
INTRODUCTION
Cells exposed to genotoxic stress protect their genome integrity by activation of a conserved DNA damage response pathway that orchestrates DNA repair and represents an intrinsic barrier preventing genome instability and tumorigenesis [1, 2]. A core component of this pathway is the tumor suppressor p53 that controls cell fate decisions. Depending on the amplitude and duration of its activation, p53 promotes temporary cell cycle arrest (checkpoint), permanent withdrawal from the cell cycle (senescence) or programmed cell death (apoptosis) [3–5]. Under basal conditions, function of the p53 is suppressed by an E3 ubiquitin ligase MDM2 and its enzymatically inactive homologue MDMX that control p53 stability and transcriptional activity, respectively [6, 7]. Genotoxic stress triggers activation of ATM/ATR, Chk1/Chk2 and other kinases that extensively phosphorylate the N-terminal domain of p53, MDM2 and MDMX allowing stabilization of the p53 and promoting expression of its target genes [8–11]. One of the p53 target genes is PPM1D that codes for a Protein phosphatase 2C isoform delta (hereafter referred to as WIP1) [12]. Expression of WIP1 is induced by genotoxic stress and forming a negative feedback loop, WIP1 efficiently inhibits the p53 pathway by a direct dephosphorylation of p53 at Ser15 and also by dephosphorylation of its negative regulators MDM2 and MDMX [13–16]. By inactivating the p53 pathway, WIP1 promotes recovery from the G2 checkpoint [17, 18]. Moreover, WIP1 dephosphorylates other proteins including ATM, Chk1, Chk2, p38 and γH2AX which contributes to the termination of the DNA damage response [19–24]. In addition, WIP1 was reported to prevent premature senescence in various cell types and tissue compartments [21, 25, 26].
Chromosomal locus 17q23 carrying the PPM1D gene is commonly amplified in various human tumors including breast, ovarian and gastric cancer, neuroblastoma and lung adenocarcinoma [27–34]. In particular, amplification of the PPM1D occurs in approximately 10 % of breast tumors, typically those that retain wild type p53 [31, 35, 36]. In addition, about one third of breast tumors with amplified PPM1D locus also contain amplification of the ERBB2/HER2 oncogene suggesting that both genes may jointly promote tumor development [36]. Indeed, MMTV-driven overexpression of Ppm1d potentiated Erbb2-induced breast tumor development in mice [37]. Comparably less common than PPM1D amplifications are rare nonsense mutations in the exon 6 of PPM1D that result in expression of abnormally stable WIP1 and promote development of breast and ovary cancer [38–40].
Reactivation of the p53 function by various MDM2 or MDMX antagonists and other small molecule p53 activators has been proposed as promising strategy for treatment of cancers with the wild-type p53 [41–45]. Nutlin-3 is a potent and selective antagonist of the interaction between MDM2 and p53 (IC50 of 90 nM) [46]. Treatment with nutlin-3 activates the p53 pathway and depending on the dose induces cell cycle arrest or cell death [46]. RG7388, an orally available analogue of nutlin-3, efficiently suppressed tumor growth in vivo [47]. Clinical trials are currently ongoing to prove clinical efficacy of MDM2 antagonists in cancer therapy. Reactivation of p53 pathway can be also achieved by inhibition of WIP1 and indeed WIP1 was proposed a potential pharmacological target in cancer therapy [21, 48]. Loss of Ppm1d dramatically delayed the development of Erbb2-induced breast cancer, MYC-induced lymphoma and APCmin-induced intestinal tumors in mice [49–52]. In addition, depletion of WIP1 using RNA interference has been shown to efficiently suppress growth of various human cancer cells [30, 53–55]. However, translation of these observations into clinics is challenging due to the lack of suitable WIP1 inhibitors with sufficient specificity and favourable pharmacokinetic properties. Cyclic phosphopeptides that mimic substrates of WIP1 can block its phosphatase activity in vitro, but their efficiency in cells still remains to be addressed [56, 57]. A high-throughput screening identified a small molecule CCT007093 that inhibited WIP1 in vitro (IC50 = 8.4 μM) and eradicated WIP1 overexpressing tumor cells [58]. However, the specificity of CCT007093 towards WIP1 may be low in cells [59]. Small molecules SPI-001 and its analogue SL-176 inhibited WIP1 in vitro (IC50 = 86.9 nM and 110 nM and, respectively) and supressed growth of cells with the C-terminally truncated or overexpressed WIP1 but their efficiency at organismal level still needs to be tested [60–62]. Novel orally available inhibitor of WIP1 phosphatase GSK2830371 has recently been shown to selectively inhibit WIP1 in vitro (IC50 = 6 nM) and to efficiently suppress growth of a subset of hematopoietic tumor cell lines and neuroblastoma cells with overexpression of WIP1 [63, 64].
Here we aimed to validate the specificity and efficiency of the commercially available WIP1 inhibitors in blocking proliferation of the breast cancer cells. We have found that GSK2830371 suppressed growth of breast cancer cells with amplified PPM1D gene in a p53-dependent manner which is in good agreement with previous RNAi-based studies. In addition, we have found that inhibition of WIP1 is not sufficient to induce cell death in cancer cells but rather slows down proliferation by extending G1 and G2 phases of the cell cycle. However, breast cancer cells treated with WIP1 inhibitor are more sensitive to DNA damage-inducing chemotherapy and to MDM2 antagonist nutlin-3. Combined treatment with these drugs triggers senescence or programmed cell death and can efficiently eradicate p53 positive breast cancer cells. Our data validate GSK2830371 as potent and selective inhibitor of WIP1 that sensitizes breast cancer cells to chemotherapy.
RESULTS
WIP1 inhibition impairs proliferation of breast cancer cells with amplified PPM1D and wt-p53
To test the specificity of the novel WIP1 inhibitors in a cellular model, we generated U2OS-PPM1D-KO cells with the CRISPR-mediated knock-out of the PPM1D gene and determined the effect of CCT007093 or GSK2830371 compounds on cell growth (Figure 1A). Surprisingly, we have found that the effect of CCT007093 was not dependent on the presence of WIP1. In contrast, GSK2830371 showed a dose-dependent suppression of cell growth in parental U2OS but not in U2OS-PPM1D-KO cells. Next, we compared the ability of both compounds to potentiate a DNA damage-induced phosphorylation of two established substrates of WIP1 phosphatase, histone variant H2AX phosphorylated at Ser139 (referred to as γH2AX) and p53 phosphorylated at Ser15 (Figure 1B and 1C). We have not observed any significant differences in cells treated with DMSO and CCT007093 (10 μM) suggesting that CCT007093 does not block the activity of WIP1 in cells. In contrast, levels of γH2AX and pS15-p53 were increased in cells treated with GSK2830371 (0.5 μM) consistent with the expected inhibition of WIP1 activity. In accordance with a previous report we also observed a reduced level of WIP1 in the presence of an allosteric inhibitor GSK2830371 [63]. To further assess the efficiency of WIP1 inhibition, we compared responses to ionizing radiation in U2OS-PPM1D-KO cells and U2OS cells treated with GSK2830371 (Figure 1D). We found that treatment with GSK2830371 (0.5 μM) increased the phosphorylation of γH2AX and pS15-p53 and expression of p21 to comparable levels as the knock-out of PPM1D strongly indicating that GSK2830371 efficiently blocks WIP1 activity in cells.
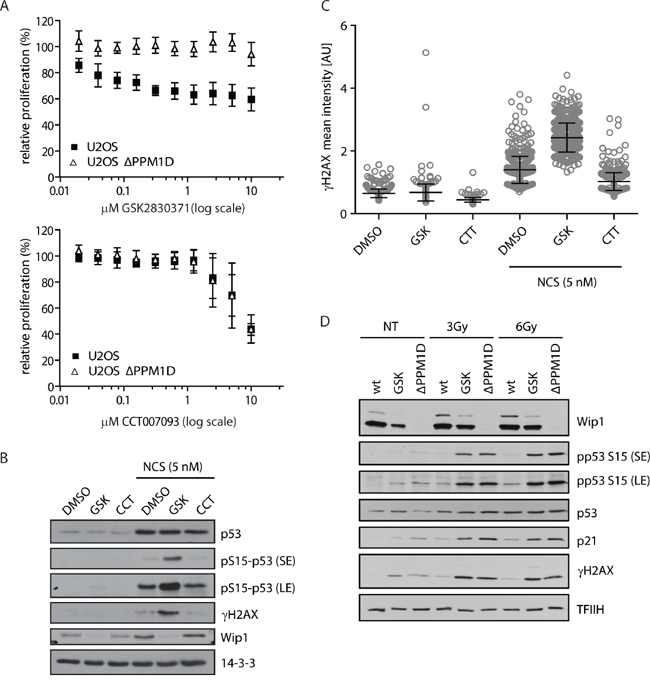
Figure 1: Validation of WIP1 inhibitors in U2OS-PPM1D-KO cells. A. U2OS or U2OS-PPM1D-KO cells were treated with DMSO, CCT007093 or GSK2830371 at indicated doses and relative cell proliferation was measured after 7 days. Error bars represent SD. B, C. U2OS cells were treated with DMSO, CCT007093 (10 μM, CCT) or GSK2830371 (0.5 μM, GSK) and DNA damage was induced by 5 nM neocarzinostatin (NCS) for 5 h. Cells were analyzed by immunoblotting (B) or fixed and nuclear γH2AX intensity was determined by immunofluorescent staining and microscopy analysis C. Dots represent individual cells. Error bars represent SD. D. U2OS or U2OS-PPM1D-KO (ΔPPM1D) cells were treated with DMSO or GSK2830371 (0.5 μM), exposed to ionizing radiation (3 and 6 Gy) and analysed by immunoblotting using indicated antibodies. Short exposure (SE) or long exposure (LE) is shown.
