
INTRODUCTION
The majority of human melanomas express mutations in BRAF [1], and because of this mutation, much effort has gone into targeting BRAF and downstream pathways [2, 3]. New targeted therapies have been developed to inhibit BRAF signaling and downstream pathways. These drugs often produce dramatic responses, but unfortunately, these responses usually last for a few months. Some melanomas with BRAF mutations demonstrate intrinsic resistance, in that there is very little response even initially to BRAF inhibition. [4] Several mechanisms of resistance have already been described, including NRAS, MAP2K1, NF1, and BRAF amplification [5–8]. In addition, splicing mutations in BRAF have also been described [4]. Combination therapy of BRAF and MEK has led to the development of the MEK2Q60P mutation [9, 10]. Reactivation of ERK signaling appears to be a common thread in many forms of BRAF and MEK activation. Finally, many melanomas do not express BRAF mutations but have high levels of ERK activation [11, 12]. Thus, there is an unmet need for additional therapies to overcome resistance to targeted therapies.
Honokiol is a small molecular weight compound derived from the tree Magnolia grandiflora. We were the first to demonstrate antitumor activity of honokiol in vivo [13]. Since then, we have found honokiol has been found to inhibit ras signaling and induce endoplasmic reticulum stress in tumor cells in multiple tumor types [14, 15]. Most recently, we have demonstrated that honokiol is an inducer of the mitochondrial gene Sirt3 [16]. In order to make honokiol more lipophilic and patentable, we synthesized a honokiol prodrug, honokiol bis-dichloroacetate (honokiol DCA) (Figure 1). In addition, we synthesized a novel honokiol derivative, bis-trifluoromethyl-bis-(4-hydroxy-3-allylphenyl)methane (hexafluoro) (Figure 1). Both honokiol DCA and hexafluoro demonstrated in vivo activity against A375 melanoma in vivo. In order to determine whether honokiol DCA had activity against vemurafenib resistant melanoma, we assessed the ability of honokiol DCA in vivo against LM36, a BRAF mutant melanoma and LM36R, a vemurafenib resistant clone of LM36 [17]. Honokiol DCA showed significant activity against the vemurafenib resistant LM36R but not against the parental LM36 cells. Unexpectedly, honokiol DCA induces Akt phosphorylation in the honokiol DCA sensitive A375 and LM36R cells, but not in the LM36 cells which are resistant to honokiol DCA. In order to determine the mechanism of this difference, we examined the ability of our compounds to induce superoxide in the tumor cell lines, since superoxide is a well known inducer of Akt phosphorylation [18]. LM36R cells, which are sensitive to honokiol DCA, lack expression of the major superoxide detoxifying gene manganese superoxide dismutase, while LM36, which is resistant to honokiol DCA, expresses high levels of MnSOD. Given that elevated Akt and superoxide are major players in advanced melanoma, honokiol analogs may be useful in treating these highly resistant subsets of melanoma.
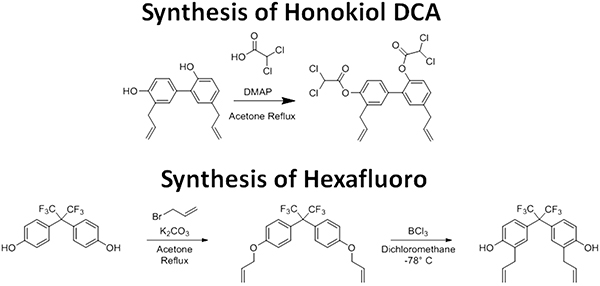
Figure 1: The synthetic scheme for Honokiol DCA and Hexafluoro.
RESULTS
Honokiol DCA and hexafluoro does not inhibit proliferation in vitro
We initially tested the antiproliferative activity of honokiol DCA and hexafluoro against A375 melanoma in vitro (Supplementary Figure S1). We found no significant difference in terms of in vitro proliferation. We have consistently observed that honokiol and derivatives tend to be more potent in vivo than in vitro, and in vitro inhibition of proliferation is not predictive of in vivo behavior with this family of compounds.
Honokiol DCA and hexafluoro inhibits tumor growth in melanoma in vivo
We then assessed the in vivo activity of honokiol DCA and hexafluoro against A375 melanoma in vivo. A375 is a commonly used model of melanoma associated with mutant BRAF. Both honokiol DCA and hexafluoro demonstrated activity against A375 in vivo, but the activity of honokiol DCA, not hexafluoro, demonstrated significant tumor growth inhibition compared to the control group (Figure 2).
