
INTRODUCTION
Many web sites of hospital radiology or radiotherapy departments state that nuclear DNA (nucDNA) is the most important radiosensitive target within cells, as it is involved in cell death, the initiation and therapy of cancer, and other biological effects. Supportive of the nucleus-centric view is the widely accepted premise that radiation-induced reproductive death of mammalian cells is principally due to lesions in nucDNA, particularly double-strand breaks (DSBs) [1]. These lesions are thought to lead to enhanced cell death in the presence of oxygen — the oxygen effect [2].
The best known explanation of the oxygen effect is the oxygen fixation hypothesis developed in the 1950s, which posited that radiation-induced non-restorable nucDNA lesions are lethal to cells in the presence of diatomic oxygen (O2) [3]. In the same decade, doubts arose that radiation damage was primarily genetic, based on studies that included, for example, the X-ray irradiation of giant amoebae, which showed that cell division is enhanced if these eukaryotes are fused with unirradiated mitochondria rather than nuclei [4]. Goldfeder [5] then proposed that mitochondria played an important role in determining cell radiosensitivity; this was based on the observation that cells highly endowed with mitochondria can function even if irradiation eliminates a substantial fraction of the mitochondria. Doubts have since emerged concerning the oxygen fixation hypothesis [3], not the least because the hypothesis fails to take into account nitric oxide (NO) as a radiosensitizer with similar effects as O2 (see Discussion).
Beyond their widely recognized roles in energy metabolism, mitochondria play important roles in calcium buffering, cell signaling and cell death. Normally, during glucose oxidation in the cytosol, electrons/reducing equivalents are released and reduced nicotinamide adenine dinucleotide, ATP and protons (H+) are produced. In anaerobic glycolysis, each glucose molecule results in the net production of two adenosine triphosphate (ATP) molecules. In aerobic glycolysis, pyruvate enters mitochondria to support Krebs cycle activity and, subsequently, far greater ATP production by the oxidative phosphorylation system is achieved (Fig. 1). This system comprises the ATP synthase, the phosphate carrier, the adenine nucleotide translocator and the electron transport chain, in which electrons provide sufficient free energy to pump protons out of the mitochondrial matrix and into the mitochondrial intermembrane space. This generates a membrane potential ∆ψ and a proton gradient ∆pH, which together are referred to as proton motive force, which in turn is used to drive the activity of ATP synthase. Oxygen is consumed when the electron transport chain is active; as electrons are transferred to oxygen by Complex IV (cytochrome c oxidase) and water is produced.
Ionizing radiation produces radicals, such as the hydrogen radical (H•) and hydroxyl radical (OH•), from the radiolysis of water as well as reactive oxygen species (ROS) from the irradiation of oxygenated aqueous media. Nevertheless, most ROS in cells, including ROS from low-dose radiation, are thought to originate from mitochondria, due to leakage of electrons from the electron transport chain. It has been estimated that 0.2%–2% of the O2 consumed by cells incubated at air saturation (21% O2, 160 mmHg, corresponding to ~203 µmol/L O2 solubility in cells at 37°C) form superoxide or other forms of ROS in mitochondria [6, 7]. O2 is first reduced to the superoxide anion (•O2-), two of which can then be dismutated by the enzyme superoxide dismutase to hydrogen peroxide (H2O2). If endogenous ROS production exceeds antioxidative mechanisms, oxidative damage to macromolecules ensues.
Although there are other sources of ROS, e.g. cytoplasmic NADPH oxidase (NOX), unfolding protein response and peroxisomes, mitochondria not only produce most of a cell’s ROS, but they may also be more susceptible than the nucleus to their damaging effects. One reason for this susceptibility is that is that the half-lives of ROS and reactive nitrogen species (RNS) in cells are generally very short (≤1 µs), severely limiting their diffusion distances. For example, OH• is most reactive and migrates short distances (1 nm), •O2- has moderate range, and H2O2 can migrate over relatively long distances (1 µm or more). Most mammalian cells, typically 10 – 20 µm in diameter, contain hundreds of mitochondria <1–10 µm long, each with 2–10 copies of mitochondrial DNA (mtDNA). Normal oxidative lesions of DNA, such as the adduct 8-hydroxydeoxyguanosine, have steady state levels 16-fold higher in mtDNA than in nucDNA of rat liver [8]. In addition, mitochondria lack histones, which protect nucDNA from damage, and they have limited DNA repair mechanisms compared with the nucleus [9]. In slowly proliferating tissues, mitochondria have surprisingly rapid turnover rates, characterized by half-lives as short as a few days; and turnover can be accelerated by ionizing radiation [10].
Ionizing radiation causes a transient increase in ROS, from the reduction of oxygen to form •O2- and H2O2, and in RNS, derived from NO. Ward et al [11] noted that low-dose, low-linear energy transfer (LET) radiation produces primary ionization events (electron tracks) and secondary reactive products that are insignificant compared with those produced by endogenous ROS (likely from mitochondria — “mtROS”). A very low-dose gamma-radiation exposure of 1 mGy leads to nucDNA DSBs in only ~3% of cells, and this effect is proportional to dose [12]. However, cells have highly efficient repair mechanisms, including non-homologous end joining in the G1 phase of the cell cycle and homologous recombination during the S and G2 phases of cell replication. Consequently, cell killing being a stochastic effect, the probability of one DSB (or a pair of breaks) causing nucDNA DSB-related cell killing is unlikely at low doses, as 20–80 DSBs are needed to produce cell death [13].
Here, we quantitatively analyze the oxygen effect to explain the variation in ionizing radiation’s therapeutic or detrimental properties with oxygen level. In the process, we examine and quantify the biological mechanisms underlying the oxygen enhancement ratio (OER): the ratio of a chosen measure of radiation-induced damage (e.g., measures including DSBs, chromosomal aberrations and subsequent apoptosis, necrosis and mitotic cell death) when O2 is present to that when it is not (Fig. 2A). We demonstrate that damage by both low-LET (e.g., β-, X- or γ-ray exposures) and high-LET (e.g., α-particle) radiations to cells in well-oxygenated tissues is related to the mitochondrial O2 consumption and production of ROS, and that repair in hypoxic tissues is limited by deficiencies in ATP. In addition, we investigate the hypothesis that the radiation-induced adaptive response of normal cells involves uncoupling of oxidative phosphorylation providing a short-term protective effect. In view of the augmented uncoupling exhibited by some cancers, we speculate that this constitutes a maladaptive response to radiotherapy treatments. Finally, we contend that these analyses, supported by other published studies, provide evidence that, especially for low-LET radiations, the primary radiation targets in oxygenated tissues are mitochondria, which produce ROS that in turn indirectly targets nucDNA.