Having established efficient concentration of GSK2830371 that specifically affects growth of U2OS cells, we continued with testing the sensitivity of breast cancer cells to GSK2830371. First, we tested the effect of WIP1 inhibition on cell proliferation in MCF7 cells that have massively amplified PPM1D locus at 17q22/q23 and harbouring wild-type p53 [31, 65]. Using cell proliferation and colony formation assays we observed dramatic reduction of cell growth after inhibition of WIP1 (Figure 2A and 2B). Reduction of cell proliferation by GSK2830371 showed EC50=0.3 μM in MCF7 cells which is in good agreement with a previous report [63]. In contrast, we have found that MCF7 cells with knocked-out TP53 were less sensitive to GSK2830371 (Figure 2A and 2C). Similarly, we observed only a minor effect of GSK2830371 in BT-474 cells that contain amplification of the PPM1D but have inactivating mutation in TP53 [65] (Figure 2D). Thus the effect of WIP1 inhibition on breast cancer cell proliferation depends on the intact p53 pathway as previously reported for haematological cancer cells [63]. Next we tested the sensitivity of CAL-51 breast cancer cells that contain a normal number of PPM1D alleles and wild type p53 (Figure 2D). We have found that CAL-51 cells were resistant to the treatment with GSK2830371 suggesting that cells with amplified PPM1D might be addicted to the high WIP1 activity whereas cells with normal levels of WIP1 can tolerate inhibition of WIP1 and proliferate also in the presence of GSK2830371. Finally, we tested the impact of GSK2830371 on proliferation of nontransformed cells. A dose of GSK2830371 that efficiently supressed growth of U2OS and MCF7 cells did not affect proliferation of BJ fibroblasts, hTERT-immortalized human retinal pigment epithelial cells (RPE) or SV40-immortalized human colon epithelia cells (HCE) indicating that inhibition of WIP1 is well tolerated by nontransformed cells (Figure 2E)
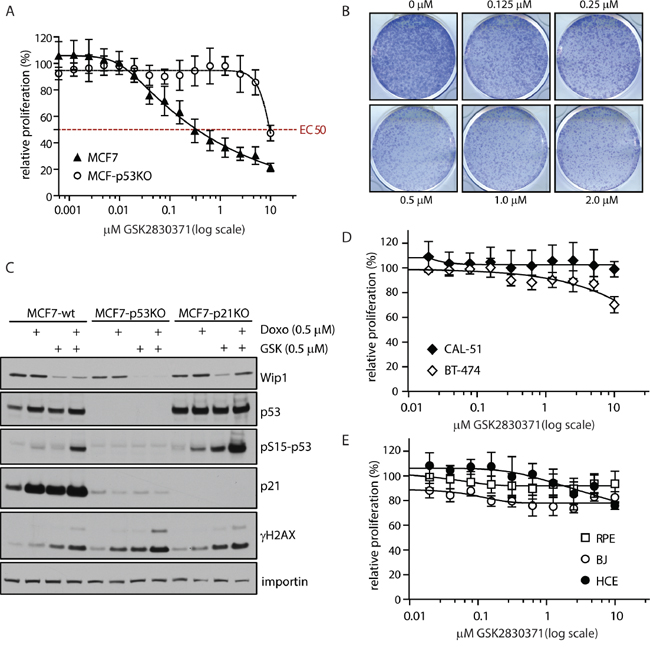
Figure 2: Inhibition of WIP1 impairs proliferation of cancer cells with amplified PPM1D. A. MCF7 or MCF7-P53-KO cells were treated with indicated doses of GSK2830371 and relative cell proliferation was measured after 7 days. Error bars represent SD. B. MCF7 cells were treated with indicated doses of GSK2830371 and cell proliferation was determined by colony formation assay after 7 days. Representative image from three independent experiments is shown. C. MCF7, MCF7-P53-KO or MCF7-P21-KO cells were treated with DMSO, GSK2830371 (0.5 μM), doxorubicin (0.5 μM) or combination of both and cells were analyzed by immunoblotting after 24 h. D. BT-474 or CAL-51 cells were treated with indicated doses of GSK2830371 and relative cell proliferation was measured after 7 days. Error bars represent SD. E. BJ fibroblasts, hTERT-RPE1 cells or human colon epithelia cells (HCE) were treated with indicated doses of GSK2830371 and relative cell proliferation was measured after 7 days. Error bars represent SD.
WIP1 inhibition delays progression through G1 and G2 phases of the cell cycle
Since we observed a strong reduction of the proliferating breast cancer cells population following WIP1 inhibition, we asked what the fate of the cells treated with GSK2830371 was. We found that GSK2830371 did not significantly affect the viability of MCF7 cells, suggesting that inhibition of WIP1 is not sufficient to induce cell death (Figure 3A). Instead we found that inhibition of WIP1 slowed down proliferation of MCF7 cells monitored by a dilution of CFSE dye in daughter cells (Figure 3B). The effect of GSK2830371 on the proliferation rate was fully dependent on p53 and p21 since we observed no differences in dilution of CFSE dye in MCF7-P53-KO or MCF7-P21-KO cells treated with WIP1 inhibitor (Figure 3B). Next we determined the effect of GSK2830371 on the cell cycle progression in MCF7 and BT-474 cells (Figure 3C). We have noted an accumulation of MCF7 cells in G1 phase 24 h after treatment with GSK2830371 (0.5 μM), whereas fraction of G2 cells was enriched in the later time points (48-72 h). This suggests that progression through G1 is slowed down in MCF7 cells early after addition of GSK2830371. Eventually cells progress through S phase to the G2 where they also progress more slowly compared to control cells. We did not observe any enrichment in the fraction of mitotic cells in the presence of GSK2830371 indicating that progression through mitosis was not affected by inhibition of WIP1 which is in good agreement with described degradation of WIP1 during prometaphase [66]. In contrast, no effect on the cell cycle progression was observed in BT-474, suggesting that observed extension of G1 and G2 phases depends on the ability to activate the p53 pathway (Figure 3C). Immunoblot analysis of MCF7 cells revealed that addition of GSK2830371 resulted in a rapid phosphorylation of p53 at Ser15 (Figure 3D). Two days after addition of GSK2830371, MCF7 cells showed increased levels of p21 which indicated a strong activation of the p53 pathway (Figure 3D). Consistent with no effect on the cell cycle progression and with the impaired p53 pathway, BT-474 cells did not show any induction of p21 levels after GSK2830371 administration (Figure 3E). Finally, we have found no effect on the cell cycle distribution in MCF7-P53-KO and MCF7-P21-KO cells treated with GSK2830371 further confirming that the effect of WIP1 inhibition on the progression through the cell cycle fully depends on the p53/p21 pathway (Figure 3F).
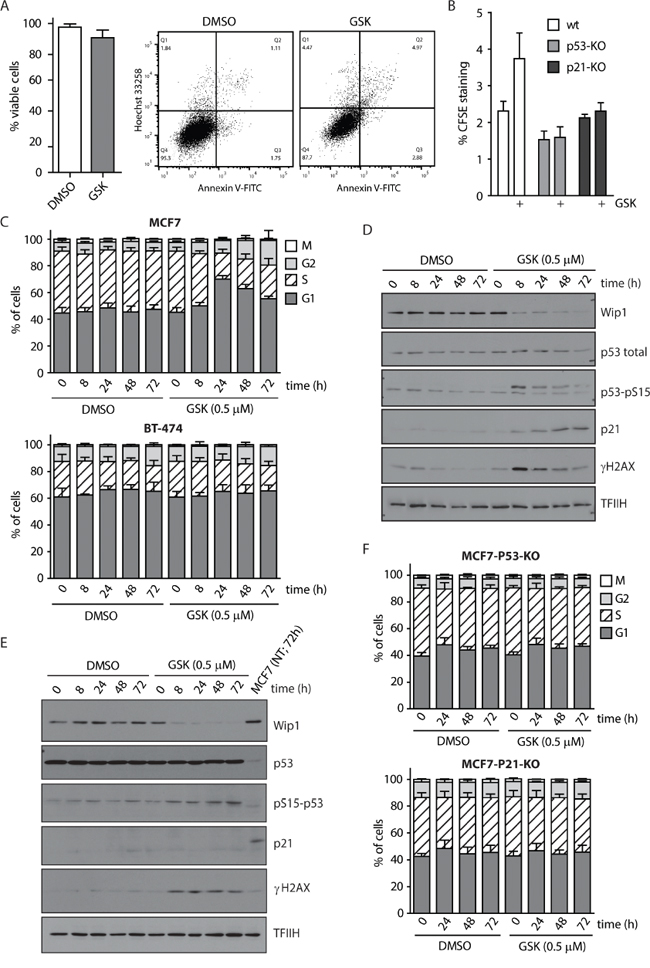
Figure 3: WIP1 inhibition leads to G1 and G2 phase accumulation in MCF7 cells. A. MCF7 cells were treated with DMSO or GSK2830371 (0.5 μM) for 5 days and percentage of living cells (Hoechst/Annexin V negative) was determined by flow cytometry. Error bars represent SD. B. MCF7, MCF7-P53-KO or MCF7-P21-KO cells were incubated with CFSE and subsequently treated with DMSO or GSK2830371 (0.5 μM) for 3 days. Fluorescent signal of CFSE was measured by flow cytometry. Plotted is the CFSE signal relative to the signal measured at day 0. Error bars represent SD. C. MCF7 or BT-474 cells were treated with DMSO or GSK2830371 (0.5 μM) for indicated times, pulsed with BrdU before fixation and distribution of cell cycle phases was determined by flow cytometry. BrdU incorporation was used as a marker of replication and pS10-H3 as a marker of mitotic cells. Error bars represent SD. D. MCF7 cells were treated as in C and analyzed by immunoblotting. E. BT-474 cells were treated as in C and analyzed by immunoblotting. F. MCF7-P53-KO or MCF7-P21-KO cells were treated with DMSO or GSK2830371 (0.5 μM) for indicated times, pulsed with BrdU before fixation and distribution of cell cycle phases was determined as in C. Error bars represent SD.
