO, a compound that has been shown to be well tolerated in man [168].
Piperlongumine has been shown to be selectively toxic to cancer cells and its mechanism of action was proposed to involve enhancing ROS in cancer cells by binding to a wide number of anti-oxidative stress proteins [169]. It is therefore plausible that piperlongumine could be useful in treating cancers that rely on anti-oxidative stress proteins that are induced by NRF2 and p53. Using 80 piperlongumine analogs it was recently demonstrated that the toxic effect of piperlongumine is due to its activity in crosslinking glutathione to proteins and depletion of cellular glutathione [170].
A more direct approach could be the targeting of NRF2 itself using brusatol, a natural compound that was shown to inhibit NRF2 by promoting its degradation [171]. The same study showed that brusatol synergized with chemotherapeutic agents in vitro and in xenograft models and to induce death in tumors that have acquired drug resistance through NRF2.
It is now clear that the cancer cells will utilize endogenous protective mechanisms to evolve chemoresistance [172-174]. It is therefore important that we take a close look at what we believe are tumor suppressor proteins and pathways as they might paradoxically be hijacked by cancer cells to promote their growth and survival. NRF2 and p53 may well be the two-faced Januses of cancer.
Acknowledgments:
This work has been supported by the Medical Research Council, United Kingdom; MIUR, MinSan, RF73, RF57, ACC12; Odysseus Grant (G.0017.12) to GM.
Reference
1. Malinin NL, West XZ and Byzova TV. Oxidation as “the stress of life”. Aging (Albany NY). 2011; 3(9):906-910.
2. Hwang AB and Lee SJ. Regulation of life span by mitochondrial respiration: the HIF-1 and ROS connection. Aging (Albany NY). 2011; 3(3):304-310.
3. Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011; 194(1):7-15.
4. Maccarrone M and Brune B. Redox regulation and the metabolic syndrome. Cell Death Differ. 2011; 18(7):1234-1236.
5. Gough DR and Cotter TG. Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell Death Dis. 2011; 2:e213.
6. Finkel T and Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000; 408(6809):239-247.
7. Suh JH, Shenvi SV, Dixon BM, Liu H, Jaiswal AK, Liu RM and Hagen TM. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci U S A. 2004; 101(10):3381-3386.
8. Vigneron A and Vousden KH. p53, ROS and senescence in the control of aging. Aging (Albany NY). 2010; 2(8):471-474.
9. Akhtar MW, Sunico CR, Nakamura T and Lipton SA. Redox Regulation of Protein Function via Cysteine S-Nitrosylation and Its Relevance to Neurodegenerative Diseases. Int J Cell Biol. 2012; 2012:463756.
10. Berry C, La Vecchia C and Nicotera P. Paraquat and Parkinson’s disease. Cell Death Differ. 2010; 17(7):1115-1125.
11. Centonze D, Muzio L, Rossi S, Furlan R, Bernardi G and Martino G. The link between inflammation, synaptic transmission and neurodegeneration in multiple sclerosis. Cell Death Differ. 2010; 17(7):1083-1091.
12. Corona C, Pensalfini A, Frazzini V and Sensi SL. New therapeutic targets in Alzheimer’s disease: brain deregulation of calcium and zinc. Cell Death Dis. 2011; 2:e176.
13. Ames BN. Dietary carcinogens and anticarcinogens. Oxygen radicals and degenerative diseases. Science. 1983; 221(4617):1256-1264.
14. Chen ZX and Pervaiz S. Involvement of cytochrome c oxidase subunits Va and Vb in the regulation of cancer cell metabolism by Bcl-2. Cell Death Differ. 2010; 17(3):408-420.
15. Choi K, Ryu SW, Song S, Choi H, Kang SW and Choi C. Caspase-dependent generation of reactive oxygen species in human astrocytoma cells contributes to resistance to TRAIL-mediated apoptosis. Cell Death Differ. 2010; 17(5):833-845.
16. Rushmore TH and Pickett CB. Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene. Characterization of a xenobiotic-responsive element controlling inducible expression by phenolic antioxidants. J Biol Chem. 1990; 265(24):14648-14653.
17. Kletzien RF, Harris PK and Foellmi LA. Glucose-6-phosphate dehydrogenase: a “housekeeping” enzyme subject to tissue-specific regulation by hormones, nutrients, and oxidant stress. Faseb J. 1994; 8(2):174-181.
18. Prestera T, Zhang Y, Spencer SR, Wilczak CA and Talalay P. The electrophile counterattack response: protection against neoplasia and toxicity. Adv Enzyme Regul. 1993; 33:281-296.