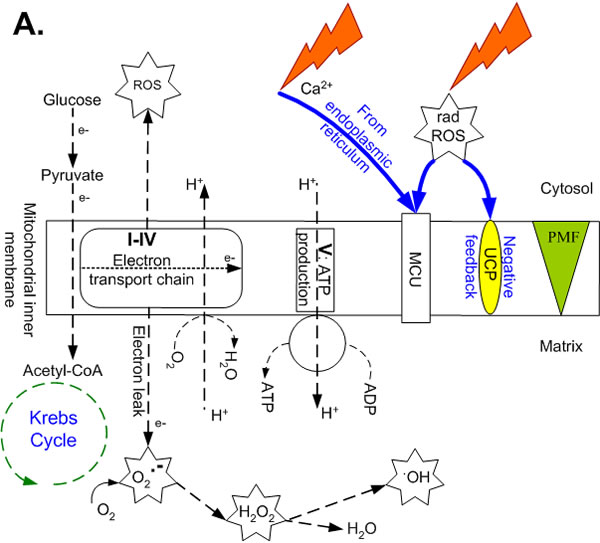
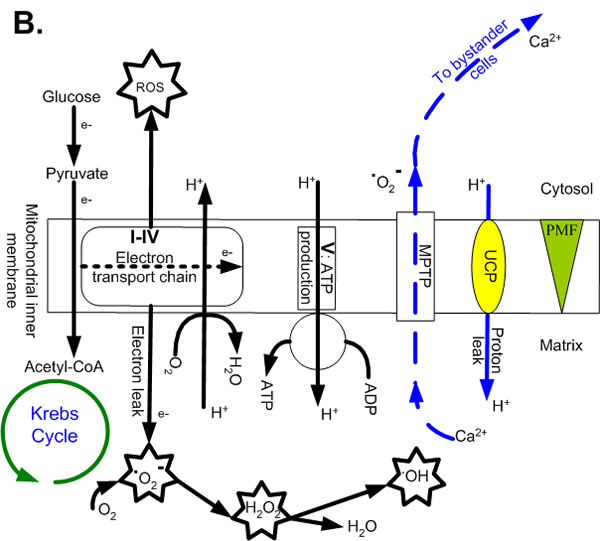
Figure 1: Ionic pathways across the mitochondrial inner membrane just before and after irradiation. Pathways and components include the electron transport chain (complexes I-IV), ATP synthase (complex V), uncoupling proteins (UCP), mitochondrial Ca2+ uniporter (MCU), mitochondrial permeability transition pore (MPTP) and proton motive force (PMF, made up of membrane potential and the proton gradient). A. The moment just before ionizing radiation-induced ROS (radROS) and Ca2+ arrive at and cross the membranes (blue pathways), showing normal, coupled respiration at moderate levels of O2 uptake and ATP synthesis, accompanied by moderate electron leak and mtROS. B. The moment just after radROS and Ca2+ (at physiological levels) stimulate transient depolarization, ATP synthesis and O2 uptake, leading to electron emission from the electron transport chain, mtROS surge on both sides of membrane and UCP partial uncoupling (blue pathway). Overload Ca2+ influx and excessive mtROS cause Ca2+ to leave the mitochondrion via the MPTP and lead to apoptosis (blue dashed pathway).
RESULTS
OER variation with O2 concentration
Our explanation of the oxygen effect is based on the Howard-Flanders and Alper formula [14] for OER, which can be rearranged as two components (Fig. 2A) for nominal cell parameter values (see Materials and Methods).
![]() 1)
1)
where OER of radiation R, varies with O2 % concentration (![]() ), maximum OER value (OERRmax), and half-maximal O2 concentration (
), maximum OER value (OERRmax), and half-maximal O2 concentration (![]() ).
).
We define the mtROS component of the effect as radiation-induced oxidative damage caused by endogenous mtROS that varies in accordance with hyperbolic increase in O2 consumption or flux by mitochondria. The mtROS component shows saturation characteristics similar to those of OER curves when O2 levels rise from anoxia (0% O2) to severe hypoxia (≤0.1%), hypoxia (≤1%) to normoxia (21% O2) [15, 16]. Considering only low-LET exposures in this section, the mtROS component dominates in hypoxia to normoxia (Table 1). When there is greater O2 available, the disadvantage conferred by more mtROS is countered by the advantage of greater ATP production capacity.
We define the low-ATP component as inversely proportional to the O2 content (%) and as equal to unity in anoxic conditions, when mtROS levels are zero (Table 1), primarily because of the lack of sufficient O2-derived ATP for mitotic DNA repair. The low-ATP component drops rapidly with rising O2, making up around a quarter of OER in hypoxic conditions when mtROS increases with O2 content.
Radiation damage can be classified as indirect, mainly due to biologic molecules inactivated by radiolysis products of water, and as direct, due to radiation-induced breakage of bonds in macromolecules such as DNA. In the presence of O2, radiation-induced mtROS species include highly reactive OH• [17]. Our interpretation of the oxygen effect is that radiolysis products induce mtROS, and this endogenous ROS production by mitochondria is the principal indirect cause of radiation damage observed in nucDNA (e.g., 8-oxo-deoxyguanosine). This explanation aligns with a finding by Chapman et al [18], who employed the OH• scavenger dimethyl sulfoxide and showed that the indirect action of OH• generated by X-rays accounted for ~30% of the lethal effect on a Chinese hamster fibroblast cell line during anoxia/severe hypoxia (in N2), and 62% of cell death under air-saturated conditions.
Two mtROS trends with O2 consumption are considered, with the total OER unchanged (Table 1): first, radiation-induced “mtROS proportional to O2 flux” (Fig. 2A), and second, “mtROS decreasing with O2 flux” (Fig. 2B). The second mtROS trend was evaluated following reports of greater mtROS generation as a percentage of electron flux through the respiration chain in hypoxic conditions [15]. In Fig. 2B, fractional mtROS decreases with O2 flux partially based on the data of Hoffman and Brookes [16]. We employ half the mtROS trend modified to produce the typical hyperbolic function for mtROS dependence on O2 level. In Table 1 the mtROS:O2-flux ratio is designated as unity at the 21% O2 level, for mtROS decreasing with O2 flux: accordingly, the proportion of mtROS produced under severe hypoxia is virtually twice that at 21% O2. Comparing mtROS proportional to O2 flux (Fig. 2A) with mtROS decreasing with O2 flux (Fig. 2B), the latter exhibits a significant increase in the mtROS component, especially for low-LET exposures in hypoxic conditions.
Table 1: Influence of low- and high-LET radiation on OER components and mtROS:O2-flux ratio*
Oxygen level (%) |
MtROS proportional to O2 flux |
MtROS decreasing with O2 flux |
||||||||
mtROS: O2-flux ratio |
Low-LET |
High-LET |
mtROS: O2-flux ratio |
Low-LET |
High-LET |
|||||
MtROS (%) |
Low-ATP (%) |
MtROS (%) |
Low-ATP (%) |
MtROS (%) |
Low-ATP (%) |
MtROS (%) |
Low-ATP (%) |
|||
0 |
1.00 |
0 |
100 |
0 |
100 |
1.99 |
0 |
100 |
0 |
100 |
0.1 |
1.00 |
33 |
67 |
15 |
85 |
1.61 |
54 |
46 |
25 |
75 |
1.0 |
1.00 |
83 |
17 |
65 |
35 |
1.16 |
96 |
4 |
75 |
25 |
10 |
1.00 |
98 |
2 |
95 |
5 |
1.01 |
99 |
1 |
96 |
4 |
21 |
1.00 |
99 |
1 |
97 |
3 |
1.00 |
99 |
1 |
97 |
3 |
*Designated as unity at 21% O2 level for mtROS decreasing with O2 flux.
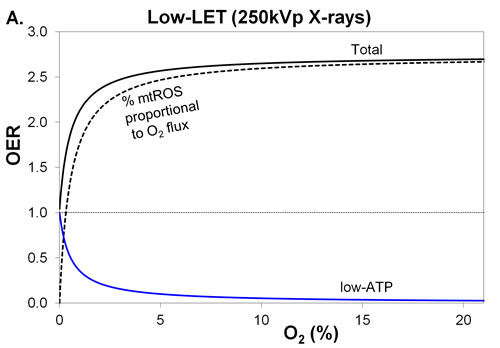
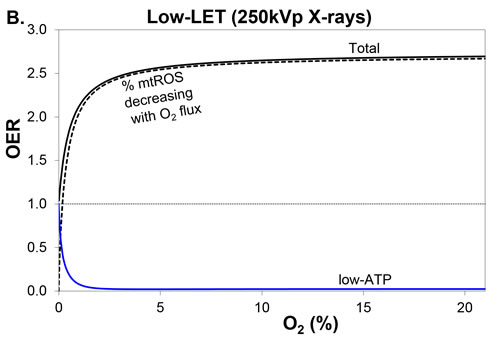
Figure 2: Variation in total OER and its components, with percentage O2 in air, for low-LET 250 kVp X-rays. The blue line and black-dashed line are the 2 OER components, low-ATP and mtROS, comprising the black line for total OER. MtROS trends shown are A. radiation-induced mtROS proportional to O2 flux and B. fractional mtROS decreasing with O2 flux (total OER is unchanged), where mtROS has a more pronounced effect than A. particularly in hypoxic conditions.