WIP1 inhibition promotes DNA damage-induced checkpoint arrest
We have previously shown that WIP1 is required for recovery from the DNA damage-induced G2 checkpoint [17]. Therefore, we tested the effect of GSK2830371 inhibitor on the ability of MCF7 cells to establish the G2 checkpoint. Whereas about 70 % of the control cells progressed to mitosis at 20 h after exposure to ionizing radiation, cells treated with GSK2830371 remained arrested in the G2 (Figure 4A). It has been reported that normal diploid RPE cells do not require WIP1 activity for recovery from the G1 checkpoint [18]. In the same time, C-terminally truncated WIP1 present in U2OS and HCT116 cells impairs activation of the G1 checkpoint [39]. To determine the contribution of the overexpressed WIP1 in suppression of the G1 checkpoint in MCF7 cells we compared fractions of cells remaining in G1 after exposure to ionizing radiation. Following exposure to a low dose of ionizing radiation (3 Gy, IR), MCF7 cells treated with GSK2830371 showed stronger accumulation in the G1 checkpoint compared to untreated cells (Figure 4B). To test how long these effects of WIP1 inhibition can persist we followed MCF7 cells for 3 to 6 days after irradiation and treatment with GSK2830371. We have found that cells with inhibited WIP1 did not incorporate BrdU three days after irradiation and that a substantial fraction of cells was arrested in the G2 checkpoint (Figure 4C and 4D). At 6 days after irradiation, we noted a dramatically reduced growth of cells exposed to a low dose (3 Gy) of IR and GSK2830371 (Figure 4E and 4F). Comparably smaller differences were observed after high dose of IR (6 Gy) when similar fractions of cells remained arrested regardless of the activity of WIP1 (Figure 4E and 4F).
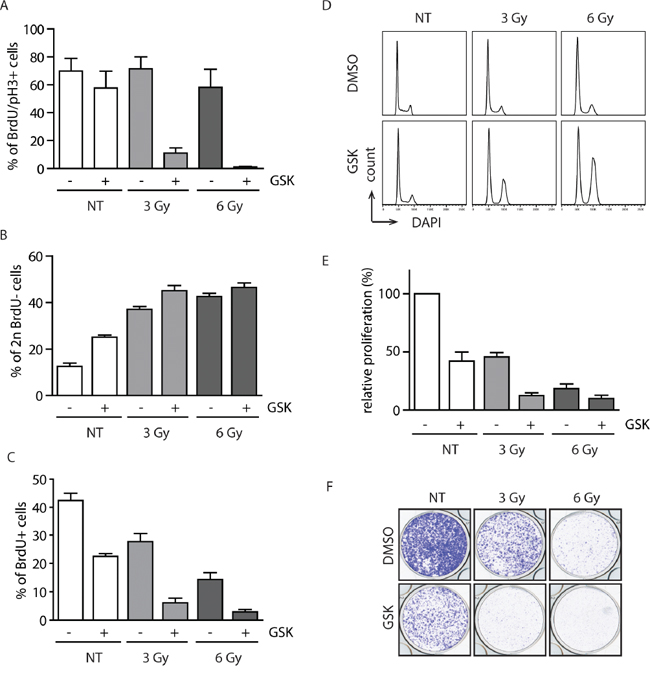
Figure 4: Inhibition of WIP1 potentiates the checkpoint through activation of the p53 pathway. A. MCF7 cells were pulsed with BrdU, treated with DMSO or GSK2830371 (0.5 μM) and exposed to IR. Cells were incubated in the presence of nocodazole and collected after 20 h. Fraction of BrdU positive cells that progressed to mitosis (pH3 marker) was determined by flow cytometry. Error bars represent SD. B. MCF7 cells were treated as in A. Fraction of BrdU negative cells with 2n DNA content (corresponding to G1) was determined by flow cytometry 20 h after treatment. Error bars represent SD. C, D. MCF7 cells were treated with DMSO or GSK2830371 (0.5 μM), exposed to IR and BrdU incorporation (C) or cell cycle profile (D) was determined after 3 days. Error bars represent SD. E. MCF7 cells were treated with DMSO or GSK2830371 (0.5 μM), exposed to IR and cell proliferation was analyzed after 6 days. Error bars represent SD. F. MCF7 cells were treated as in E and cell proliferation was determined by colony formation assay after 6 days. Representative image from three independent experiments is shown.
WIP1 inhibition sensitizes cells to genotoxic stress and to MDM2 inhibitor nutlin-3
Since we observed potentiation of the IR-induced checkpoint arrest after inhibition of WIP1 we decided to test the combination of GSK2830371 with various chemotherapeutics causing genotoxic stress. High dose of doxorubicin (0.5 μM) strongly suppressed proliferation of MCF7 cells, which is consistent with extensive DNA damage caused by inhibition of topoisomerase II (Figure 4A). In contrast, low dose of doxorubicin (0.05 μM) caused only mild activation of p53 pathway and was relatively well tolerated in MCF7 cells (Figure 5A and 5B). Combined treatment with doxorubicin (0.05 μM) and GSK2830371 increased activation of the p53 pathway and significantly reduced proliferation of MCF7 cells (Figure 5A and 5B). Similar potentiation was observed also in combination of GSK2830371 and low doses of etoposide and bleomycin (data not shown). Together with the observed response to ionizing radiation (Figure 4E and 4F) this suggests that loss of WIP1 activity can potentiate DNA damage response to the low level of genotoxic stress whereas extensive DNA damage can trigger activation of this signaling cascade leading to a sustained growth arrest despite high expression levels of WIP1 present in MCF7 cells.
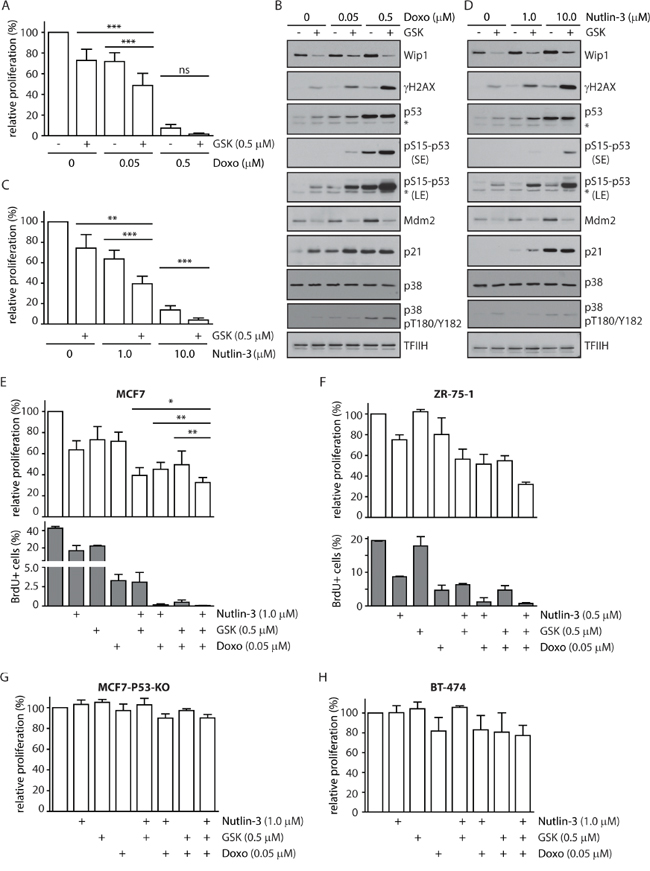
Figure 5: Inhibition of WIP1 increases sensitivity of cells to DNA damage and to nutlin-3. A. MCF7 cells were incubated with indicated doses of doxorubicin in combination with DMSO or GSK2830371 and relative fraction of proliferating cells was determined after 3 days. Error bars represent SD. B. MCF7 cells were incubated as in A and analysed by immunoblotting. Staining for TFIIH was used as loading control. Asterisk indicates an unspecific reactivity band. Short exposure (SE) or long exposure (LE) is shown. C. MCF7 cells were incubated with indicated doses of nutlin-3 in combination with DMSO or GSK2830371 and relative fraction of proliferating cells was determined after 3 days. Error bars represent SD. D. MCF7 cells were incubated with indicated doses of nutlin-3 and GSK2830371 for 1 day and analysed by immunoblotting. Staining for TFIIH was used as loading control. Asterisk indicates an unspecific reactivity band. Short exposure (SE) or long exposure (LE) is shown. E. MCF7 cells were incubated for 3 days with indicated doses of doxorubicin, nutlin-3 and GSK2830371 and fraction of proliferating cells was determined by cell survival assay (top) or by incorporation of BrdU (bottom). Error bars represent SD. F. ZR-75-1 cells were incubated for 6 days with indicated doses of doxorubicin, nutlin-3 and GSK2830371 and fraction of proliferating cells was determined by cell proliferation assay (top) or by incorporation of BrdU (bottom). Error bars represent SD. G. MCF7-P53-KO cells were incubated with indicated doses of doxorubicin, nutlin-3 and GSK2830371 and relative fraction of proliferating cells was determined after 3 days. Error bars represent SD. H. BT-474 cells were incubated with indicated doses of doxorubicin, nutlin-3 and GSK2830371 and relative fraction of proliferating cells was determined after 6 days. Error bars represent SD.
Transcriptional activity of the tumor suppressor p53 is regulated at multiple levels, including extensive phosphorylation in the transactivation and oligomerization domains and MDM2-dependent ubiquitination and degradation [67, 68]. Since inhibition of WIP1 increases phosphorylation of p53 at Ser15, we decided to test whether GSK2830371 could potentiate the effect of an MDM2 antagonist nutlin-3 that increases the total level of p53 [46]. As expected, treatment with high dose of nutlin-3 (10 μM) strongly suppressed cell proliferation of MCF7 cells (Figure 5C). Low dose of nutlin-3 (1 μM) showed an intermediate effect on cell proliferation of MCF7 cells that was further enhanced by simultaneous inhibition of WIP1 (Figure 5C). Consistent with an expected mode of action, we observed increased levels of total p53 after treatment with nutlin-3, increased phosphorylation of p53 at Ser15 after treatment with GSK2830371 and both effects after combined treatment with both inhibitors (Figure 5D). Efficient inhibition of WIP1 is documented by increased basal phosphorylation of γH2AX which is an established substrate of WIP1 and also by decreased levels of MDM2 which is destabilized in the absence of WIP1 activity (Figure 5B and 5D) [14, 20, 22]. Although inhibition of WIP1 slightly increased the basal phosphorylation of p38 at Thr180/Tyr182 (established substrate of WIP1), we did not observe any further increase of p38 activity in combination of GSK2830371 with doxorubicin or nutlin (Figure 5B and 5D). This suggests that p38 does not potentiate the cytotoxic effect of WIP1 and WIP1 impacts on p53 independently on the p38 pathway.