19. Prestera T, Holtzclaw WD, Zhang Y and Talalay P. Chemical and molecular regulation of enzymes that detoxify carcinogens. Proc Natl Acad Sci U S A. 1993; 90(7):2965-2969.
20. Kensler TW, Davidson NE, Groopman JD, Roebuck BD, Prochaska HJ and Talalay P. Chemoprotection by inducers of electrophile detoxication enzymes. Basic Life Sci. 1993; 61:127-136.
21. Uejima Y, Fukuchi Y, Teramoto S, Tabata R and Orimo H. Age changes in visceral content of glutathione in the senescence accelerated mouse (SAM). Mech Ageing Dev. 1993; 67(1-2):129-139.
22. Nakata K, Kawase M, Ogino S, Kinoshita C, Murata H, Sakaue T, Ogata K and Ohmori S. Effects of age on levels of cysteine, glutathione and related enzyme activities in livers of mice and rats and an attempt to replenish hepatic glutathione level of mouse with cysteine derivatives. Mech Ageing Dev. 1996; 90(3):195-207.
23. Hagen TM, Vinarsky V, Wehr CM and Ames BN. (R)-alpha-lipoic acid reverses the age-associated increase in susceptibility of hepatocytes to tert-butylhydroperoxide both in vitro and in vivo. Antioxid Redox Signal. 2000; 2(3):473-483.
24. Laborde E. Glutathione transferases as mediators of signaling pathways involved in cell proliferation and cell death. Cell Death Differ. 2010; 17(9):1373-1380.
25. Sporn MB and Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012; 12(8):564-571.
26. Vanlangenakker N, Vanden Berghe T, Bogaert P, Laukens B, Zobel K, Deshayes K, Vucic D, Fulda S, Vandenabeele P and Bertrand MJ. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ. 2011; 18(4):656-665.
27. Lee JJ, Kim BC, Park MJ, Lee YS, Kim YN, Lee BL and Lee JS. PTEN status switches cell fate between premature senescence and apoptosis in glioma exposed to ionizing radiation. Cell Death Differ. 2011; 18(4):666-677.
28. Ling LU, Tan KB, Lin H and Chiu GN. The role of reactive oxygen species and autophagy in safingol-induced cell death. Cell Death Dis. 2011; 2:e129.
29. Zong D, Haag P, Yakymovych I, Lewensohn R and Viktorsson K. Chemosensitization by phenothiazines in human lung cancer cells: impaired resolution of gammaH2AX and increased oxidative stress elicit apoptosis associated with lysosomal expansion and intense vacuolation. Cell Death Dis. 2011; 2:e181.
30. Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD and Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999; 13(1):76-86.
31. Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K and Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004; 24(16):7130-7139.
32. Motohashi H and Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends in molecular medicine. 2004; 10(11):549-557.
33. Niture SK and Jaiswal AK. INrf2 (Keap1) targets Bcl-2 degradation and controls cellular apoptosis. Cell Death Differ. 2011; 18(3):439-451.
34. Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M and Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. 2002; 99(18):11908-11913.
35. Levonen AL, Landar A, Ramachandran A, Ceaser EK, Dickinson DA, Zanoni G, Morrow JD and Darley-Usmar VM. Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem J. 2004; 378(Pt 2):373-382.
36. Itoh K, Wakabayashi N, Katoh Y, Ishii T, O’Connor T and Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells. 2003; 8(4):379-391.
37. Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K and Yamamoto M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol. 2006; 26(1):221-229.
38. Kwak MK, Itoh K, Yamamoto M, Sutter TR and Kensler TW. Role of transcription factor Nrf2 in the induction of hepatic phase 2 and antioxidative enzymes in vivo by the cancer chemoprotective agent, 3H-1, 2-dimethiole-3-thione. Mol Med. 2001; 7(2):135-145.
39. Alam J, Stewart D, Touchard C, Boinapally S, Choi AM and Cook JL. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem. 1999; 274(37):26071-26078.
40. Venugopal R and Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci U S A. 1996; 93(25):14960-14965.
41. Wild AC, Moinova HR and Mulcahy RT. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. The Journal of biological chemistry. 1999; 274(47):33627-33636.
42. Moinova HR and Mulcahy RT. Up-regulation of the human gamma-glutamylcysteine synthetase regulatory subunit gene involves binding of Nrf-2 to an electrophile responsive element. Biochemical and biophysical research communications. 1999; 261(3):661-668.
43. Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M and Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochemical and biophysical research communications. 1997; 236(2):313-322.