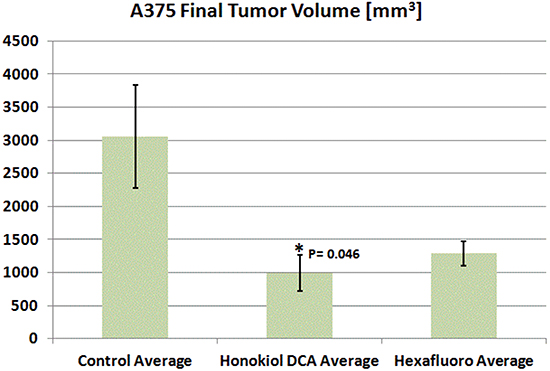
Figure 2: Honokiol DCA demonstrated significant antitumor activity in vivo against A375 malignant melanoma p <0.05. Animals treated with Hexafluoro showed marked but not significant tumor inhibition, p = 0.069. (n = 4).
In order to gain further insight into the mechanism of action of these small molecules, we harvested vehicle control and drug treated tumors and subjected them to gene array analysis (Supplementary Figure S3). A limited number of genes were commonly upregulated by both compounds in comparison to control and were confirmed by qRT-PCR (Figure 3). Honokiol DCA tended to show greater induction of genes than hexafluoro, and these genes may be useful as biomarkers for honokiol activity. Of interest, one of the genes that was commonly upregulated is succinate dehydrogenase B (SDHB), a tumor suppressor gene and respiratory enzyme [19, 20]. This is consistent with the known effect of honokiol induction of Sirt3.
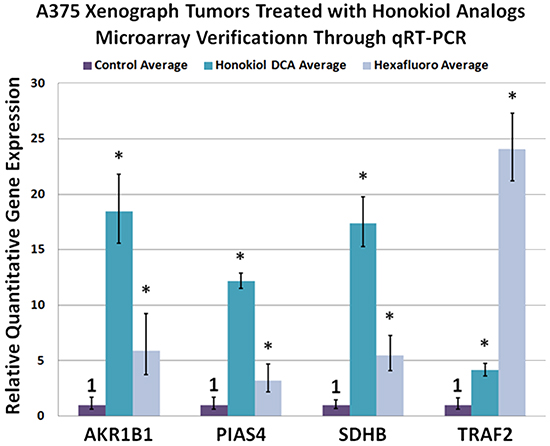
Figure 3: A375 xenograph tumors treated with honokiol analogs microarray verification through qRT-PCR. Upregulation of SDHB (succinate dehydrogenase B), AKR1B10 (aldo ketoreductase 1B10), PIAS4 (E3 SUMO-protein ligase PIAS4 also known as protein inhibitor of activated STAT protein 4 (PIAS4) or protein inhibitor of activated STAT protein gamma) and TRAF2 (TNF receptor-associated factor 2) was observed via microarray analysis and further verified through qRT-PCR. (p < 0.05; n = 3)
A major unmet need in treatment of human melanoma is drugs that have activity against vemurafenib resistant melanoma [21, 22]. We tested the ability of honokiol DCA to inhibit the growth in vitro (Supplementary Figure S2) as well as in vivo (Figure 4) of LM36 and LM36R, a pair of cell lines with BRAFV600E mutations that are sensitive and resistant to vemurafenib respectively [17]. LM36R, the more aggressive cell line, was inhibited in vivo by honokiol DCA significantly, while there was little effect on the sensitive and less aggressive LM36 parental cell line (Figure 4).
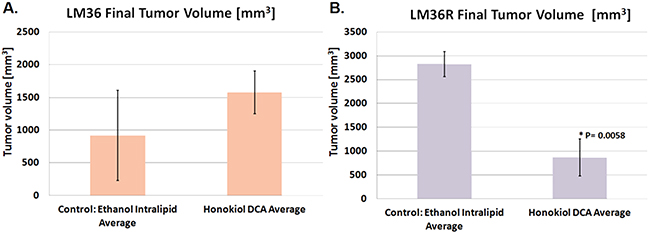
Figure 4: Honokiol DCA demonstrates significant antitumor activity in vivo against vemurafenib-resistant melanoma LM36R. Honokiol DCA did not demonstrate significant antitumor activity in vivo against parental model LM36 A. despite having activity against the derived vemurafenib-resistant melanoma LM36R B. suggesting acquired signaling differences through resistance of BRAF inhibition. (p < 0.05; n = 4)
We assessed the signaling effects of honokiol derivatives against the melanoma cell lines, with particular attention to MAP kinase and Akt signaling, since these have been shown to be important mediators of melanoma growth (Figure 5). Surprisingly, honokiol DCA induced phosphorylation of Akt in the more aggressive but sensitive LM36R and A375 cells, but not in the less aggressive LM36 cells. The increase in Akt phosphorylation despite the effective antitumor activity suggested that ROS generated from the mitochondria might be activating Akt [23]. In addition, both phosphorylation of MAP kinase and levels of Rac1b were reduced.