Finally, we tested the potentiation of the cytostatic effect by combining the GSK2830371 with low doses of nutlin-3 and doxorubicin. We found that this triple combination further decreased cell proliferation of MCF7 cells compared to treatments with individual drugs or with the double inhibitor combinations (Figure 5E). Triple combination of GSK2830371, nutlin-3 and doxorubicin also potentiated the cytostatic effect in ZR-75-1 cells that contain amplification of the PPM1D locus and harbour wild-type p53 (Figure 5F). In contrast no potentiation was observed in BT-474 and MCF7-P53-KO cells strongly indicating that status of p53 plays a key role in determining the cell sensitivity to WIP1 inhibition (Figure 5G and 5H).
Inhibition of WIP1 potentiates activation of p53 pathway
To quantify activation of the p53 pathway after treatment of MCF7 cells with combination of WIP1 inhibitor and chemotherapeutics we analyzed the expression profiles of selected established p53 target genes. As expected, expression of CDKN1A increased 3-5 fold after treatment with GSK2830371, nutlin-3 or doxorubicin administered individually (Figure 6A). Double combination of GSK2830371 with nutlin-3 or doxorubicin resulted in approximately 20 fold increase in CDKN1 expression. The highest induction of CDKN1A expression (about 50 fold) was observed after triple combination of GSK2830371, nutlin-3 and doxorubicin. Similarly, expression of p53 up-regulated modulator of apoptosis (PUMA) or pro-apoptotic regulator BAX showed the strongest induction after triple combination of GSK2830371, nutlin-3 and doxorubicin. In contrast, we did not observe any significant change in expression of an apoptosis-promoting gene NOXA. Inversely, we observed a strongly reduced expression of BIRC5 (coding for survivin), an anti-apoptotic gene that was reported to be suppressed in a p53-dependent manner [69, 70]. In addition, we have found strongly increased expression of PPM1D and MDM2 after triple combination of GSK2830371, nutlin-3 and doxorubicin, which is consistent with the described transcriptional regulation of both genes by p53. Although expression of PPM1D mRNA was increased after triple combination of the drugs, protein levels of WIP1 were decreased (Figure 6B) due to the destabilization of WIP1 caused by binding of GSK2830371 to its catalytic domain [63]. After 3 days of GSK2830371 treatment we did not observe increased total levels of p53; however p53 was heavily phosphorylated at Ser15 known to stimulate its transcriptional activity [11].
Inhibition of WIP1 promotes induction of senescence and apoptosis
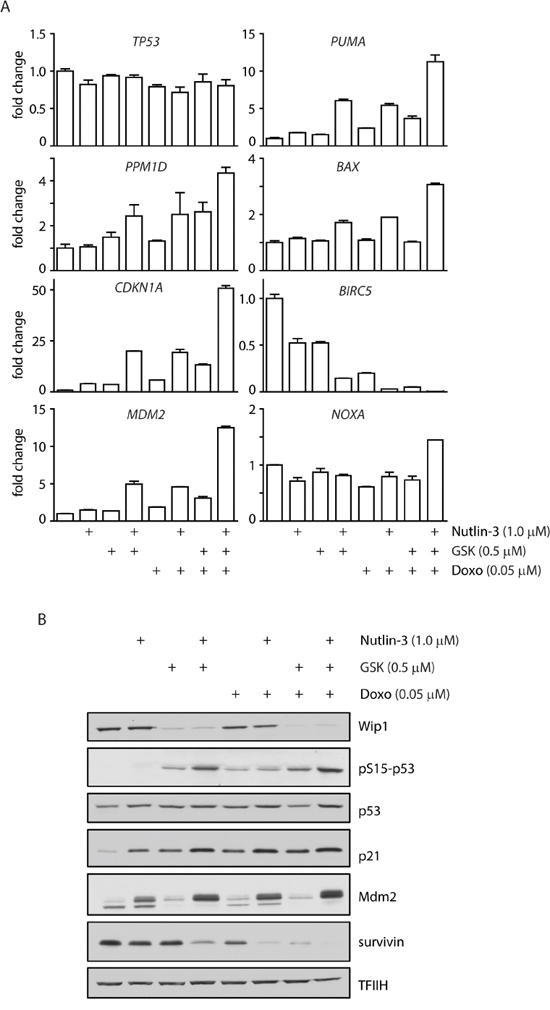
Figure 6: Inhibition of WIP1 increases transcription of p53 target genes. A. MCF7 cells were incubated for 3 days with indicated doses of doxorubicin, nutlin-3 and GSK2830371 and expression of indicated genes was determined by qRT-PCR. Levels are presented as the ratio of mRNA to GAPDH mRNA and are normalized to untreated cells. Error bars correspond to SEM. B. MCF7 cells were incubated as in A and expression of selected proteins was analysed by immunoblotting.
Since the expression profiling showed induction of the checkpoint and pro-apoptotic genes, we asked what the fate of cells treated with WIP1 inhibitor alone or in combination with other chemotherapeutics was. Although, cell proliferation was suppressed in MCF7 cells treated with GSK2830371, we observed only mild reduction in the fraction of viable cells compared to the control cells (Figure 3A). In contrast, GSK2830371 significantly decreased viability of MCF7 cells when administered concomitantly with a high dose of doxorubicin (0.5 μM) while having only mild effect when administered together with low dose of doxorubicin (0.05 μM) (Figure 7A, 7B). Similarly, GSK2830371 decreased viability of MCF7 cells treated with a high dose of nutlin-3 (10.0 μM) (Figure 7B). Consistent with a previous report, nutlin-3 increased sensitivity of cells to the low dose of doxorubicin (0.05 μM) [71]. Moreover, we have observed that GSK2830371 further increased the sensitivity of MCF7 cells to a combined treatment with nutlin-3 and doxorubicin (Figure 7B). This suggests that inhibition of WIP1 can potentiate cytotoxic effects of doxorubicin and the MDM2 antagonist nutlin-3. In addition, we observed induction of caspase 9 activity after combined treatment with GSK2830371, nutlin-3 and doxorubicin which is consistent with activation of an intrinsic apoptotic pathway (Figure 7C) [72].
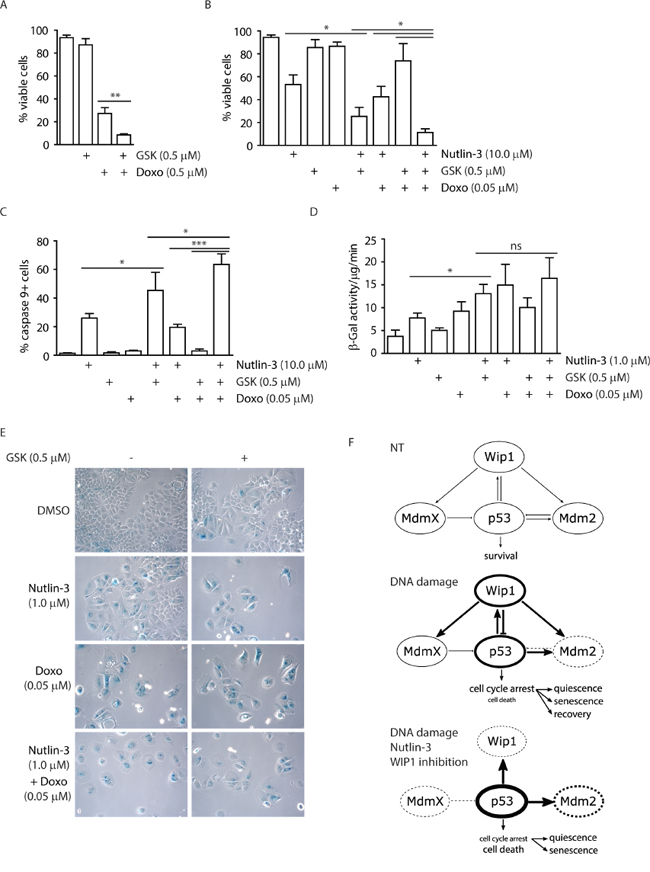
Figure 7: Inhibition of WIP1 potentiates induction of senescence or apoptosis. A. MCF7 cells were incubated with GSK2830371 (0.5 μM) and doxorubicin (0.5 μM) for 3 days and fraction of viable cells (Hoechst/Annexin V negative) was determined by flow cytometry. Error bars represent SD. B. MCF7 cells were incubated with indicated combinations of GSK2830371 (0.5 μM), nutlin-3 (10.0 μM) and doxorubicin (0.05 μM) for 3 days and fraction of viable cells (Hoechst/Annexin V negative) was determined by flow cytometry. Error bars represent SD. C. MCF7 cells were treated as in (B) and fraction of cells with active caspase 9 was determined by flow cytometry. Error bars represent SD. D. MCF7 cells were incubated with indicated combinations of GSK2830371, nutlin-3 (1.0 μM) and doxorubicin (0.05 μM) for 7 days. Activity of SA-β-galactosidase was measured in cell extracts using fluorimetric assay. Error bars represent SD. E. MCF7 cells were incubated as in D and SA-β-galactosidase activity was evaluated by light microscopy. F. Model for outcomes of treatment with p53/mdm2/Wip1 pathway modulators. Under non-treated conditions, p53 activity is tightly controlled by MDM2 and MDMX. Upon mild DNA damage, MDM2 is inhibited and destabilized leading to stabilization of p53 that in turn leads to increased transcription of its targets including WIP1 phosphatase. Subsequently WIP1 inactivates p53 pathway by direct dephosphorylation of p53 Ser15 and through activation of MDM2 and possibly also MDMX by their dephosphorylation. When MDM2-p53 interaction inhibitor nutlin-3 and WIP1 inhibitor are combined with DNA damage, MDM2 cannot ubiquitinate and thus degrade p53 and WIP1 cannot oppose activation of p53. This leads to further increase of p53 protein levels and its phosphorylation at Ser15 and results mainly in cell death. Thickness of the circle lines represents protein levels; dashed lines mean inhibition of the protein activity.