44. Ray PD, Huang BW and Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012; 24(5):981-990.
45. Gullett NP, Ruhul Amin AR, Bayraktar S, Pezzuto JM, Shin DM, Khuri FR, Aggarwal BB, Surh YJ and Kucuk O. Cancer prevention with natural compounds. Semin Oncol. 2010; 37(3):258-281.
46. Talalay P, Batzinger RP, Benson AM, Bueding E and Cha YN. Biochemical studies on the mechanisms by which dietary antioxidants suppress mutagenic activity. Adv Enzyme Regul. 1978; 17:23-36.
47. Ramos-Gomez M, Dolan PM, Itoh K, Yamamoto M and Kensler TW. Interactive effects of nrf2 genotype and oltipraz on benzo[a]pyrene-DNA adducts and tumor yield in mice. Carcinogenesis. 2003; 24(3):461-467.
48. Fahey JW, Haristoy X, Dolan PM, Kensler TW, Scholtus I, Stephenson KK, Talalay P and Lozniewski A. Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzo[a]pyrene-induced stomach tumors. Proc Natl Acad Sci U S A. 2002; 99(11):7610-7615.
49. Conaway CC, Wang CX, Pittman B, Yang YM, Schwartz JE, Tian D, McIntee EJ, Hecht SS and Chung FL. Phenethyl isothiocyanate and sulforaphane and their N-acetylcysteine conjugates inhibit malignant progression of lung adenomas induced by tobacco carcinogens in A/J mice. Cancer Res. 2005; 65(18):8548-8557.
50. Dinkova-Kostova AT, Jenkins SN, Fahey JW, Ye L, Wehage SL, Liby KT, Stephenson KK, Wade KL and Talalay P. Protection against UV-light-induced skin carcinogenesis in SKH-1 high-risk mice by sulforaphane-containing broccoli sprout extracts. Cancer Lett. 2006; 240(2):243-252.
51. Gills JJ, Jeffery EH, Matusheski NV, Moon RC, Lantvit DD and Pezzuto JM. Sulforaphane prevents mouse skin tumorigenesis during the stage of promotion. Cancer Lett. 2006; 236(1):72-79.
52. Hu R, Khor TO, Shen G, Jeong WS, Hebbar V, Chen C, Xu C, Reddy B, Chada K and Kong AN. Cancer chemoprevention of intestinal polyposis in ApcMin/+ mice by sulforaphane, a natural product derived from cruciferous vegetable. Carcinogenesis. 2006; 27(10):2038-2046.
53. Shen G, Khor TO, Hu R, Yu S, Nair S, Ho CT, Reddy BS, Huang MT, Newmark HL and Kong AN. Chemoprevention of familial adenomatous polyposis by natural dietary compounds sulforaphane and dibenzoylmethane alone and in combination in ApcMin/+ mouse. Cancer Res. 2007; 67(20):9937-9944.
54. Jeong WS, Jun M and Kong AN. Nrf2: a potential molecular target for cancer chemoprevention by natural compounds. Antioxid Redox Signal. 2006; 8(1-2):99-106.
55. Baird L and Dinkova-Kostova AT. The cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol. 2011; 85(4):241-272.
56. Meixner A, Karreth F, Kenner L, Penninger JM and Wagner EF. Jun and JunD-dependent functions in cell proliferation and stress response. Cell Death Differ. 2010; 17(9):1409-1419.
57. Vurusaner B, Poli G and Basaga H. Tumor suppressor genes and ROS: complex networks of interactions. Free Radic Biol Med. 2012; 52(1):7-18.
58. Wang X. p53 regulation: teamwork between RING domains of Mdm2 and MdmX. Cell Cycle. 2011; 10(24):4225-4229.
59. Tsvetkov P, Reuven N and Shaul Y. Ubiquitin-independent p53 proteasomal degradation. Cell Death Differ. 2010; 17(1):103-108.
60. Marchenko ND, Hanel W, Li D, Becker K, Reich N and Moll UM. Stress-mediated nuclear stabilization of p53 is regulated by ubiquitination and importin-alpha3 binding. Cell Death Differ. 2010; 17(2):255-267.
61. Marine JC and Lozano G. Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ. 2010; 17(1):93-102.
62. Lee JT and Gu W. The multiple levels of regulation by p53 ubiquitination. Cell Death Differ. 2010; 17(1):86-92.
63. Tang Y, Chen Y, Jiang H and Nie D. Short-chain fatty acids induced autophagy serves as an adaptive strategy for retarding mitochondria-mediated apoptotic cell death. Cell Death Differ. 2011; 18(4):602-618.