Oxygen effect and LET
A high-LET exposure, compared with the same low-LET absorbed dose measured in grays (Gy), has a greater potential to cause tissue damage. The differing characteristics of the oxygen effect for low- and high-LET exposures [19] are assessed by comparing 250 kVp X-rays (LET, 1.3 keV/µm) and 2.5 MeV (LET, 166 keV/µm) α-particles with maximum OER values of 2.74 and 1.00, respectively, and half-maximal O2 concentration at 0.55% O2 for both. High-LET α-particle tracks produce greater clusters of chemical spurs and radiolysis events in water that lead to the formation of highly oxidising H2O2 tracks [20]. With increasing LET, the short-range OH• diminish, accompanied by enhanced numbers of long-range •O2- and H2O2 [17]. In the presence of O2, while low-LET exposures produce OH• that kill about 60%–90% of mammalian cells, for very high-LET exposures only 25%–30% indirectly succumb [21, 22]. For both low- and high-LET, at 1%–21% O2 levels, the mtROS component dominates the low-ATP component (Figs. 2, 3).
Unlike mtROS damage, low-ATP damage is independent of LET. For a particular O2 level, the mtROS:low-ATP ratio decreases with higher LET. Consequently, at 1%–21% O2 levels, the low-ATP component, due to deficiencies in O2-related ATP is two-fold or more for high LET than for lower LET (Table 1). Subsequently, the low-ATP component is greater for high-LET exposures, which reflects the inability to repair more complex DNA damage than that sustained from a low-LET exposure (Table 1).
Radiotherapy fraction-induced mtROS followed by uncoupling of respiration
The initial contribution to the oxygen effect is from the radiolysis of water, which produces free radicals that stimulate the endoplasmic reticulum to release calcium ions, Ca2+. It is our hypothesis that this physiological Ca2+ (relatively low Ca2+) in turn, enters the mitochondrial matrix and stimulates ATP synthesis [23] as well as the production of mtROS; the latter then triggers a transient, partial uncoupling of oxidative phosphorylation via uncoupling protein 2 (UCP2) [24-26]. To explore the importance of the mitochondrial-centric oxygen effect, we study the biological mechanisms involved in an acute 2 Gy, low-LET exposure under aerobic conditions: allied to a typical curative dose of epithelial tumors being 60–80 Gy in fractions of ~2 Gy. Specifically, we examine post-irradiation parameters for mitochondria: first, the temporal behavior expected between radiotherapy fractions, based on irradiation studies; and, second, the lower effectiveness of radiotherapy on hypoxic tissue, based on uncoupling studies.
These descriptions are more illustrative than quantitative, since the selected and unmatched parameters are derived from published reports of experiments with exposures of different doses in cultured cells (malignant and non-malignant) and in isolated mitochondria of different organs and species (Table 2). We assume a high- dose (2 Gy), low-LET exposure of non-malignant cells with coupled respiration.
Post irradiation-based studies indicate that a high dose of 2 Gy causes an immediate transient increase of radiolysis-induced ROS/RNS levels in a cell that for minutes, is at a ~3.2-fold higher rate than background levels (Table 2) [27]. This produces a surge in cytosolic Ca2+, released by the endoplasmic reticulum (Fig. 1A), and a subsequent uptake of Ca2+ into the mitochondrial matrix via the mitochondrial Ca2+ uniporter (Fig. 1B). The radiation-induced calcium influx at normal physiological levels into mitochondria also results in an immediate increase in mitochondrial electron transport activity.
Within minutes after an acute exposure, depolarization of the membrane potential occurs (∆ψ; ~ -5 to -15% change), which can last minutes [27], hours [28], or more [10]. Radiation, and similarly increased mtROS, has been shown to trigger UCP2 activity (proton leak uncoupling) and UCP2 expression increases then plateaus beginning 1 hour after exposure (Table 2) [24, 29]. By depressing ∆ψ, UCP2 acts in a negative feedback loop to decrease mtROS and ATP production for hours or more [25, 26, 30]. This small decrease in ∆ψ has been observed to revert to nearly normal levels within 12 hours if there is some cell recovery [28], but it can persist if accompanied by genomic instability [10]. Importantly though, an overload of Ca2+ influx and excessive mtROS causes mitochondrial permeability transition pore activation and apoptosis (Fig. 1B).
Table 2: Estimated percentage difference in mitochondrial-related parameter values before and after a 2 Gy, low-LET exposure under aerobic conditions*
Parameters |
Time after exposure |
Change post- exposure (%) |
Cell and mitochondrial types and reference values |
ROS/RNS vs. dose |
0-3 min |
+220% ROS/RNS |
3 Gy 90Sr β-dose leads to 4.25-fold ↑ ROS gradient – human squamous carcinoma [27] |
∆ψ vs. dose |
10 min–1 h |
-15% ∆ψ |
4 Gy γ-rays leads to 30% ↓ ∆ψ – human fibroblasts [28]: similarly, depolarization occurs 3-5 mins [27] |
UCP2 vs. time |
1 h |
+16% UCP2 |
5 Gy leads to 40% ↑ UCP2 – apoptosis-sensitive murine B cell lymphoma [29] |
∆ψ vs. dose |
12 h |
-7% ∆ψ |
4 Gy γ-rays leads to 14% ↓ ∆ψ – human fibroblasts [28] |
∆ψ vs. dose |
Chronic |
-5% ∆ψ |
10 Gy X-rays leads to 26% ↓ ∆ψ – 3 hamster fibroblast clones [10] |
O2 consumption vs. dose |
Chronic |
+30% O2 consumption |
10 Gy X-rays leads to 2.5-fold ↑ O2 consumption – 3 hamster fibroblast clones [10] |
*Experiments utilized cultured cells or isolated mitochondria. Average percentage increase and decrease of cell types indicated by %↑ and %↓, respectively.
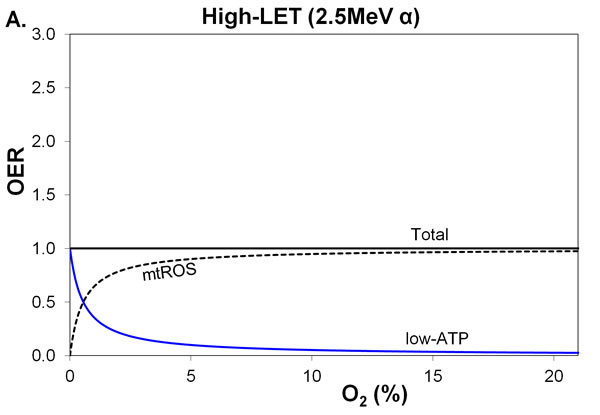
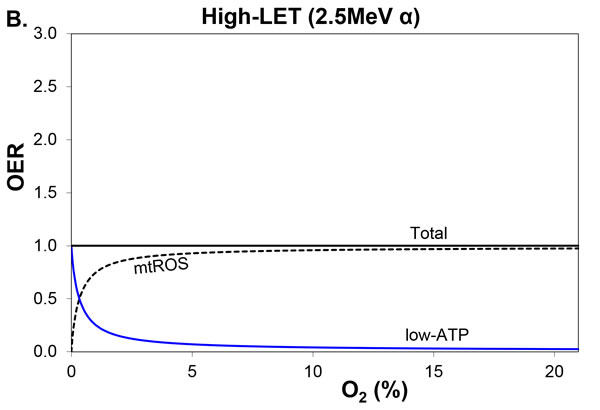
Figure 3: Variation in total OER and its components, with percentage O2 in air, for high-LET 2.5 MeV α-particles. The blue line and black-dashed line are the 2 OER components, low-ATP and mtROS, comprising the black line for unchanged total OER. MtROS trends appear virtually the same for A. radiation-induced mtROS proportional to O2 flux and B. fractional mtROS decreasing with O2 flux (total OER is unchanged).