Whereas combination of the high dose of nutlin-3 and GSK2830371 efficiently induced cell death, most cells survived treatment with the low dose of nutlin-3. Since these cells did not incorporate BrdU (Figure 5E), we hypothesized that they corresponded to the population of cells permanently withdrawn from the cell cycle. Indeed, MCF7 cells treated with GSK2830371 and 1.0 μM nutlin-3 exhibited flattened and enlarged morphology; and showed induction of β-galactosidase activity, both established markers of cellular senescence (Figure 7D and 7E) [73].
In summary, we have validated GSK2830371 as potent and specific inhibitor of WIP1 phosphatase. Our data suggest that mild activation of p53 pathway caused by a partial stabilization (through low levels of nutlin-3) or phosphorylation of p53 (through inhibition of WIP1) is sufficient to slow down proliferation and eventually promotes cellular senescence. Conversely, full activation of p53 pathway achieved by combined effects of genotoxic stress with inhibition of two negative regulators of p53, MDM2 and WIP1 can potentiate cell death in breast cancer cells (Figure 7F).
DISCUSSION
Taking advantage of the U2OS cells with knocked-out PPM1D, we compared effects of the two commercially available inhibitors of WIP1 phosphatase in a cellular model. Data presented here and also by others strongly suggest that CCT007093 compound suppresses the cell growth independently of WIP1 inhibition [59]. It is possible that CCT007093 stimulates the p38 pathway as originally reported, however caution should be taken when interpreting these effects as a result of WIP1 inhibition. In contrast, our cellular model confirmed the specificity of the novel allosteric inhibitor GSK2830371 that interfered with dephosphorylation of γH2AX (an established substrate of WIP1) and suppressed cell growth in a WIP1-dependent manner. Notably, an impact of GSK2830371 on activation of the DNA damage response pathway was comparable to that of the PPM1D knock out indicating that GSK2830371 can efficiently inhibit WIP1 in cells.
We have found that GSK2830371 administered at doses that specifically block WIP1 activity does not affect proliferation of nontransformed cells but impairs proliferation of breast cancer cells with amplified PPM1D. MCF7 cells treated with GSK2830371 accumulate over time in the G2 phase of the cell cycle. This observation is in good agreement with the higher ratio of the G2 cells reported in the population of PPM1D-/- MEFs compared to the wild type MEFs and also with the increased expression level of WIP1 during the G2 in human cells [66, 74]. Analyzis of the MCF7-P53-KO and MCF7-P21-KO cells has shown that this effect of WIP1 on the cell cycle progression is mediated by the p53/p21 pathway. Level of p21 present during G2 was recently identified as an important factor that determines the fate of proliferating cells [75, 76]. Low level of p21 in G2 allows immediate building up of the CDK2 activity following mitotic exit and results in continuous proliferation. In contrast, cells with high level of p21 during G2 remain temporarily arrested in a quiescence after completing cell division and do not proliferate unless stimulated with excessive dose of growth factors [75]. It is plausible that these cells eventually become senescent after long period of sustained p21-dependent inhibition of cyclin dependent kinases. It appears that cells progressing through G2 phase are very sensitive to activation of the p53/p21 pathway. Indeed, short activation of p53 during G2 triggered nuclear retention and subsequent degradation of Cyclin B1 and was sufficient to induce a permanent withdrawal from the cell cycle [77, 78]. Here we have shown that inhibition of WIP1 potentiates an effect of a low dose of nutlin-3 resulting in increased induction of senescence in breast cancer cells.
Although GSK2830371 efficiently suppressed growth of breast cancer cells with amplified PPM1D and wild type TP53, it did not affect viability of MCF7 cells suggesting that inhibition of WIP1 alone may not be sufficient to eradicate tumor cells. On the other hand, we have found that inhibition of WIP1 by GSK2830371 potentiated doxorubicin-induced cell death in breast cancer cells. This data is consistent with previously reported high sensitivity of Wip1-depleted MCF7 cells to doxorubicin [79]. Similar potentiation of the cytotoxic effect of doxorubicin by WIP1 inhibition has recently been reported in neuroblastoma cells and in a colorectal carcinoma cells with a C-terminally truncated PPM1D [61, 64]. In addition, we have found that inhibition of WIP1 potentiated cell death induced by nutlin-3. Synergistic effect of nutlin-3 and doxorubicin has been reported in B-cell leukemia and in breast cancer cells [71, 80]. Here we show that combination of GSK2830371 with doxorubicin and nutlin-3 further increased activation of the p53 pathway and resulted in massive cell death. Clinical outcome of doxorubicin therapy can be impaired by induction of senescence in breast cancer cells with wild-type p53 [81, 82]. Strong induction of p53 function by concomitant inhibition of WIP1 and/or MDM2 could increase the fraction of cells eliminated by cell death and thus could improve the response to doxorubicin. In addition, therapeutic effect of doxorubicin is limited by a cumulative, dose-related cardiotoxicity [83]. Possible reduction of the doxorubicin dose administered in combination with WIP1 inhibitor could be beneficial for breast cancer patients by decreasing undesired side effects of chemotherapy.
WIP1 has been reported to directly target several proteins implicated in apoptosis (including BAX and RUNX2) in p53 negative cells [84–86]. However, suppression of cell growth and induction of cell death by WIP1 depletion or inhibition fully depends on the p53 pathway. In addition, inhibition of WIP1 efficiently affects growth of cells with amplified or truncated PPM1D whereas little effect is observed in cells with normal levels of WIP1. This suggests that determination of the status of TP53 and PPM1D in the tumors will be important for predicting the therapeutical outcome of WIP1 inhibitors. Further research is needed to identify additional factors determining the sensitivity of cancer cells to WIP1 inhibitors. Response of cancer cells to nutlin-3 depends on the level of MDM2 and is commonly impaired by overexpression of MDMX [71, 87, 88]. Since GSK2830371 potentiates the cytotoxic effect of nutlin-3, we hypothesize that MDMX overexpressing tumors might be attractive candidates for testing the sensitivity to WIP1 inhibition.
MATERIALS AND METHODS
Cell lines
Human osteosarcoma U2OS and breast cancer MCF7 cells were generous gifts from Dr. Medema (NKI, Amsterdam), BT474 from Dr. Truksa (IBT, Prague), CAL-51 and BJ fibroblasts (population doubling 40-50) from Dr. Bartek (IMG, Prague). ZR-75-1 cells were obtained from European Collection of Cell Cultures, hTERT-RPE1 from ATCC and human SV40-immortalized colon epithelia HCE cells from Applied Biological Materials (ABM, #T0570). Cells were grown at 37°C and 5% CO2 in DMEM, RPMI (ZR-75-1 and BT-474) or Prigrow III media (HCE cells) supplemented with 6-10% FBS (Gibco), penicillin (100 U/ml), and streptomycin (0.1 mg/ml). All cell lines were regularly checked for absence of mycoplasma infection using MycoAlert Plus reagent (Lonza). To knock-out TP53 or CDKN1A gene, MCF7 cells were transfected with a combination (1:1) of p53 CRISPR/Cas9 KO Plasmid (Santa Cruz, sc-416469) or p21 CRISPR/Cas9 KO Plasmid (sc-400013) and corresponding HDR Plasmids and stable clones were selected by puromycin (10 μg/ml). Integration of the HDR cassette to genomic loci was confirmed by sequencing and loss of protein expression by immunoblotting. To generate PPM1D knock-out cells, U2OS cells were transfected with a CAS9-2A-GFP plasmid expressing the gRNA corresponding to the tgagcgtcttctccgaccaggg sequence in exon 1 of the human PPM1D (Sigma). Individual GFP positive clones were expanded and loss of WIP1 expression was determined by immunoblotting. Transfection of plasmid DNA was performed using Lipofectamine LTX according to recommendations of manufacturer (Life Technologies). Where indicated, cells grown on culture plates were exposed to ionizing radiation generated by X-ray instrument T-200 (16.5 Gy/min, Wolf-Medizintechnik).
Antibodies and chemicals
The following antibodies were used: WIP1 (sc-130655), p53 (sc-6243), TFIIH (sc-293), importin (sc-137016), p21 (sc-397) from Santa Cruz; pSer15-p53 (#9284), γH2AX (#9718), p38 MAPK Thr180/Tyr182 (#9216S) and p38 MAPK (#9212) from Cell Signaling Technology); γH2AX (05-636, Millipore); MDM2 (Calbiochem); Alexa Fluor-labelled secondary antibodies (Life Technologies); anti-BrdU FITC-conjugated antibody (#347583, BD Biosciences) and anti-pSer10-H3 antibody (Upstate). Doxorubicin hydrochloride (Sigma), GSK2830371 and nutlin-3 (both MedChem Express) were diluted in DMSO and used at indicated doses. Resazurin, neocarzinostatin (NCS) and carboxyfluorescein diacetate succinimidyl ester (CFSE) were purchased from Sigma.
Cell proliferation assay
MCF7 or BT-474 cells were seeded into 96-well plates at 2x103 or 0.5x103 cells/well, treated with a compound dilution series and analyzed after 3 or 7 days, respectively. CAL-51, RPE, HCE, BJ or ZR-75-1 cells were seeded into 96 well plates at 0.02-2x104 cells/well and grown for 7 days. Resazurin (30 μg/mL) was added to growth media and fluorescence signal (excitation wavelength 560 nm, emission wavelength 590 nm) was measured after 1 to 5 h using EnVision plate reader (PerkinElmer).