64. Stindt MH, Carter S, Vigneron AM, Ryan KM and Vousden KH. MDM2 promotes SUMO-2/3 modification of p53 to modulate transcriptional activity. Cell Cycle. 2011; 10(18):3176-3188.
65. Dastidar SG, Raghunathan D, Nicholson J, Hupp TR, Lane DP and Verma CS. Chemical states of the N-terminal “lid” of MDM2 regulate p53 binding: simulations reveal complexities of modulation. Cell Cycle. 2011; 10(1):82-89.
66. Wu H and Leng RP. UBE4B, a ubiquitin chain assembly factor, is required for MDM2-mediated p53 polyubiquitination and degradation. Cell Cycle. 2011; 10(12):1912-1915.
67. Pani G and Galeotti T. Role of MnSOD and p66shc in mitochondrial response to p53. Antioxid Redox Signal. 2011; 15(6):1715-1727.
68. Budanov AV, Lee JH and Karin M. Stressin’ Sestrins take an aging fight. EMBO Mol Med. 2010; 2(10):388-400.
69. Budanov AV and Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008; 134(3):451-460.
70. Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE and Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005; 11(12):1306-1313.
71. Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, Lokshin M, Hosokawa H, Nakayama T, Suzuki Y, Sugano S, Sato E, Nagao T, Yokote K, Tatsuno I and Prives C. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U S A. 2010; 107(16):7461-7466.
72. Hu W, Zhang C, Wu R, Sun Y, Levine A and Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A. 2010; 107(16):7455-7460.
73. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S and Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007; 104(49):19345-19350.
74. Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, Li J, Peng B, Fleming JB, Wang H, Liu J, Lemischka IR, Hung MC and Chiao PJ. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012; 21(1):105-120.
75. DeBerardinis RJ, Lum JJ, Hatzivassiliou G and Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008; 7(1):11-20.
76. Reitzer LJ, Wice BM and Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem. 1979; 254(8):2669-2676.
77. Erickson JW and Cerione RA. Glutaminase: a hot spot for regulation of cancer cell metabolism? Oncotarget. 2010; 1(8):734-740.
78. Maddocks OD and Vousden KH. Metabolic regulation by p53. J Mol Med (Berl). 2011; 89(3):237-245.
79. Vousden KH. Alternative fuel--another role for p53 in the regulation of metabolism. Proc Natl Acad Sci U S A. 2010; 107(16):7117-7118.
80. Vousden KH and Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009; 137(3):413-431.
81. Feng Z and Levine AJ. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol. 2010; 20(7):427-434.
82. Marcel V, Dichtel-Danjoy ML, Sagne C, Hafsi H, Ma D, Ortiz-Cuaran S, Olivier M, Hall J, Mollereau B, Hainaut P and Bourdon JC. Biological functions of p53 isoforms through evolution: lessons from animal and cellular models. Cell Death Differ. 2011; 18(12):1815-1824.
83. Madan E, Gogna R, Bhatt M, Pati U, Kuppusamy P and Mahdi AA. Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor. Oncotarget. 2011; 2(12):948-957.
84. Melino G. p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ. 2011; 18(9):1487-1499.
85. Aoubala M, Murray-Zmijewski F, Khoury MP, Fernandes K, Perrier S, Bernard H, Prats AC, Lane DP and Bourdon JC. p53 directly transactivates Delta133p53alpha, regulating cell fate outcome in response to DNA damage. Cell Death Differ. 2011; 18(2):248-258.
86. Marine JC. p53 stabilization: the importance of nuclear import. Cell Death Differ. 2010; 17(2):191-192.
87. Jung JH, Bae S, Lee JY, Woo SR, Cha HJ, Yoon Y, Suh KS, Lee SJ, Park IC, Jin YW, Lee KH, An S and Lee JH. E3 ubiquitin ligase Hades negatively regulates the exonuclear function of p53. Cell Death Differ. 2011; 18(12):1865-1875.
88. Kruse JP and Gu W. Modes of p53 regulation. Cell. 2009; 137(4):609-622.
89. Lee MK, Tong WM, Wang ZQ and Sabapathy K. Serine 312 phosphorylation is dispensable for wild-type p53 functions in vivo. Cell Death Differ. 2011; 18(2):214-221.
90. MacLaine NJ and Hupp TR. How phosphorylation controls p53. Cell Cycle. 2011; 10(6):916-921.
91. Furlan A, Stagni V, Hussain A, Richelme S, Conti F, Prodosmo A, Destro A, Roncalli M, Barila D and Maina F. Abl interconnects oncogenic Met and p53 core pathways in cancer cells. Cell Death Differ. 2011; 18(10):1608-1616.