Studies of mitochondrial uncoupling in cells exposed to different O2 levels, such as those by Gnaiger et al [15, 31], demonstrate changes in O2 consumption, and mtROS production that are dependent on O2 levels arising from uncoupling of oxidation phosphorylation in surviving cells [24]. The reciprocal of the half-maximal O2 concentration is an important parameter, as it is an index for the O2 affinity of the respiratory system. Before radiation exposure, it is assumed that the coupled half-maximal O2 value is 0.55% [32] and the maximum O2 flow is 40 pmol/s/106 cells. On the basis that a 2 Gy exposure causes total uncoupling, with no change in the respiratory capacity, the half-maximal O2 concentration drops to 0.20%, based on data for human endothelial cells [31]. Therefore, in addition to a post-irradiation increase in O2 consumption of cells in aerobic conditions [10], compared with coupled respiration, uncoupling causes a more rapid climb in the O2 consumption profile (hence mtROS emission) for low O2 levels. Specifically, at a low O2 level of 0.1% O2, radiation-induced uncoupling nearly doubles O2 consumption, hence the mtROS component (modelled: fractional mtROS decreases with O2 flux, Table 1), which may explain the genetic instability and poorer outcomes sometimes observed after radiotherapy of hypoxic tumors [33].
A known consequence of the uncoupling of oxidative phosphorylation is lower mtROS emissions observed in cells expressing UCP2 [24, 34, 35]. After exposure, this may be accompanied by an increased affinity for O2 that, on the one hand, produces more mtROS [10], (especially in hypoxic conditions) [31], yet, on the other hand, provides a beneficial radiotherapeutic environment for well-oxygenated non-malignant cells to repair the sub-lethal damage that usually occurs within 24 hours of exposure. Similarly, uncoupling in malignantly transformed cells has been observed for moderate doses (0.4 and 0.8 Gy) and high doses (9 and 10 Gy) of X- and α-radiation, evidenced by modified oxidation phosphorylation activity, cytochrome c oxidase and O2 consumption per mtDNA copy number [36, 37]. Likewise, Voehringer et al. [29] showed radiation-induced UCP2 activation lead to mitochondrial uncoupling and even cell death, in an irradiated murine B cell lymphoma model, although the hypothesized radiation-induced loss of membrane potential was observed by others [27, 28]. Even though radiation-induced uncoupling has been demonstrated under particular cellular conditions [38], there is plainly a need to experimentally verify in the one study that radiation exposures result in UCP2 activation plus a loss of membrane potential. Albeit, even allowing for the diverse and somewhat incompatible nature of the studies reported, this analysis suggests that uncoupled respiration is an important mechanism involved in sub-lethal and lethal radiation exposures of both malignant and non-malignant cells.
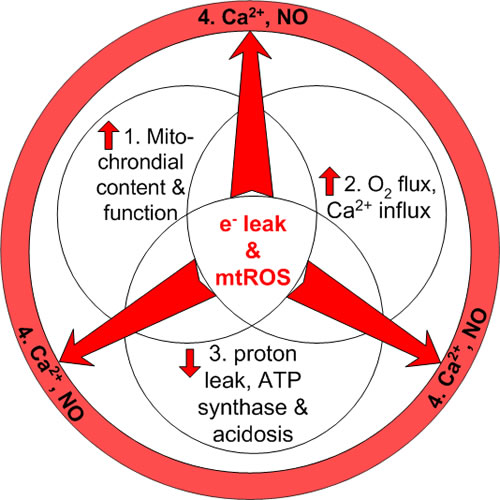
Figure 4: Mitochondrial factors modified by irradiation influence mtROS emission. The first three factors are endogenous: greater mitochondrial content and function, greater O2 consumption and Ca2+ influx, and decreased proton leak, ATP synthase and acidosis. These factors are accompanied by greater electron leakage and resulting mtROS generation. The fourth factor is exogenous, illustrating the egress of Ca2+ and NO influencing the mtROS emission in bystander cells.
DISCUSSION
Factors influencing radiation-induced mtROS emission
We propose a major role for mitochondria in radiation-induced cell death and the oxygen effect. While the exact nature of the processes involved are not fully understood, it is useful to consider the known factors that affect radiation-induced mtROS emission. Two factors that increase mtROS are a higher number of dysfunctional mitochondria in a cell, and a greater local O2 concentration. A third factor that decreases mtROS emission is proton leak, or the uncoupling of electron transport chain activity from ATP synthase activity (Fig. 4). A fourth factor is that mtROS can be amplified by the generation and migration of NO and Ca2+, impinging upon neighboring mitochondria and cells.
Higher mitochondrial content and functional capacity
Mitochondrial mass in peripheral lymphocytes increased 2.3-fold after a 4.5 Gy low-LET radiation dose, and increased by a larger factor after a 9 Gy dose, in patients undergoing total body irradiation therapy [39]. Similarly, fractional increases in mitochondrial fusion or mitochondrial mass have been seen after lower doses of low-LET and high-LET radiation to neurons and lung tumor cells [36, 40]. In an effort to aid repair after irradiation, a cell acquires a greater capacity to produce ATP, offset by mitochondrial dysfunction denoted by more mtROS production [10, 38].
Greater O2 consumption that coincides with ATP demand
Howard-Flanders and Alper [14] first quantified OER using gram-negative E. coli bacteria. This OER formulation is also applicable to malignant and non-malignant human cells (eukaryotic cells); relevant here is the fact that mitochondria have gram-negative bacterial origins, and first invaded cells to establish an endosymbiotic relationship. Although there are many influences (e.g., membrane potential) on mtROS emission, we model the mtROS generated as proportional to, or fractional decrease with, O2 consumption, showing saturation kinetic characteristics, rather than a linear regression, with O2 tension. Mitochondrial-derived ROS are produced on both the matrix side and cytoplasmic side of the mitochondrial inner membrane, which is impermeable to •O2- but not H2O2. (Fig. 1B) [7]. MtROS O2-dependent trends also depend on other issues, such as metabolic conditions, antioxidant activity, mitochondrial site of ROS origin and drop in phosphorylation efficiency during ADP limitation at high O2 levels [6, 16, 41]. The mitochondrial sites of ROS generation by irradiation are comparatively uncharacterized and may involve pro-apoptotic, enzymatic events requiring cytochrome c release [17, 42]. Mitochondria usually have a high reserve capacity to produce ATP in times of need, such as when cells are irradiated. Stable clones of hamster fibroblasts increased their O2 consumption two-fold after being subjected to 10 Gy low-LET X-rays [10]. A similar elevation in O2 consumption was measured for a lower dose (0.8 Gy) of high-LET α-radiation [33].