Alternatively, rate of cell proliferation was determined using CFSE Cell Proliferation assay as previously described [89]. Cells were stained with 50 μM CFSE in complete media for 15 min in 37°C, washed with complete media and seeded to 12-well plate at 2.5x104 cells/well. Where indicated, GSK2830371 (0.5 μM) was added to the media. Cells were harvested and fixed by 4% paraformaldehyde 3 days after treatment. Percentage of the remaining CFSE staining compared to the cells harvested immediately after staining was determined by flow cytometry.
Clonogenic assay
Cells were seeded in 6-well plates at 2x104 cells/well. Cells were treated with a compound dilution series on day 1. After 6-7 days, cells were washed with PBS, fixed by 70 % ethanol for 15 min and stained with crystal violet dye.
Cell cycle assay
Cells were grown for indicated times in the presence of DMSO or GSK2830371 (0.5 μM), pulsed with BrdU (10 μM for 30 min; Sigma), harvested by trypsinization and fixed in ice-cold 70 % ethanol. Following the protocol from manufacturer, cells were stained with anti-BrdU-FITC (replication marker, BD Biosciences), anti-pSer10H3 (mitotic marker) and DAPI and analyzed by flow cytometry using LSRII (BD Biosciences) and FlowJo software (FlowJo).
Checkpoint analysis
Evaluation of the cell cycle checkpoints was performed as described previously with minor modifications [39]. Cells were pulsed with BrdU (10 μM for 30 min) and treated with GSK2830371 (0.5 μM) or DMSO before irradiation with 3 or 6 Gy and were grown for further 20 h in the presence of nocodazole (250 ng/ml). Cells were processed as mentioned above and analyzed by flow cytometry. BrdU-positive cells were assayed for progression through the G2 phase to mitosis (4n DNA content, pH3+). BrdU-negative cells with 2n content were used for quantification of cells arrested in G1 checkpoint.
Cell viability assay
MCF7 cells were seeded into 12-well plates at 2x104 cells/well, treated with Nutlin-3 (10 μM or 1 μM), GSK2830371 (0.5 μM) and doxorubicin (0.05 μM or 0.05 μM) and grown for 3 days. Cells were trypsinized, washed with PBS and incubated with FITC-conjugated Annexin V (BD Biosciences) and Hoechst-33258 for 15 minutes. Fraction of living cells was determined as Annexin V negative and Hoechst negative population analyzed by flow cytometry.
β-galactosidase assay
Senescence-associated β-galactosidase activity was quantified in cell extracts as previously described [90]. Briefly, MCF7 cells were seeded into 6 cm plates at 0.5x105 cells/plate and grown in media supplemented with indicated combinations of nutlin-3 (1 μM), GSK2830371 (0.5 μM) and doxorubicin (0.05 μM) for 7 days. Cells were washed in PBS, collected to ice cold lysis buffer (5 mM CHAPS, 40 mM citric acid, 40 mM sodium phosphate, 0.5 μM benzamidine and 0.25 mM PMSF, pH 6.0), vortexed and centrifuged for 5 min at 12,000g. Cell extract was mixed 1:1 with 2x reaction buffer supplemented with 4-MUG (1.7 mM, Sigma) and MgCl2 (4 mM) and incubated at 37°C for 0.5 – 4 hours. Reaction was stopped by addition of sodium carbonate (400 mM) and fluorescence signal was measured at excitation wavelength 360 nm and emission wavelength 465 nm using EnVision plate reader. β-galactosidase activity was determined as the rate of 4-MUG conversion to the fluorescent 4-MU and normalized to the protein concentration measured by BCA assay. Alternatively, cells were grown on coverslips, fixed by 0.2 % glutaraldehyde 7 days after treatment with indicated combinations of nutlin-3 (1 μM), GSK2830371 (0.5 μM) and doxorubicin (0.05 μM) and β-galactosidase activity was determined by colorimetric staining as previously [73].
Caspase assay
Activity of caspase-9 was measured using SR-FLICA Caspase-9 assay according to manufacturer protocol (Immunochemistry Technologies). Briefly, cells were seeded to 12-well plates, treated as indicated, harvested by trypsinization after 48 h and re-suspended in complete media containing SR-FLICA caspase-9 and incubated 1 h at 37°C. After incubation, cells were washed with Apoptosis wash buffer for 10 min at 37°C. Percentage of cells positive for caspase-9 activity was determined by flow cytometry.
Immunofluorescence microscopy
U2OS cells grown on coverslips were treated with DMSO, CCT007093 or GSK2830371 for 1 h and DNA damage was induced by neocarzinostatin for 5 h. Cells were fixed by 4 % formaldehyde (10 min at RT), permeabilized by ice-cold methanol and stained with antibody against γH2AX and with DAPI. Average nuclear intensity of γH2AX signal was quantified using Scan^R high-content screening station as described previously [66].
Quantitative real-time PCR (qPCR)
Total RNA was isolated using RNeasy mini kit (Qiagen). cDNA was synthetized using 0.5 μg RNA, random hexamer, and RevertAid H Minus Reverse Transcriptase (Thermo Scientific). RT-qPCR was performed using LightCycler 480 SYBR Green I Master mix; Light Cycler LC480 (Roche) and following cycle conditions: initial denaturation 95°C for 7 min, followed by 45 cycles of denaturation 95°C for 15 s, annealing 60°C for 15s and extension 72°C for 15s. A melting curve analysis was used to confirm the specificity of amplification, and Ct values were determined using LigtCycler480 software. All data are presented as the ratio of the tested mRNA to GAPDH mRNA. Primers are listed in the table.
Gene |
Forward sequence |
Reverse sequence |
|---|---|---|
PPM1D |
CTGAACCTGACTGACAGCCC |
CTTGGCCATGGATCCTCCTC |
BIRC5 |
CTGCCTGGTCCCAGAGTG |
GTGGCACCAGGGAATAAACC |
MDM2 |
TCGACCTAAAAATGGTTGCAT |
GGCAGGGCTTATTCCTTTTC |
PUMA |
TCTCGGTGCTCCTTCACTCT |
ACGTTTGGCTCATTTGCTCT |
BAX |
GCTGGACATTGGACTTCCTC |
GTCTTGGATCCAGCCCAAC |
CDKN1A |
GGCGGCAGACCAGCATGACA |
CCTCGCGCTTCCAGGACTGC |
TP53 |
CAGCACATGACGGAGGTTGT |
TCATCCAAATACTCCACACGC |
NOXA |
GCTGGGGAGAAACAGTTCAG |
AATGTGCTGAGTTGGCACTG |
Statistical analysis
Statistical analysis was performed in GraphPad Prism 5.04 software. Statistical significance was determined from at least three independent experiments using a paired two-tailed T-test (* corresponds to p-value < 0.05; ** p-value < 0.005; *** p-value < 0.0005). Error bars indicate standard deviations. EC50 was calculated using Richard’s five-parameter dose-response curve for non-linear fitting analysis.
ACKNOWLEDGMENTS
We are thankful to Rene Medema (NKI, Amsterdam), Jaroslav Truksa (IBT, Prague) and Jiri Bartek (IMG, Prague) for providing essential reagents.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This work has been supported by the Grant Agency of the Czech Republic (P305-12-2485), Worldwide Cancer Research (14-1176) and IMG ASCR (RVO: 68378050).
REFERENCES
1. Jackson SP and Bartek J. The DNA-damage response in human biology and disease. Nature. 2009; 461: 1071-1078.
2. Halazonetis TD, Gorgoulis VG, and Bartek J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science. 2008; 319:1352-1355.
3. Leontieva O, Gudkov A, and Blagosklonny M. Weak p53 permits senescence during cell cycle arrest. Cell Cycle. 2010; 9: 4323-7.
4. Kracikova M, Akiri G, George A, Sachidanandam R, and Aaronson SA. A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death and Differentiation. 2013; 20:576-588.
5. Zhang X-P, Liu F, Cheng Z, and Wang W. Cell fate decision mediated by p53 pulses. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106:12245-12250.
6. Pei D, Zhang Y, and Zheng J. Regulation of p53: a collaboration between Mdm2 and MdmX. Oncotarget. 2012; 3:228-235. doi: 10.18632/oncotarget.443.
7. Popowicz G, Czarna A, Rothweiler U, Szwagierczak A, Krajewski M, Weber L, and Holak T. Molecular basis for the inhibition of p53 by Mdmx. Cell Cycle. 2007; 6:2386-92.
8. Meulmeester E, Pereg Y, Shiloh Y, and Jochemsen A. ATM-mediated phosphorylations inhibit Mdmx/Mdm2 stabilization by HAUSP in favor of p53 activation. Cell Cycle. 2005; 4:1166-70.
9. Shieh SY, Ikeda M, Taya Y, and Prives C. DNA Damage-Induced Phosphorylation of p53 Alleviates Inhibition by MDM2. Cell. 1997; 91:325-334.
10. Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh S-Y, Taya Y, Prives C, and Abraham RT. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes & Development. 1999; 13:152-157.
11. Jenkins LMM, Durell SR, Mazur SJ, and Appella E. p53 N-terminal phosphorylation: a defining layer of complex regulation. Carcinogenesis. 2012; 33:1441-1449.
12. Fiscella M, Zhang H, Fan S, Sakaguchi K, Shen S, Mercer WE, Vande Woude GF, O Connor PM, and Appella E. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci USA. 1997; 94:6048-6053.
13. Lu X, Nannenga B, and Donehower L. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes & Development. 2005; 19:1162-74.
14. Lu X, Ma O, Nguyen T-A, Jones SN, Oren M, and Donehower LA. The Wip1 Phosphatase Acts as a Gatekeeper in the p53-Mdm2 Autoregulatory Loop. Cancer Cell. 2007; 12:342-354.