92. Ahmed A, Yang J, Maya-Mendoza A, Jackson DA and Ashcroft M. Pharmacological activation of a novel p53-dependent S-phase checkpoint involving CHK-1. Cell Death Dis. 2011; 2:e160.
93. Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R and Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007; 445(7128):661-665.
94. Wiman KG. Pharmacological reactivation of mutant p53: from protein structure to the cancer patient. Oncogene. 2010; 29(30):4245-4252.
95. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C and Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007; 445(7128):656-660.
96. Cheok CF, Kua N, Kaldis P and Lane DP. Combination of nutlin-3 and VX-680 selectively targets p53 mutant cells with reversible effects on cells expressing wild-type p53. Cell death and differentiation. 2010; 17(9):1486-1500.
97. Koster R, Timmer-Bosscha H, Bischoff R, Gietema JA and de Jong S. Disruption of the MDM2-p53 interaction strongly potentiates p53-dependent apoptosis in cisplatin-resistant human testicular carcinoma cells via the Fas/FasL pathway. Cell Death Dis. 2011; 2:e148.
98. Spinnler C, Hedstrom E, Li H, de Lange J, Nikulenkov F, Teunisse AF, Verlaan-de Vries M, Grinkevich V, Jochemsen AG and Selivanova G. Abrogation of Wip1 expression by RITA-activated p53 potentiates apoptosis induction via activation of ATM and inhibition of HdmX. Cell Death Differ. 2011; 18(11):1736-1745.
99. Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS and McMahon SB. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006; 24(6):841-851.
100. Tang Y, Luo J, Zhang W and Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006; 24(6):827-839.
101. Li X, Wu L, Corsa CA, Kunkel S and Dou Y. Two mammalian MOF complexes regulate transcription activation by distinct mechanisms. Mol Cell. 2009; 36(2):290-301.
102. Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R and Gu W. Tumor Suppression in the Absence of p53-Mediated Cell-Cycle Arrest, Apoptosis, and Senescence. Cell. 2012; 149(6):1269-1283.
103. Chao SK, Horwitz SB and McDaid HM. Insights into 4E-BP1 and p53 mediated regulation of accelerated cell senescence. Oncotarget. 2011; 2(1-2):89-98.
104. Kuribayashi K, Finnberg N, Jeffers JR, Zambetti GP and El-Deiry WS. The relative contribution of pro-apoptotic p53-target genes in the triggering of apoptosis following DNA damage in vitro and in vivo. Cell Cycle. 2011; 10(14):2380-2389.
105. Lane DP, Verma C and Fang CC. The p53 inducing drug dosage may determine quiescence or senescence. Aging (Albany NY). 2010; 2(11):748.
106. Galluzzi L, Kepp O and Kroemer G. TP53 and MTOR crosstalk to regulate cellular senescence. Aging (Albany NY). 2010; 2(9):535-537.
107. Darzynkiewicz Z. Another “Janus paradox” of p53: induction of cell senescence versus quiescence. Aging (Albany NY). 2010; 2(6):329-330.
108. Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E and Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006; 126(1):107-120.
109. Taguchi K, Motohashi H and Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011; 16(2):123-140.
110. Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, Asamura H, Yamamoto M and Hirohashi S. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci U S A. 2008; 105(36):13568-13573.
111. Kim YR, Oh JE, Kim MS, Kang MR, Park SW, Han JY, Eom HS, Yoo NJ and Lee SH. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J Pathol. 2010; 220(4):446-451.
112. Shibata T, Kokubu A, Gotoh M, Ojima H, Ohta T, Yamamoto M and Hirohashi S. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology. 2008; 135(4):1358-1368, 1368 e1351-1354.
113. Padmanabhan B, Tong KI, Ohta T, Nakamura Y, Scharlock M, Ohtsuji M, Kang MI, Kobayashi A, Yokoyama S and Yamamoto M. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell. 2006; 21(5):689-700.
114. Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, Herman JG, Baylin SB, Sidransky D, Gabrielson E, Brock MV and Biswal S. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006; 3(10):e420.
115. Ohta T, Iijima K, Miyamoto M, Nakahara I, Tanaka H, Ohtsuji M, Suzuki T, Kobayashi A, Yokota J, Sakiyama T, Shibata T, Yamamoto M and Hirohashi S. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008; 68(5):1303-1309.