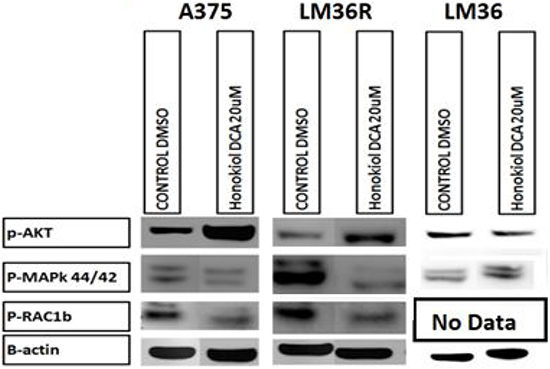
Figure 5: Western blot analysis show increased levels of p-AKT S473 on human melanoma cell lines A375 and vemurafenib resistant LM36R when treated for 24hrs at 20 μM. AKT activation was unchanged in the parental melanoma cell line LM36. The effect on Honokiol DCA sensitive A375 and LM36R may be due to downregulation of p-42/44 MAPK, while LM36 is insensitive to Honokiol DCA. B-actin was used as a loading control. This experiment represents a single western blot analysis for p-Rac1B and triplicate experiments for p-AKT S473 and p-42/44 MAPK.
Since Akt phosphorylation is often induced by reactive oxygen, we decided to assess whether reactive oxygen levels were affected by drug treatment. Two major sources of ROS in tumor cells are NADPH oxidases, which are rac dependent, and mitochondrial ROS, which is not rac dependent. Given that drug treatment decreased rac expression, if ROS is induced, the likely source would be mitochondrial rather than NADPH oxidase based.
Honokiol DCA and hexafluoro induce elevated mitochondrial ROS production
Using DHE fluorescence, we assessed superoxide expression in response to treatment. Hexafluoro induced superoxide in all three cell lines, while honokiol DCA induced superoxide in 2 out of 3 cell lines (Figure 6A). We then examined mitochondrial derived superoxide using MitoSox fluorescence (Figure 6B). Both honokiol derivatives induced mitochondrial superoxide in all three cell lines. Induction of superoxide was more robust in the LM36R cells compared with LM36, so we examined expression of MnSOD in LM36 and LM36R. Expression of MnSOD was nearly absent in LM36R, while it was highly expressed in LM36 (Figure 7). Decreased phosphorylation of DRP1at S616 was also observed with treatment of both honokiol DCA and Hexafluoro (Figure 8). Given that phosphorylation of DRP1 at S616 is required for mitochondrial fission, the decreased phosphorylation of this site indicates a transition to mitochondrial fusion. While all three cell lines have elevated levels of phosphorylated DRP1, the honokiol DCA sensitive LM36R and A375 appeared to have a greater decrease in phosphorylation than the honokiol DCA resistant (and less aggressive) LM36 (Figure 8). The combination of induction of mitochondrial fusion in the absence of MnSOD may cause selective inhibitory effects on aggressive tumors lacking MnSOD.
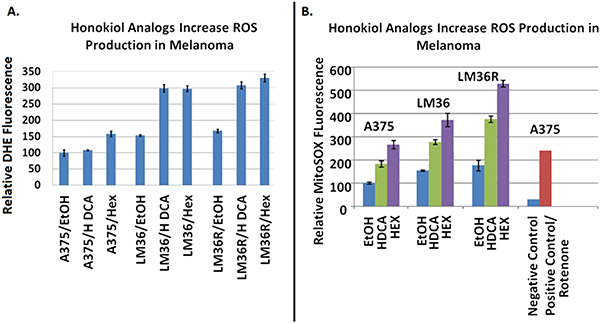
Figure 6: DHE fluorescence assays show Honokiol DCA and Hexafluoro increase overall superoxide levels. A. Through the MitoSox Fluorescence assay Honokiol DCA and Hexafluoro demonstrate increased mitochondrial derived reactive oxygen production, indicating that they may act against melanoma through a reversion of the Warburg phenomenon. B. The combination of increased mitochondrial ROS plus NFkB inhibition may lead to selective tumor cell death.