15. Lu X, Nguyen T, Zhang X, and Donehower L. The Wip1 phosphatase and Mdm2: cracking the “Wip” on p53 stability. Cell Cycle. 2008; 7:164-168.
16. Zhang X, Lin L, Guo H, Yang J, Jones SN, Jochemsen A, and Lu X. Phosphorylation and Degradation of MdmX Is Inhibited by Wip1 Phosphatase in the DNA Damage Response. Cancer Research. 2009; 69:7960-7968.
17. Lindqvist A, de Bruijn M, Macurek L, Bras A, Mensinga A, and Bruinsma W. Wip1 confers G2 checkpoint recovery competence by counteracting p53-dependent transcriptional repression. EMBO J. 2009; 28:3196-3206.
18. Shaltiel IA, Aprelia M, Saurin AT, Chowdhury D, Kops GJPL, Voest EE, and Medema RH. Distinct phosphatases antagonize the p53 response in different phases of the cell cycle. PNAS. 2014; 111:7313-7318.
19. Shreeram S, Demidov ON, Hee WK, Yamaguchi H, Onishi N, Kek C, Timofeev ON, Dudgeon C, Fornace AJ, Anderson CW, Minami Y, Appella E, and Bulavin DV. Wip1 Phosphatase Modulates ATM-Dependent Signaling Pathways. Mol. Cell. 2006; 23:757-764.
20. Macurek L, Lindqvist A, Voets O, Kool J, Vos H, and Medema R. Wip1 phosphatase is associated with chromatin and dephosphorylates gammaH2AX to promote checkpoint inhibition. Oncogene. 2010; 15;29:2281-91.
21. Le Guezennec X and Bulavin DV. WIP1 phosphatase at the crossroads of cancer and aging. Trends in Biochemical Sciences. 2010; 35:109-114.
22. Moon S, Lin L, Zhang X, Nguyen T, Darlington Y, Waldman A, Lu X, and LA. D. Wildtype p53-induced phosphatase 1 dephosphorylates histone variant gamma-H2AX and suppresses DNA double strand break repair. J Biol Chem. 2010; 23:285:12935-47.
23. Takekawa M, Adachi M, Nakahata A, Nakayama I, Itoh F, Tsukuda H, Taya Y, and Imai K. p53-inducible Wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J., 2000; 19:6517-6526.
24. Fujimoto H, Onishi N, Kato N, Takekawa M, Xu X, and Kosugi A. Regulation of the antioncogenic Chk2 kinase by the oncogenic Wip1 phosphatase. Cell Death Differ. 2006; 13:1170-1180.
25. Lee JS, Lee MO, Moon BH, Shim SH, Fornace AJ, and Cha H-J. Senescent Growth Arrest in Mesenchymal Stem Cells Is Bypassed by Wip1-Mediated Downregulation of Intrinsic Stress Signaling Pathways. STEM CELLS. 2009; 27:1963-1975.
26. Sakai H, Fujigaki H, Mazur SJ, and Appella E. Wild-type p53-induced phosphatase 1 (Wip1) forestalls cellular premature senescence at physiological oxygen levels by regulating DNA damage response signaling during DNA replication. Cell Cycle. 2014; 13:1015-1029.
27. Satoh N, Maniwa Y, Bermudez VP, Nishimura K, Nishio W, Yoshimura M, Okita Y, Ohbayashi C, Hurwitz J, and Hayashi Y. Oncogenic phosphatase Wip1 is a novel prognostic marker for lung adenocarcinoma patient survival. Cancer Science. 2011; 102:1101-6.
28. Castellino R, De Bortoli M, Lu X, Moon S-H, Nguyen T-A, Shepard M, Rao P, Donehower L, and Kim J. Medulloblastomas overexpress the p53-inactivating oncogene WIP1/PPM1D. J. Neurooncol., 2008; 86:245-256.
29. Li J, Yang Y, Peng Y, Austin RJ, van Eyndhoven WG, Nguyen KCQ, Gabriele T, McCurrach ME, Marks JR, Hoey T, Lowe SW, and Powers S. Oncogenic properties of PPM1D located within a breast cancer amplification epicenter at 17q23. Nat Genet. 2002; 31:133-134.
30. Saito-Ohara F, Imoto I, Inoue J, Hosoi H, Nakagawara A, Sugimoto T, and Inazawa J. PPM1D Is a Potential Target for 17q Gain in Neuroblastoma. Cancer Res. 2003; 63:1876-1883.
31. Bulavin DV, Demidov ON, Saito Si, Kauraniemi P, Phillips C, Amundson SA, Ambrosino C, Sauter G, Nebreda AR, Anderson CW, Kallioniemi A, Fornace AJ, and Appella E. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat Genet. 2002; 31:210-215.
32. Tan DSP, Iravani M, McCluggage WG, Lambros MB, Milanezi F, Mackay A, Gourley C, Geyer FC, Vatcheva R, Millar J, Thomas K, Natrajan R, Savage K, et al. Genomic Analysis Reveals the Molecular Heterogeneity of Ovarian Clear Cell Carcinomas. Clinical Cancer Research. 2011; 17:1521-1534.
33. Liang C, Guo E, Lu S, Wang S, Kang C, Chang L, Liu L, Zhang G, Wu Z, Zhao Z, Ma S, Wang L, and Jiao B-h. Over-expression of Wild-type p53-induced phosphatase 1 confers poor prognosis of patients with gliomas. Brain Research. 2012; 1444:65-75.
34. Rauta J, Alarmo E-L, Kauraniemi P, Karhu R, Kuukasjärvi T, and Kallioniemi A. The serine-threonine protein phosphatase PPM1D is frequently activated through amplification in aggressive primary breast tumours. Breast Cancer Research and Treatment. 2006; 95:257-263.
35. The Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumors. Nature. 2012; 490:61-70.
36. Emelyanov A and Bulavin DV. Wip1 phosphatase in breast cancer. Oncogene. 2015; 34:4429-4438.
37. Demidov ON, Kek C, Shreeram S, Timofeev O, Fornace AJ, Appella E, and Bulavin DV. The role of the MKK6//p38 MAPK pathway in Wip1-dependent regulation of ErbB2-driven mammary gland tumorigenesis. Oncogene. 2006; 26:2502-2506.
38. Ruark E, Snape K, Humburg P, Loveday C, Bajrami I, Brough R, Rodrigues DN, Renwick A, Seal S, Ramsay E, Duarte SDV, Rivas MA, Warren-Perry M, et al. Mosaic PPM1D mutations are associated with predisposition to breast and ovarian cancer. Nature. 2013; 493:406-410.
39. Kleiblova P, Shaltiel IA, Benada J, Sevčík J, Pecháčková S, Pohlreich P, Voest EE, Dundr P, Bartek J, Kleibl Z, Medema RH, and Macurek L. Gain-of-function mutations of PPM1D/Wip1 impair the p53-dependent G1 checkpoint. The Journal of Cell Biology. 2013; 201:511-521.
40. Dudgeon C, Shreeram S, Tanoue K, Mazur SJ, Sayadi A, Robinson RC, Appella E, and Bulavin DV. Genetic variants and mutations of PPM1D control the response to DNA damage. Cell Cycle. 2013; 12:2656-2664.
41. Hoe KK, Verma CS, and Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014; 13:217-236.
42. Brown CJ, Lain S, Verma CS, Fersht AR, and Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer. 2009; 9:862-873.
43. Dong P, Ihira K, Hamada J, Watari H, Yamada T, Hosaka M, Hanley SJB, Kudo M, and Sakuragi N. Reactivating p53 functions by suppressing its novel inhibitor iASPP: a potential therapeutic opportunity in p53 wild-type tumors. Oncotarget. 2015; 6:19968-75. doi: 10.18632/oncotarget.4847.
44. Qin J-J, Wang W, Voruganti S, Wang H, Zhang W-D, and Zhang R. Identification of a new class of natural product MDM2 inhibitor: In vitro and in vivo anti-breast cancer activities and target validation. Oncotarget. 2015; 6:2623-40. doi: 10.18632/oncotarget.3098.
45. Reed D, Shen Y, Shelat AA, Arnold LA, Ferreira AM, Zhu F, Mills N, Smithson DC, Regni CA, Bashford D, Cicero SA, Schulman BA, Jochemsen AG, et al. Identification and Characterization of the First Small Molecule Inhibitor of MDMX. Journal of Biological Chemistry. 2010; 285:10786-10796.
46. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, and Liu EA. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science. 2004; 303:844-848.
47. Ding Q, Zhang Z, Liu J-J, Jiang N, Zhang J, Ross TM, Chu X-J, Bartkovitz D, Podlaski F, Janson C, Tovar C, Filipovic ZM, Higgins B, et al. Discovery of RG7388, a Potent and Selective p53–MDM2 Inhibitor in Clinical Development. Journal of Medicinal Chemistry. 2013; 56:5979-5983.
48. Harrison M, Li J, Degenhardt Y, Hoey T, and Powers S. Wip1-deficient mice are resistant to common cancer genes. Trends Mol Med., 2004; 10:359-361.
49. Bulavin DV, Phillips C, Nannenga B, Timofeev O, Donehower LA, Anderson CW, Appella E, and Fornace AJ. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16Ink4a-p19Arf pathway. Nat Genet. 2004; 36:343-350.
50. Shreeram S, Hee WK, Demidov ON, Kek C, Yamaguchi H, Fornace AJ, Jr., Anderson CW, Appella E, and Bulavin DV. Regulation of ATM/p53-dependent suppression of myc-induced lymphomas by Wip1 phosphatase. J. Exp. Med., 2006; 203:2793-2799.
51. Demidov ON, Timofeev O, Lwin HNY, Kek C, Appella E, and Bulavin DV. Wip1 Phosphatase Regulates p53-Dependent Apoptosis of Stem Cells and Tumorigenesis in the Mouse Intestine. Cell Stem Cell. 2007; 1:180-190.
52. Demidov ON, Zhu Y, Kek C, Goloudina AR, Motoyama N, and Bulavin DV. Role of Gadd45a in Wip1-dependent regulation of intestinal tumorigenesis. Cell Death and Differentiation. 2012; 19:1761-1768.