116. Jiang T, Chen N, Zhao F, Wang XJ, Kong B, Zheng W and Zhang DD. High levels of Nrf2 determine chemoresistance in type II endometrial cancer. Cancer Res. 2010; 70(13):5486-5496.
117. Kim SK, Yang JW, Kim MR, Roh SH, Kim HG, Lee KY, Jeong HG and Kang KW. Increased expression of Nrf2/ARE-dependent anti-oxidant proteins in tamoxifen-resistant breast cancer cells. Free Radic Biol Med. 2008; 45(4):537-546.
118. Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, Chen W, Yi X, Zheng W, Wondrak GT, Wong PK and Zhang DD. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008; 29(6):1235-1243.
119. Homma S, Ishii Y, Morishima Y, Yamadori T, Matsuno Y, Haraguchi N, Kikuchi N, Satoh H, Sakamoto T, Hizawa N, Itoh K and Yamamoto M. Nrf2 enhances cell proliferation and resistance to anticancer drugs in human lung cancer. Clin Cancer Res. 2009; 15(10):3423-3432.
120. Shim GS, Manandhar S, Shin DH, Kim TH and Kwak MK. Acquisition of doxorubicin resistance in ovarian carcinoma cells accompanies activation of the NRF2 pathway. Free Radic Biol Med. 2009; 47(11):1619-1631.
121. Hong YB, Kang HJ, Kwon SY, Kim HJ, Kwon KY, Cho CH, Lee JM, Kallakury BV and Bae I. Nuclear factor (erythroid-derived 2)-like 2 regulates drug resistance in pancreatic cancer cells. Pancreas. 2010; 39(4):463-472.
122. Zhang P, Singh A, Yegnasubramanian S, Esopi D, Kombairaju P, Bodas M, Wu H, Bova SG and Biswal S. Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol Cancer Ther. 2010; 9(2):336-346.
123. DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, Scrimieri F, Winter JM, Hruban RH, Iacobuzio-Donahue C, Kern SE, Blair IA, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011; 475(7354):106-109.
124. Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C, Shyr C, Wakabayashi N, Kensler TW, Wasserman WW and Biswal S. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010; 38(17):5718-5734.
125. Munoz-Pinedo C, El Mjiyad N and Ricci JE. Cancer metabolism: current perspectives and future directions. Cell Death Dis. 2012; 3:e248.
126. Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M and Motohashi H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012; 22(1):66-79.
127. Engelman JA, Luo J and Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006; 7(8):606-619.
128. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Montalto G, Cervello M, Nicoletti F, Fagone P, Malaponte G, Mazzarino MC, Candido S, Libra M, Basecke J, Mijatovic S, Maksimovic-Ivanic D, Milella M, et al. Mutations and Deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Cascades. Oncotarget. 2012; 3(9):954-987.
129. Collavin L, Lunardi A and Del Sal G. p53-family proteins and their regulators: hubs and spokes in tumor suppression. Cell Death Differ. 2010; 17(6):901-911.
130. Lu WJ, Lee NP, Kaul SC, Lan F, Poon RT, Wadhwa R and Luk JM. Mortalin-p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy. Cell Death Differ. 2011; 18(6):1046-1056.
131. Kogan-Sakin I, Tabach Y, Buganim Y, Molchadsky A, Solomon H, Madar S, Kamer I, Stambolsky P, Shelly A, Goldfinger N, Valsesia-Wittmann S, Puisieux A, Zundelevich A, Gal-Yam EN, Avivi C, Barshack I, et al. Mutant p53(R175H) upregulates Twist1 expression and promotes epithelial-mesenchymal transition in immortalized prostate cells. Cell Death Differ. 2011; 18(2):271-281.
132. Botcheva K, McCorkle SR, McCombie WR, Dunn JJ and Anderson CW. Distinct p53 genomic binding patterns in normal and cancer-derived human cells. Cell Cycle. 2011; 10(24):4237-4249.
133. Afanasyeva EA, Mestdagh P, Kumps C, Vandesompele J, Ehemann V, Theissen J, Fischer M, Zapatka M, Brors B, Savelyeva L, Sagulenko V, Speleman F, Schwab M and Westermann F. MicroRNA miR-885-5p targets CDK2 and MCM5, activates p53 and inhibits proliferation and survival. Cell Death Differ. 2011; 18(6):974-984.
134. Barlev NA, Sayan BS, Candi E and Okorokov AL. The microRNA and p53 families join forces against cancer. Cell Death Differ. 2010; 17(2):373-375.