Proton leak uncoupling of the electron transport chain from ATP synthase activity
Mitochondrial uncoupling, through proton leaks, lowers both ROS emission and the efficiency of mitochondrial ATP production [25, 31]. There are two general classes of proton leak: a basal leak and an inducible form that employs uncoupling proteins and other mitochondrial inner membrane proteins, including the ADP/ATP translocator [43]. The acute activation mechanisms of the uncoupling proteins involve ROS, ROS byproducts (e.g., 4-HNE) and protein post-translational mechanisms, notably glutathionylation [25]. Superoxide was shown to stimulate UCP1, 2, 3 activities in the presence of fatty acids [26]; likewise, high- dose γ- rays stimulate UCP2 activity, which thus lowers mitochondrial membrane potential [24]. Consequently, UCPs may increase radioresistance by lowering membrane potential and thereby minimizing ROS emission [24-26].
MtROS can be amplified by the generation and migration of radiolysis-initiated ROS, Ca2+ and NO
Subjecting cells, including tumor cells, to 1% NO in an inert gas has a sensitizing effect on radiation-induced cell death, similar to that of oxygenation [44]. Our analysis of the oxygen effect accommodates this fundamental finding. In both O2 and NO atmospheres, mitochondria play an important role in the radiation effects on cells [27]. Of particular pertinence to the biological mechanisms underlying the oxygen effect, the HIF1α gene is regulated not only by O2, but also by NO and ROS [45]. This is especially relevant at low O2 levels, at which the low-ATP component dominates. Support for the mtROS OER component includes the finding that, in the presence of O2, the radiation activation of mitochondrial inducible NO synthases and the radiolysis of water are major sources of ROS/RNS, with higher LET being more likely to generate long-range radicals capable of reaching nucDNA [17, 46].
Particularly at high doses, severe radiation damage of mitochondria results in mutated mtDNA, catastrophic mitochondrial Ca2+ influx, the release of cytochrome c, and the onset of an irreversible mitochondrial permeability transition associated with apoptosis and necrosis. An initial ionizing event can be amplified by Ca2+ signaling [27] across the cytoplasm to other mitochondria of the affected cell or even to bystander cells [17, 38, 47]. While further research is clearly needed, we have examined whether low-LET-generated Ca2+ and mtROS are more signal-related than overwhelmingly detrimental, and whether the converse is true for high-LET clustered ionizations [48]. Our initial conclusion is that this OER analysis better relates to low-LET than to the unrepairable damage of high-LET exposures.
Oxygen effect and dose
While the relationships of cell survival curves to dose are beyond the scope of this analysis, experimental studies report differences in the cellular effects of low- and high-dose exposures for both low- and high-LET radiation [49]. No analysis of the differing effects of low-dose or high-dose on OER curves has been pursued, although the review of Wenzl and Wilkens [49] noted variable OER behavior. Indeed, OER values can increase, decrease or remain nearly constant with increasing radiation doses, depending on the cell line and radiation exposure used.
Notwithstanding, our mitochondrial-related analysis has a bearing on the debate over the origin of the “shoulder” region of survival curves, especially evident in doses of <10 Gy of low-LET radiation under aerobic conditions. This shoulder region is exploited in split dose or multi-fractionated radiotherapy regimes with intervals, leading to a decreased response to a second exposure of aerated tissue. On the basis of our analysis, there is clearly a need to experimentally explore whether radiation-induced UCP2 activates uncoupling of the oxidative phosphorylation system (proton leak) in mitochondria. In particular, where a first radiation exposure and uncoupling modifies the oxidative stress generated by a second exposure and its subsequence relevance to fractionated/chronic exposures and to radiation adaptive responses.
One explanation for the success of fractionated therapy is that hypoxic tumor cells have time to reoxygenate after apoptotic cells have been removed following the initial exposure. There are difficulties in explaining the shoulder region in terms of the DSB dose response, which is generally linear for a very wide range of doses [50]. However, the shoulder region of cell survival curves is usually interpreted in one of two ways. First, dose-dependent detrimental effects to cells may eventually lead to lethal damage at high dose. Second, enzymatic repair, aided by low dose promoting greater mitochondrial fusion and mitochondrial mass [40], becomes ineffective at high dose.
Repair of sub-lethal radiation damage occurs preferentially in different phases of the cell cycle, with checkpoints that monitor ATP before allowing the cell cycle to progress [51]. Especially relevant to the low-ATP OER component is that mitotic cell death after irradiation is more prevalent in hypoxia, when ATP is in short supply. This mitotic cell death pre-empts DNA repair by mitotic homologous recombination.
The multi-hit cell damage of X-irradiation at high dose (4 Gy) and dose rates (3.3 Gy/min) compromise mitochondrial maintenance and regeneration, as mitochondrial protein import decreases in human fibroblasts [28], whereas at low dose (0.1 Gy) and dose rates (2 mGy/h), membrane potential was increased and protein import was found to be enhanced compared to controls. This appears to have a knock-on effect: in one study in rat splenocytes, low-dose exposure (~0.01–0.1 Gy) demonstrated a transitory inhibition of mitosis followed by an increase in cell division, likely linked to sub-lethal repair [52]. At higher doses, cell proliferation was reduced.
Assuming a linear dose dependency, for relatively low-dose, low-LET exposures, mtROS production would be close to basal levels (Table 2). This may imply that the mtROS component of OER curves is more closely related to the controlled progression of Ca2+-initiated signaling [48], cell cycle arrest and apoptosis than to an immediate, catastrophic mitochondrial permeability transition, large depolarization and release of cytochrome c into the cytosol. Therefore, based on the mtROS component having the dual traits of oxidative damage and greater ATP production, we suggest, particularly for low-dose low-LET exposures, that the shoulder region should not be interpreted as exclusively damage- or repair-related, but as a combination of these two mechanisms.
Radiosensitivity varies with supplies of O2, glucose and ATP
Levels of anaerobic glycolysis versus aerobic oxidative phosphorylation seesaw, depending on the availability of O2, nutrients and other factors. Oxygen tension has a strong influence on wound healing, where epithelial growth is proportional to O2 tension up to normoxic and even higher levels, although cell replication is rare at <2.6% O2 and virtually non-existent at <0.7% O2 [53]. Hypoxia inducible factors (HIFs), a small group of master transcription factors have central roles in the adaptation to hypoxia, perhaps even up to levels of 20% O2 [54]. Under normal O2 conditions, when the mtROS OER component dominates, glycolytic metabolism is inhibited at the expense of oxidative phosphorylation activity. By contrast, in hypoxia, when the low-ATP OER component dominates, there is increased protein unfolding, yet decreased protein turnover, ATP demand and oxidative stress. Cellular mitochondrial content is reduced in anaerobic compared with aerobic cultures, measured by citrate synthase activity in human primary fibroblasts, which supports a lesser role for mitochondrial damage in low O2 conditions [55]. The shift from oxidative phosphorylation to anaerobic glycolysis is accompanied by a far lower ATP production capacity and an energy deficit, which affects a cell’s ability to repair and replicate after stress, including that from radiation.
Of the myriad tumor driver genes, only the high-frequency mutations in the p53 tumor suppressor gene are significantly associated with rising cancer incidence rates in aging adults [56]. It is well known that stresses such as ionizing radiation increase nuclear p53 protein levels as a consequence of nucDNA damage and that therapy-level exposure can accelerate the processes of aging and cancer [57]. Deficiency in the p53 protein confers not only apoptotic evasion, but also a preference for a glycolytic metabolism [58]. Nevertheless, most of the ATP in tumor cells is thought to be produced through glutamine-driven oxidative phosphorylation, in both normoxic and low O2 conditions [59]. Cells have evolved coping strategies at low O2 levels such as increasing p53 activation of a G1-phase cell cycle checkpoint distinct from nucDNA damage pathways [60]. In addition, hypoxia induces anaerobic glycolysis, and the consequential increase in lactate production and secondary effects (Fig. 4), such as extra-mitochondrial, extra-cellular acidosis. These may bring about a greater proton burden, loss of membrane potential and mitochondrial remodeling, minimizing the effects of radiotherapy [61].