53. Pärssinen J, Alarmo E-L, Karhu R, and Kallioniemi A. PPM1D silencing by RNA interference inhibits proliferation and induces apoptosis in breast cancer cell lines with wild-type p53. Cancer Genetics and Cytogenetics. 2008; 182:33-39.
54. Tan DSP, Lambros MBK, Rayter S, Natrajan R, Vatcheva R, Gao Q, Marchiò C, Geyer FC, Savage K, Parry S, Fenwick K, Tamber N, Mackay A, et al. PPM1D Is a Potential Therapeutic Target in Ovarian Clear Cell Carcinomas. Clinical Cancer Research. 2009; 15:2269-2280.
55. Wang P, Rao J, Yang H, Zhao H, and Yang L. PPM1D silencing by lentiviral-mediated RNA interference inhibits proliferation and invasion of human glioma cells. Journal of Huazhong University of Science and Technology. 2011; 31:94-99.
56. Yamaguchi H, Durell SR, Feng H, Bai Y, Anderson CW, and Appella E. Development of a Substrate-Based Cyclic Phosphopeptide Inhibitor of Protein Phosphatase 2Cδ, Wip1†. Biochemistry. 2006; 45:13193-13202.
57. Hayashi R, Tanoue K, Durell SR, Chatterjee DK, Miller Jenkins LM, Appella DH, and Appella E. Optimization of a Cyclic Peptide Inhibitor of Ser/Thr Phosphatase PPM1D (Wip1). Biochemistry. 2011; 50:4537-4549.
58. Rayter S, Elliott R, Travers J, Rowlands MG, Richardson TB, Boxall K, Jones K, Linardopoulos S, Workman P, Aherne W, Lord CJ, and Ashworth A. A chemical inhibitor of PPM1D that selectively kills cells overexpressing PPM1D. Oncogene. 2007; 27:1036-1044.
59. Lee JS, Park JR, Kwon OS, Kim H, Fornace AJ, and Cha HJ. Off-target response of a Wip1 chemical inhibitor in skin keratinocytes. Journal of Dermatological Science. 2014; 73:125-134.
60. Yagi H, Chuman Y, Kozakai Y, Imagawa T, Tkahashi Y, Yoshimura F, Tanino K, and Sakaguchi K. A small molecule inhibitor of p53-inducible protein phosphatase PPM1D. Bioorg Med Chem Lett. 2012; 22:729-32.
61. Kozakai Y, Kamada R, Kiyota Y, Yoshimura F, Tanino K, and Sakaguchi K. Inhibition of C-terminal truncated PPM1D enhances the effect of doxorubicin on cell viability in human colorectal carcinoma cell line. Bioorganic & Med. Chem. Lett. 2014; 24:5593-5596.
62. Ogasawara S, Kiyota Y, Chuman Y, Kowata A, Yoshimura F, Tanino K, Kamada R, and Sakaguchi K. Novel inhibitors targeting PPM1D phosphatase potently suppress cancer cell proliferation. Bioorganic & Medicinal Chemistry. 2015; 23:6246-6249.
63. Gilmartin AG, Faitg TH, Richter M, Groy A, Seefeld MA, Darcy MG, Peng X, Federowicz K, Yang J, Zhang S-Y, Minthorn E, Jaworski J-P, Schaber M, et al. Allosteric Wip1 phosphatase inhibition through flap-subdomain interaction. Nat Chem Biol. 2014; 10:181-187.
64. Richter M, Dayaram T, Gilmartin AG, Ganji G, Pemmasani SK, Van Der Key H, Shohet JM, Donehower LA, and Kumar R. WIP1 Phosphatase as a Potential Therapeutic Target in Neuroblastoma. PLoS ONE. 2015; 10:e0115635.
65. Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Research. 2015; 43:D805-D811.
66. Macurek L, Benada J, Müllers E, Halim VA, Krejčíková K, Burdová K, Pecháčková S, Hodný Z, Lindqvist A, Medema RH, and Bartek J. Downregulation of Wip1 phosphatase modulates the cellular threshold of DNA damage signaling in mitosis. Cell Cycle. 2013; 12:251-262.
67. Hock AK and Vousden KH. The role of ubiquitin modification in the regulation of p53. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2014; 1843:137-149.
68. Kruse J-P and Gu W. Modes of p53 Regulation. Cell. 2009; 137:609-622.
69. Mirza A, McGuirk M, Hockenberry T, Wu Q, Ashar H, Black S, Wen S, Wang L, Kirschmeier P, Bishop W, Nielsen L, Pickett C, and Liu S. Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene. 2002; 21:2613-2622.
70. Hoffman WH, Biade S, Zilfou JT, Chen J, and Murphy M. Transcriptional Repression of the Anti-apoptoticsurvivin Gene by Wild Type p53. Journal of Biological Chemistry. 2002; 277:3247-3257.
71. Lam S, Lodder K, Teunisse AFAS, Rabelink MJWE, Schutte M, and Jochemsen AG. Role of Mdm4 in drug sensitivity of breast cancer cells. Oncogene. 2010; 29:2415-2426.
72. Johnson C and Jarvis W. Caspase-9 regulation: an update. Apoptosis. 2004; 9:423-7.
73. Debacq-Chainiaux F, Erusalimsky JD, Campisi J, and Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protocols. 2009; 4:1798-1806.
74. Choi J, Nannenga B, Demidov O, Bulavin D, Cooney A, and Brayton C. Mice deficient for the wild-type p53-induced phosphatase gene (Wip1) exhibit defects in reproductive organs, immune function, and cell cycle control. Mol Cell Biol. 2002; 22:1094-1105.
75. Spencer Sabrina L, Cappell Steven D, Tsai F-C, Overton KW, Wang Clifford L, and Meyer T. The Proliferation-Quiescence Decision Is Controlled by a Bifurcation in CDK2 Activity at Mitotic Exit. Cell. 2013; 155:369-383.
76. Overton KW, Spencer SL, Noderer WL, Meyer T, and Wang CL. Basal p21 controls population heterogeneity in cycling and quiescent cell cycle states. Proceedings of the National Academy of Sciences. 2014; 111:E4386-E4393.
77. Krenning L, Feringa Femke M, Shaltiel Indra A, van den Berg J, and Medema René H. Transient Activation of p53 in G2 Phase Is Sufficient to Induce Senescence. Molecular Cell. 2014; 55:59-72.
78. Müllers E, Cascales HS, Jaiswal H, Saurin AT, and Lindqvist A. Nuclear translocation of Cyclin B1 marks the restriction point for terminal cell cycle exit in G2 phase. Cell Cycle. 2014; 13:2733-2743.
79. Kong W, Jiang X, and Mercer W. Downregulation of Wip-1 phosphatase expression in MCF-7 breast cancer cells enhances doxorubicin-induced apoptosis through p53-mediated transcriptional activation of Bax. Cancer Biol Ther. 2009; 8:555-63.
80. Coll-Mulet L, Iglesias-Serret D, Santidrián AF, Cosialls AM, de Frias M, Castaño E, Campàs C, Barragán M, de Sevilla AF, Domingo A, Vassilev LT, Pons G, and Gil J. MDM2 antagonists activate p53 and synergize with genotoxic drugs in B-cell chronic lymphocytic leukemia cells. Blood. 2006; 107:4109-4114.
81. Bertheau P, Lehmann-Che J, Varna M, Dumay A, Poirot B, Porcher R, Turpin E, Plassa L-F, de Roquancourt A, Bourstyn E, de Cremoux P, Janin A, Giacchetti S, et al. p53 in breast cancer subtypes and new insights into response to chemotherapy. The Breast. 2013; 22, Supplement 2:S27-S29.
82. Jackson JG, Pant V, Li Q, Chang LL, Quintás-Cardama A, Garza D, Tavana O, Yang P, Manshouri T, Li Y, El-Naggar AK, and Lozano G. p53 mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell. 2012; 21:793-806.
83. Rochette L, Guenancia C, Gudjoncik A, Hachet O, Zeller M, Cottin Y, and Vergely C. Anthracyclines/trastuzumab: new aspects of cardiotoxicity and molecular mechanisms. Trends in Pharmacological Sciences. 2015; 36:326-348.
84. Song JY, Ryu SH, Cho YM, Kim YS, Lee BM, Lee SW, and Choi J. Wip1 suppresses apoptotic cell death through direct dephosphorylation of BAX in response to γ-radiation. Cell Death & Disease. 2013; 4:e744.
85. Goloudina AR, Tanoue K, Hammann A, Fourmaux E, Le Guezennec X, Bulavin DV, Mazur SJ, Appella E, Garrido C, and Demidov ON. Wip1 promotes RUNX2-dependent apoptosis in p53-negative tumors and protects normal tissues during treatment with anticancer agents. Proceedings of the National Academy of Sciences. 2012; 109:E68–E75.
86. Goloudina AR, Mazur SJ, Appella E, Garrido C, and Demidov ON. Wip1 sensitizes p53-negative tumors to apoptosis by regulating the Bax/Bcl-xL ratio. Cell Cycle. 2012; 11:1883-1887.
87. Hu B, Gilkes DM, Farooqi B, Sebti SM, and Chen J. MDMX Overexpression Prevents p53 Activation by the MDM2 Inhibitor Nutlin. J Biol Chem. 2006; 281:33030-33035.
88. Xia M, Knezevic D, Tovar C, Huang B, Heimbrook DC, and Vassilev LT. Elevated MDM2 boosts the apoptotic activity of p53-MDM2 binding inhibitors by facilitating MDMX degradation. Cell Cycle. 2008; 7:1604-1612.
89. Quah BJC, Warren HS, and Parish CR. Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nat. Protocols. 2007; 2:2049-2056.
90. Gary RK and Kindell SM. Quantitative assay of senescence-associated β-galactosidase activity in mammalian cell extracts. Analytical Biochemistry. 2005; 343:329-334.