135. Spizzo R, Nicoloso MS, Lupini L, Lu Y, Fogarty J, Rossi S, Zagatti B, Fabbri M, Veronese A, Liu X, Davuluri R, Croce CM, Mills G, Negrini M and Calin GA. miR-145 participates with TP53 in a death-promoting regulatory loop and targets estrogen receptor-alpha in human breast cancer cells. Cell Death Differ. 2010; 17(2):246-254.
136. Goodman RH and Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000; 14(13):1553-1577.
137. Christoffersen NR, Shalgi R, Frankel LB, Leucci E, Lees M, Klausen M, Pilpel Y, Nielsen FC, Oren M and Lund AH. p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ. 2010; 17(2):236-245.
138. Bertheau P, Turpin E, Rickman DS, Espie M, de Reynies A, Feugeas JP, Plassa LF, Soliman H, Varna M, de Roquancourt A, Lehmann-Che J, Beuzard Y, Marty M, Misset JL, Janin A and de The H. Exquisite sensitivity of TP53 mutant and basal breast cancers to a dose-dense epirubicin-cyclophosphamide regimen. PLoS Med. 2007; 4(3):e90.
139. Bertheau P, Espie M, Turpin E, Lehmann J, Plassa LF, Varna M, Janin A and de The H. TP53 status and response to chemotherapy in breast cancer. Pathobiology. 2008; 75(2):132-139.
140. McGill G and Fisher DE. p53 and cancer therapy: a double-edged sword. J Clin Invest. 1999; 104(3):223-225.
141. Jackson JG, Pant V, Li Q, Chang LL, Quintas-Cardama A, Garza D, Tavana O, Yang P, Manshouri T, Li Y, El-Naggar AK and Lozano G. p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell. 2012; 21(6):793-806.
142. Sinthupibulyakit C, Grimes KR, Domann FE, Xu Y, Fang F, Ittarat W, St Clair DK and St Clair W. p53 is an important factor for the radiosensitization effect of 2-deoxy-D-glucose. Int J Oncol. 2009; 35(3):609-615.
143. Sinthupibulyakit C, Ittarat W, St Clair WH and St Clair DK. p53 Protects lung cancer cells against metabolic stress. Int J Oncol. 2010; 37(6):1575-1581.
144. el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW and Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993; 75(4):817-825.
145. Pang LY, Scott M, Hayward RL, Mohammed H, Whitelaw CB, Smith GC and Hupp TR. p21(WAF1) is component of a positive feedback loop that maintains the p53 transcriptional program. Cell Cycle. 2011; 10(6):932-950.
146. Chen W, Sun Z, Wang XJ, Jiang T, Huang Z, Fang D and Zhang DD. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol Cell. 2009; 34(6):663-673.
147. Asher G, Tsvetkov P, Kahana C and Shaul Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005; 19(3):316-321.
148. Asher G, Lotem J, Cohen B, Sachs L and Shaul Y. Regulation of p53 stability and p53-dependent apoptosis by NADH quinone oxidoreductase 1. Proc Natl Acad Sci U S A. 2001; 98(3):1188-1193.
149. Asher G, Lotem J, Sachs L, Kahana C and Shaul Y. Mdm-2 and ubiquitin-independent p53 proteasomal degradation regulated by NQO1. Proc Natl Acad Sci U S A. 2002; 99(20):13125-13130.
150. Kalo E, Kogan-Sakin I, Solomon H, Bar-Nathan E, Shay M, Shetzer Y, Dekel E, Goldfinger N, Buganim Y, Stambolsky P, Goldstein I, Madar S and Rotter V. Mutant p53R273H attenuates the expression of phase 2 detoxifying enzymes and promotes the survival of cells with high ROS levels. J Cell Sci. 2012.
151. You A, Nam CW, Wakabayashi N, Yamamoto M, Kensler TW and Kwak MK. Transcription factor Nrf2 maintains the basal expression of Mdm2: An implication of the regulation of p53 signaling by Nrf2. Arch Biochem Biophys. 2011; 507(2):356-364.
152. Faraonio R, Vergara P, Di Marzo D, Pierantoni MG, Napolitano M, Russo T and Cimino F. p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J Biol Chem. 2006; 281(52):39776-39784.
153. Li D, Marchenko ND and Moll UM. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011; 18(12):1904-1913.
154. Vaseva AV, Yallowitz AR, Marchenko ND, Xu S and Moll UM. Blockade of Hsp90 by 17AAG antagonizes MDMX and synergizes with Nutlin to induce p53-mediated apoptosis in solid tumors. Cell Death Dis. 2011; 2:e156.