Tumor cells generate increased mtROS, partially as a result of mitochondrial dysfunction [33]. When irradiated, some tumor cells have higher levels of the uncoupling protein UCP2 and mitochondrial proton leak, thereby lessening mtROS and conferring resistance to cytotoxic treatments. This was shown in a study by one of us [24], which also found that after a low-LET dose of 20 Gy in aerated conditions, resistant subclones of leukemia cells have elevated UCP2 activity, suppressing ROS and subsequent nucDNA damage as measured by 8-oxo-deoxyguanosine. These resistant subclones also augment their metabolism, utilizing fatty acids when glucose levels are low. Conversely, as illustrated earlier, hypoxic tumors that exhibit radiation-induced uncoupling can also be expected to exhibit enhanced mtROS generation. Similarly, inhibition of UCP2 increases mtROS and restores sensitivity to some chemotherapeutics [62]. These studies showed that uncoupling is an important aspect governing how cells adapt to a radiation exposure. It would be worth confirming in future studies the premise that a tumor’s radioresistance is due in part to an extended period of adaptive uncoupling.
Are radiation effects mitochondria-centric rather than nucleus-centric?
Published experimental research, and this analysis of the radiation oxygen effect, identify a major role for uncoupled respiration in mitochondria. They also provide strong, if not definitive, evidence for the dominance of indirect actions of radiation involving mitochondria over direct actions on the nucleus. For example, both low-LET and high-LET irradiation of fibroblasts and other cells can produce mitochondrial dysfunction accompanied by persistent ROS production and genomic instability [10, 17, 33, 46, 63]. While many would argue that nucDNA DSBs are a crucial facet of radiation damage to cells, Franken et al [64] showed that the relative biological effectiveness (RBE), based on the number of DSBs, remains about unity, no matter the level of LET, whereas chromosomal aberrations, lethal damage and reproductive death are much more LET-dependent.
Experimental evidence, acquired mainly after exposures with charged particle microbeams or radiolabels with short-range impact, shows that direct damage to the nucleus can be the most effective cause of radiation effects such as DNA damage and cell killing. Yet irradiation of the cytoplasm or mitochondria indirectly causes these same effects, sometimes with lower efficiencies [65]. For example, Tartier et al [47] irradiated cytoplasm with high-LET helium ions and showed that direct hits to the nucDNA of HeLa cells were not needed to cause damage, as measured in the form of p53 binding protein 1 (53BP1) foci at DSB sites in irradiated and bystander cells. The study showed that the damage was caused indirectly, via production of ROS and RNS.
Furthermore, Zhang and colleagues [46] induced DNA oxidative damage and DSBs by targeting the cytoplasm of wild type ρ+ (mitochondria-endowed) human small airway epithelial cells with a helium ion microbeam of 1 µm width. The findings were compared to those from mtDNA depleted (ρ0) cells. A significant increase in autophagy and micronuclei indicated that genomic instability had resulted in the ρ+ cells. These studies, conducted in oxygenated environments, increasingly point to mitochondria as essential in mediating cytoplasmic radiation-induced genotoxic damage in targeted or bystander mammalian cells.
Conversely, some studies support the view that lower O2 levels tend to more nucleus-centric damage. Low-LET 137Cs γ-exposures (0–10 Gy) under chronically hypoxic conditions have been shown to increase G1-associated nucDNA DSBs, leading to greater genetic instability and lower clonogenic survival [66]. The selection of the nucleus as the primary cellular radiation target is supported by the observing that when the Auger-emitter 125I is incorporated in close proximity to the nucleus it is much more effective at producing DSBs or cell death than 125I sited in the cytoplasm [67, 68].
Lastly, Leach et al [27] observed radiation-induced ROS/RNS in osteosarcoma ρ+ cells, but not in mitochondria-depleted ρ0 cells. There are reports that both normal and malignant irradiated ρ0 cells are more radioresistant than ρ+ cells in the presence of O2 [69, 70]. Yet in hypoxic conditions, there was little difference in the radiosensitivity of ρ+ and ρ0 cells. This would be expected in light of our hypothesis that the oxygen effect can be quantitatively explained by OER comprising two components, the most prominent being the generation of mtROS under aerobic conditions. Experimental studies are needed for confirmation of this hypothesis.
In conclusion, we acknowledge that radiation damage to the cell nucleus is of ultimate importance. However we propose that this nuclear damage generally arises indirectly as a knock-on effect, via Ca2+ flux and damage to mitochondria, which appears to be the most likely initiator of radiation-induced ROS, nucDNA damage, cell death and perhaps oncogenesis. Consequently, we suggest that further research in radiotherapy would benefit from greater emphasis on aspects involving mitochondria, the contribution of the various sources of ROS, and uncoupled oxidative phosphorylation.
MATERIALS AND METHODS
The analysis utilizes the OER, which varies from unity in anoxia to typically about three for low-LET radiation, with radiosensitivity varying most rapidly with O2 partial pressures below ~2% atmospheric (Fig. 2A). Areas in solid tumors can be anoxic, while in normal cells physiological O2 tension varies widely, ranging from ~11% concentration in the blood of large arteries to as low as 0.1% within cells [16].
We based our analytical methods on a competitive type of reaction expression developed by Howard-Flanders and Alper [14] for OER of radiation R and its variation with O2 % concentration (![]() ) (Fig. 2A), or O2 pressure in air:
) (Fig. 2A), or O2 pressure in air:
![]() 2)
2)
where the maximum OER value, (OERRmax) is obtained in the presence of 100% O2 (one atmosphere is 101.3 kPa, 760 mmHg or 760 torr). The value of (OERRmax) is very similar to that for the OER for normoxia (![]() ) conditions (Table 1). This analysis utilized
) conditions (Table 1). This analysis utilized ![]() values for the lowest and highest of nine radiation types with increasing LET derived by Barendsen et al [19]. Half-maximal O2 concentration (
values for the lowest and highest of nine radiation types with increasing LET derived by Barendsen et al [19]. Half-maximal O2 concentration (![]() ) is the percentage O2 for which the cellular radiosensitivity is midway between the anoxic and fully oxygenated responses. A value of 0.55% O2 was assumed for both low- and high-LET radiation; to our knowledge there is no discernible trend in
) is the percentage O2 for which the cellular radiosensitivity is midway between the anoxic and fully oxygenated responses. A value of 0.55% O2 was assumed for both low- and high-LET radiation; to our knowledge there is no discernible trend in ![]() with LET [32].
with LET [32].
The O2 flux variation with ![]() in mitochondria, controlled by several enzymes, can be represented by a rectangular hyperbolic function [15]:
in mitochondria, controlled by several enzymes, can be represented by a rectangular hyperbolic function [15]:
![]() 3)
3)
Assuming that radiation-induced oxidative stress (an abnormal state) is primarily linked to the leakage of electrons by the mitochondrial electron transport chain, proportional to O2 consumption ![]() (pmol/s /cm3), Eqn. 2 can be rearranged into two components as shown in Eqn. 1. The first OER component mtROS is similar in form to Eqn. 3. The second OER component, low-ATP is unity for cells in anoxia and diminishes with the O2 tension. (This does not account for uncoupled respiration under hypoxia; see Results.).
(pmol/s /cm3), Eqn. 2 can be rearranged into two components as shown in Eqn. 1. The first OER component mtROS is similar in form to Eqn. 3. The second OER component, low-ATP is unity for cells in anoxia and diminishes with the O2 tension. (This does not account for uncoupled respiration under hypoxia; see Results.).
ACKNOWLEDGEMENTS
Dr. Dudley Goodhead of the Medical Research Council (UK) and Dr. David Patten of Ottawa University are thanked for their critical and insightful comments on the paper. We acknowledge Carolyn Brown’s assistance in preparing the manuscript.
grant Support
This work was undertaken as part of the Science and Technology program of the Canadian Government, CNL Project 2.3.2-006 Improving Occupational Dosimetry. This study has benefited from the library facilities that McGill University makes available to its adjunct professors (RBR). MEH is funded by the Natural Science and Engineering Research Council of Canada, and the Canadian Institutes of Health Research.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Barendsen GW. The relationships between RBE and LET for different types of lethal damage in mammalian cells: biophysical and molecular mechanisms. Radiat Res. 1994; 139:257-270.
2. Thoday JM and Read J. Effect of oxygen on the frequency of chromosome aberrations produced by X-rays. Nature. 1947; 160:608.
3. Ewing D. The oxygen fixation hypothesis: a reevaluation. Am J Clin Oncol. 1998; 21:355-361.
4. Daniels EW and Breyer EP. Recovery of supralethally X-irradiated and nitrogen mustard (HN2)-treated amoebae. Int J Radiat Biol Relat Stud Phys Chem Med. 1970; 18:179-187.
5. Goldfeder A. (1965). The integrity of cytoplasmic ultrastructures: a factor in cellular radiosensitivtiy. Cellular Radiation Biology, Symposium on Fundamental Cancer Research. (M.D. Anderson Hospital and Tumor Institute: Baltimore: Williams and Wilkins), pp. 539-546.
6. Chance B, Sies H and Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979; 59:527-605.
7. St-Pierre J, Buckingham JA, Roebuck SJ and Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002; 277:44784-44790.
8. Richter C, Park JW and Ames BN. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci U S A. 1988; 85:6465-6467.
9. Croteau DL and Bohr VA. Repair of oxidative damage to nuclear and mitochondrial DNA in mammalian cells. J Biol Chem. 1997; 272:25409-25412.
10. Dayal D, Martin SM, Owens KM, Aykin-Burns N, Zhu Y, Boominathan A, Pain D, Limoli CL, Goswami PC, Domann FE and Spitz DR. Mitochondrial complex II dysfunction can contribute significantly to genomic instability after exposure to ionizing radiation. Radiat Res. 2009; 172:737-745.
11. Ward JF, Blakely WF and Joner EI. Mammalian cells are not killed by DNA single-strand breaks caused by hydroxyl radicals from hydrogen peroxide. Radiat Res. 1985; 103:383-392.
12. Rothkamm K and Lobrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low X-ray doses. Proc Natl Acad Sci U S A. 2003; 100:5057-5062.
13. Radford IR. Evidence for a general relationship between the induced level of DNA double-strand breakage and cell-killing after X-irradiation of mammalian cells. Int J Radiat Biol Relat Stud Phys Chem Med. 1986; 49:611-620.
14. Howard-Flanders P and Alper T. The sensitivity of microorganisms to irradiation under controlled gas conditions. Radiat Res. 1957; 7:518-540.
15. Gnaiger E, Steinlechner-Maran R, Mendez G, Eberl T and Margreiter R. Control of mitochondrial and cellular respiration by oxygen. J Bioenerg Biomembr. 1995; 27:583-596.
16. Hoffman DL and Brookes PS. Oxygen sensitivity of mitochondrial reactive oxygen species generation depends on metabolic conditions. J Biol Chem. 2009; 284:16236-16245.
17. Azzam EI, Jay-Gerin JP and Pain D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Letters. 2012; 327(1-2):48-60.
18. Chapman JD, Reuvers AP, Borsa J and Greenstock CL. Chemical radioprotection and radiosensitization of mammalian cells growing in vitro. Radiat Res. 1973; 56:291-306.
19. Barendsen GW, Koot CJ, Van Kersen GR, Bewley DK, Field SB and Parnell CJ. The effect of oxygen on impairment of the proliferative capacity of human cells in culture by ionizing radiations of different LET. Int J Radiat Biol Relat Stud Phys Chem Med. 1966; 10:317-327.
20. Goodhead DT. The initial physical damage produced by ionizing radiations. Int J Radiat Biol. 1989; 56:623-634.
21. Roots R, Chatterjee A, Chang P, Lommel L and Blakely EA. Characterization of hydroxyl radical-induced damage after sparsely and densely ionizing irradiation. Int J Radiat Biol Relat Stud Phys Chem Med. 1985; 47:157-166.
22. Hirayama R, Ito A, Tomita M, Tsukada T, Yatagai F, Noguchi M, Matsumoto Y, Kase Y, Ando K, Okayasu R and Furusawa Y. Contributions of direct and indirect actions in cell killing by high-LET radiations. Radiat Res. 2009; 171:212-218.
23. Adam-Vizi V and Starkov AA. Calcium and mitochondrial reactive oxygen species generation: How to read the facts. J Alzheimers Dis. 2010; 20:S413-S426.
24. Harper ME, Antoniou A, Villalobos-Menuey E, Russo A, Trauger R, Vendemelio M, George A, Bartholomew R, Carlo D, Shaikh A, Kupperman J, Newell EW, Bespalov IA, Wallace SS, Liu Y, Rogers JR, et al. Characterization of a novel metabolic strategy used by drug-resistant tumor cells. FASEB J. 2002; 16:1550-1557.
25. Mailloux RJ and Harper ME. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic Biol Med. 2011; 51:1106-1115.
26. Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, Stuart JA, Harper JA, Roebuck SJ, Morrison A, Pickering S, Clapham JC and Brand MD. Superoxide activates mitochondrial uncoupling proteins. Nature. 2002; 415:96-99.
27. Leach JK, Van Tuyle G, Lin PS, Schmidt-Ullrich R and Mikkelsen RB. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res. 2001; 61:3894-3901.
28. Pandey BN, Gordon DM, De Toledo SM, Pain D and Azzam EI. Normal human fibroblasts exposed to high- or low-dose ionizing radiation: differential effects on mitochondrial protein import and membrane potential. Antioxid Redox Signal. 2006; 8:1253-1261.
29. Voehringer DW, Hirschberg DL, Xiao J, Lu Q, Roederer M, Lock CB, Herzenberg LA, Steinman L and Herzenberg LA. Gene microarray identification of redox and mitochondrial elements that control resistance or sensitivity to apoptosis. P Natl Acad Sci USA. 2000; 97:2680-2685.
30. Mills EM, Xu D, Fergusson MM, Combs CA, Xu Y and Finkel T. Regulation of cellular oncosis by uncoupling protein 2. J Biol Chem. 2002; 277:27385-27392.
31. Gnaiger E, Lassnig B, Kuznetsov A, Rieger G and Margreiter R. Mitochondrial oxygen affinity, respiratory flux control and excess capacity of cytochrome c oxidase. J Exp Biol. 1998; 201:1129-1139.
32. Richardson RB. Age-dependent changes in oxygen tension, radiation dose and sensitivity within normal and diseased coronary arteries-Part C: oxygen effect and its implications on high- and low-LET dose. Int J Radiat Biol. 2008; 84:858-865.
33. Pelicano H, Carney D and Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat. 2004; 7:97-110.
34. Korshunov SS, Skulachev VP and Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997; 416(1):15-18.
35. Affourtit C, Jastroch M and Brand MD. Uncoupling protein-2 attenuates glucose-stimulated insulin secretion in INS-1E insulinoma cells by lowering mitochondrial reactive oxygen species. Free Radic Biol Med. 2011; 50:609-616.
36. Zhang S, Wen G, Huang SX, Wang J, Tong J and Hei TK. Mitochondrial alteration in malignantly transformed human small airway epithelial cells induced by alpha-particles. Int J Cancer. 2013; 132:19-28.