155. Puca R, Nardinocchi L, Porru M, Simon AJ, Rechavi G, Leonetti C, Givol D and D’Orazi G. Restoring p53 active conformation by zinc increases the response of mutant p53 tumor cells to anticancer drugs. Cell Cycle. 2011; 10(10):1679-1689.
156. Li H, Lakshmikanth T, Garofalo C, Enge M, Spinnler C, Anichini A, Szekely L, Karre K, Carbone E and Selivanova G. Pharmacological activation of p53 triggers anticancer innate immune response through induction of ULBP2. Cell Cycle. 2011; 10(19):3346-3358.
157. Vance S, Liu E, Zhao L, Parsels JD, Parsels LA, Brown JL, Maybaum J, Lawrence TS and Morgan MA. Selective radiosensitization of p53 mutant pancreatic cancer cells by combined inhibition of Chk1 and PARP1. Cell Cycle. 2011; 10(24):4321-4329.
158. Del Bufalo D, Bagnato A, Fusco A and Milella M. Lost in translation: bridging the gap between cancer research and effective therapies. Cell Death Differ. 2011; 18(6):1082-1084.
159. Brennan R, Federico S and Dyer MA. The war on cancer: have we won the battle but lost the war? Oncotarget. 2010; 1(2):77-83.
160. Demidenko ZN and McCubrey JA. Recent progress in targeting cancer. Aging (Albany NY). 2011; 3(12):1154-1162.
161. van Leeuwen IM and Lain S. Pharmacological manipulation of the cell cycle and metabolism to protect normal tissues against conventional anticancer drugs. Oncotarget. 2011; 2(4):274-276.
162. Rao B, van Leeuwen IM, Higgins M, Campbel J, Thompson AM, Lane DP and Lain S. Evaluation of an Actinomycin D/VX-680 aurora kinase inhibitor combination in p53-based cyclotherapy. Oncotarget. 2010; 1(7):639-650.
163. Azmi AS, Banerjee S, Ali S, Wang Z, Bao B, Beck FW, Maitah M, Choi M, Shields TF, Philip PA, Sarkar FH and Mohammad RM. Network modeling of MDM2 inhibitor-oxaliplatin combination reveals biological synergy in wt-p53 solid tumors. Oncotarget. 2011; 2(5):378-392.
164. Jung K and Reszka R. Mitochondria as subcellular targets for clinically useful anthracyclines. Adv Drug Deliv Rev. 2001; 49(1-2):87-105.
165. Robinson MM, McBryant SJ, Tsukamoto T, Rojas C, Ferraris DV, Hamilton SK, Hansen JC and Curthoys NP. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem J. 2007; 406(3):407-414.
166. Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, Zimmerman LJ, Liebler DC, Slebos RJ, Lorkiewicz PK, Higashi RM, Fan TW, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012; 15(1):110-121.
167. Thangavelu K, Pan CQ, Karlberg T, Balaji G, Uttamchandani M, Suresh V, Schuler H, Low BC and Sivaraman J. Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. Proc Natl Acad Sci U S A. 2012; 109(20):7705-7710.
168. Bailey HH. L-S,R-buthionine sulfoximine: historical development and clinical issues. Chem Biol Interact. 1998; 111-112:239-254.
169. Raj A, Rifkin SA, Andersen E and van Oudenaarden A. Variability in gene expression underlies incomplete penetrance. Nature. 2010; 463(7283):913-918.
170. Adams DJ, Dai M, Pellegrino G, Wagner BK, Stern AM, Shamji AF and Schreiber SL. Synthesis, cellular evaluation, and mechanism of action of piperlongumine analogs. Proc Natl Acad Sci U S A. 2012; 109(38):15115-15120.
171. Ren D, Villeneuve NF, Jiang T, Wu T, Lau A, Toppin HA and Zhang DD. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc Natl Acad Sci U S A. 2011; 108(4):1433-1438.
172. Michaelis M, Rothweiler F, Barth S, Cinatl J, van Rikxoort M, Loschmann N, Voges Y, Breitling R, von Deimling A, Rodel F, Weber K, Fehse B, Mack E, Stiewe T, Doerr HW, Speidel D, et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis. 2011; 2:e243.
173. Saxena M, Stephens MA, Pathak H and Rangarajan A. Transcription factors that mediate epithelial-mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis. 2011; 2:e179.
174. Bhatnagar N, Li X, Padi SK, Zhang Q, Tang MS and Guo B. Downregulation of miR-205 and miR-31 confers resistance to chemotherapy-induced apoptosis in prostate cancer cells. Cell Death Dis. 2010; 1:e105.