37. Yost MT, Robson HH and Yost HT. Uncoupling of oxidative phosphorylation in rat liver and spleen mitochondria by exposure to total-body irradiation. Radiat Res. 1967; 32:187-199.
38. Nugent SM, Mothersill CE, Seymour C, McClean B, Lyng FM and Murphy JE. Increased mitochondrial mass in cells with functionally compromised mitochondria after exposure to both direct gamma radiation and bystander factors. Radiat Res. 2007; 168:134-142.
39. Wen Q, Hu Y, Ji F and Qian G. Mitochondrial DNA alterations of peripheral lymphocytes in acute lymphoblastic leukemia patients undergoing total body irradiation therapy. Radiat Oncol. 2011; 6:133.
40. Chien L, Chen WK, Liu ST, Chang CR, Kao MC, Chen KW, Chiu SC, Hsu ML, Hsiang IC, Chen YJ and Chen L. Low-dose ionizing radiation induces mitochondrial fusion and increases expression of mitochondrial complexes I and III in hippocampal neurons. Oncotarget. 2015; 6:30628-30639. doi: 10.18632/oncotarget.5790.
41. Gnaiger E, Mendez G and Hand SC. High phosphorylation efficiency and depression of uncoupled respiration in mitochondria under hypoxia. Proc Natl Acad Sci U S A. 2000; 97:11080-11085.
42. Atkinson J, Kapralov AA, Yanamala N, Tyurina YY, Amoscato AA, Pearce L, Peterson J, Huang Z, Jiang J, Samhan-Arias AK, Maeda A, Feng W, Wasserloos K, Belikova NA, Tyurin VA, Wang H, et al. A mitochondria-targeted inhibitor of cytochrome c peroxidase mitigates radiation-induced death. Nat Commun. 2011; 2:497.
43. Harper ME, Green K and Brand MD. The efficiency of cellular energy transduction and its implications for obesity. Annu Rev Nutr. 2008; 28:13-33.
44. Gray LH, Green FO and Hawes CA. Effect of nitric oxide on the radiosensitivity of tumour cells. Nature. 1958; 182:952-953.
45. Chun YS, Kim MS and Park JW. Oxygen-dependent and -independent regulation of HIF-1alpha. J Korean Med Sci. 2002; 17:581-588.
46. Zhang B, Davidson MM and Hei TK. Mitochondria regulate DNA damage and genomic instability induced by high LET radiation. Life Sci Space Res (Amst). 2014; 1:80-88.
47. Tartier L, Gilchrist S, Burdak-Rothkamm S, Folkard M and Prise KM. Cytoplasmic irradiation induces mitochondrial-dependent 53BP1 protein relocalization in irradiated and bystander cells. Cancer Res. 2007; 67:5872-5879.
48. Anoopkumar-Dukie S, Conere T, Sisk GD and Allshire A. Mitochondrial modulation of oxygen-dependent radiosensitivity in some human tumour cell lines. Br J Radiol. 2009; 82:847-854.
49. Wenzl T and Wilkens JJ. Theoretical analysis of the dose dependence of the oxygen enhancement ratio and its relevance for clinical applications. Radiat Oncol. 2011; 6:171.
50. Barnard S, Bouffler S and Rothkamm K. The shape of the radiation dose response for DNA double-strand break induction and repair. Genome Integr. 2013; 4:1.
51. Azzam EI, de Toledo SM, Waker AJ and Little JB. High and low fluences of alpha-particles induce a G(1) checkpoint in human diploid fibroblasts. Cancer Research. 2000; 60:2623-2631.
52. Ishii K, Yamaoka K, Hosoi Y, Ono T and Sakamoto K. Enhanced mitogen-induced proliferation of rat splenocytes by low-dose whole-body X-irradiation. Physiol Chem Phys Med NMR. 1995; 27:17-23.
53. Niinikoski J and Hunt TK.. Oxygen and healing wounds: tissue-bone repair enhancement. In:Oriani, G et al, ed. Handbook on Hyperbaric Medicine. (Milano: Springer-Verlag). 1996; pp. 485-507.
54. Schofield CJ and Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004; 5(5):343-354.
55. Solaini G, Baracca A, Lenaz G and Sgarbi G. Hypoxia and mitochondrial oxidative metabolism. Biochim Biophys Acta. 2010; 1797:1171-1177.
56. Richardson RB. p53 mutations associated with aging-related rise in cancer incidence rates. Cell Cycle. 2013; 12:2468-2478.
57. Richardson RB. Ionizing radiation and aging: rejuvenating an old idea. Aging (Albany, NY). 2009; 1:887-902.
58. Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F and Hwang PM. p53 regulates mitochondrial respiration. Science. 2006; 312:1650-1653.
59. Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T and Rabinowitz JD. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol. 2013; 9:712.
60. Graeber TG, Peterson JF, Tsai M, Monica K, Fornace AJ, Jr. and Giaccia AJ. Hypoxia induces accumulation of p53 protein, but activation of a G1-phase checkpoint by low-oxygen conditions is independent of p53 status. Mol Cell Biol. 1994; 14:6264-6277.
61. Khacho M, Tarabay M, Patten D, Khacho P, MacLaurin JG, Guadagno J, Bergeron R, Cregan SP, Harper ME, Park DS and Slack RS. Acidosis overrides oxygen deprivation to maintain mitochondrial function and cell survival. Nat Commun. 2014; 5:3550.
62. Pfefferle A, Mailloux RJ, Adjeitey CN and Harper ME. Glutathionylation of UCP2 sensitizes drug resistant leukemia cells to chemotherapeutics. Biochim Biophys Acta. 2013; 1833:80-89.
63. Kim GJ, Chandrasekaran K and Morgan WF. Mitochondrial dysfunction, persistently elevated levels of reactive oxygen species and radiation-induced genomic instability: a review. Mutagenesis. 2006; 21:361-367.
64. Franken NAP, ten Cate R, Krawczyk PM, Stap J, Haveman J, Aten J and Barendsen GW. Comparison of RBE values of high-LET alpha-particles for the induction of DNA-DSBs, chromosome aberrations and cell reproductive death. Radiation Oncology. 2011; 6:64.
65. Zhou H, Hong M, Chai Y and Hei TK. Consequences of cytoplasmic irradiation: studies from microbeam. J Radiat Res. 2009; 50 Suppl A:A59-65.
66. Kumareswaran R, Ludkovski O, Meng A, Sykes J, Pintilie M and Bristow RG. Chronic hypoxia compromises repair of DNA double-strand breaks to drive genetic instability. J Cell Sci. 2012; 125:189-199.
67. Hofer KG, Harris CR and Smith JM. Radiotoxicity of intracellular 67Ga, 125I and 3H. Nuclear versus cytoplasmic radiation effects in murine L1210 leukaemia. Int J Radiat Biol Relat Stud Phys Chem Med. 1975; 28:225-241.
68. Balagurumoorthy P, Xu X, Wang K, Adelstein SJ and Kassis AI. Effect of distance between decaying 125I and DNA on Auger-electron induced double-strand break yield. Int J Radiat Biol. 2012; 88:998-1008.
69. Yamazaki H, Yoshida K, Yoshioka Y, Isohashi F, Ozeki S, Koizumi M, Yoneda M and Inoue T. Impact of mitochondrial DNA on hypoxic radiation sensitivity in human fibroblast cells and osteosarcoma cell lines. Oncol Rep. 2008; 19:1545-1549.
70. Cloos CR, Daniels DH, Kalen A, Matthews K, Du J, Goswami PC and Cullen JJ. Mitochondrial DNA depletion induces radioresistance by suppressing G2 checkpoint activation in human pancreatic cancer cells. Radiat Res. 2009; 171:581-587.