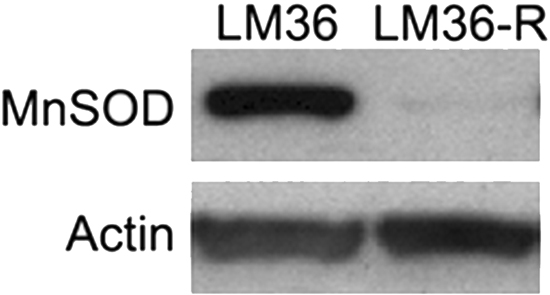
Figure 7: Western blot analysis comparing LM36 and LM36R MnSOD levels shows a loss of MnSOD expression in LM36R when compared to its parental cell line LM36. MnSOD expression levels may explain divergent sensitivity to Honokiol DCA between LM36 and LM36R. Actin was used as loading control.
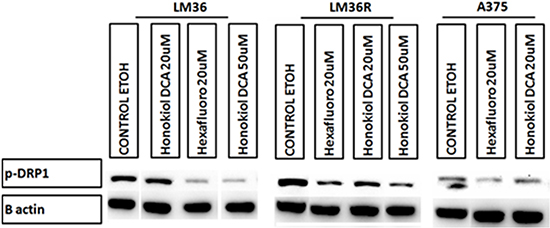
Figure 8: Western blot analysis show decrease levels of pDRP1 (S616) on human melanoma cell lines A375 and vemurafenib resistant LM36R when treated for 24hrs at 20μM. The decrease in DRP1 phosphorylation suggests a possible mechanism for Honokiol DCA and Hexafluoro may be through inhibition of mitochondria fission.
DISCUSSION
Melanoma is a common tumor that is well known for early metastasis and resistance to chemotherapy, antiangiogenic therapy [24–26], and radiation [27]. Recently, genetic subsets of melanoma have been characterized, especially with known driver mutations such as BRAF, NRAS, GNAQ, RAC1, C-KIT, and others [28]. Based upon the new knowledge of genetic alteration in melanoma, therapeutic interventions have been designed to treat melanoma. Most prominent among these are BRAF and MEK inhibitors. While BRAF mutant melanomas often respond to BRAF/MEK inhibition, in most cases the response is short lived. A major mechanism of resistance is activation of alternative signaling pathways [8].
One of the major signaling pathways that is not addressed by current therapies is superoxide. In what has been called the reactive oxygen driven tumor, superoxide can inactivate multiple tumor suppressors, such as p53, PTEN, IkB, and protein phosphatases [29]. Superoxide is a double edged sword for tumor cells, as it can serve as a tumor signaling pathway, or it can be used to kill the tumor cell [30]. Honokiol is a natural product that has attracted much attention because of its broad antitumor activity [31], and has been described as having pro-oxidant or anti-oxidant properties [32–34]. The precise context of honokiol’s activity as an anti-oxidant or pro-oxidant is not fully understood. Our group was the first to demonstrate in vivo activity against established tumors [13]. Since this finding, we and others have demonstrated antitumor activity against epithelial, hematopoietic and sarcoma types of malignancy [13, 35, 36]. Honokiol has been demonstrated to have anti-invasive properties, anti-metastatic properties, and chemopreventive properties. Most recently, honokiol has been found to be a potent activator of the mitochondrial deacetylase Sirt3 [16], which is associated with an increased respiratory phenotype and fused mitochondria. Mitofusin2, associated with mitochondrial fusion, is also an antagonist of ras activation [37, 38], and induction of mitochondrial fusion could thus potentially explain the ras antagonism seen with honokiol [15]. Finally, the full potential of ras and raf oncogenes to transform is associated with defective fission type of mitochondria [39].
Delivery of natural products is a major hurdle in their translation to the clinic. Two major barriers to translation of natural products are multiple mechanisms of activity and the difficulty on obtaining intellectual property production on natural products with known structure. We believe that the ortho-allyphenol moiety is crucial to the activity of honokiol, based upon prior structure-function studies of the native honokiol molecule [13]. In order to test this, we have synthesized a novel honokiol analog with 2 ortho-allylphenol moieties containing a single carbon spacer (hexafluoro). This molecule is synthesized in 2 steps from industrially available precursors. In addition, we modified the natural product honokiol into a prodrug through esterification, which maintains activity in vivo.
Despite lack of efficacy in in vitro proliferation assays, both honokiol DCA and hexafluoro were both active in vivo against A375 melanoma, a BRAF mutant melanoma xenograft model. The NCI 60 proliferation inhibition index is commonly used as a screening index for candidate drugs [40]. Intriguingly, the antitumor properties of our analogs would not have been discovered based upon antiproliferative studies, as our compounds are not highly active as antiproliferative agents in culture. The energy requirements for proliferating in tissue culture may be very different from those required to create a 3 dimensional tumor under hypoxic conditions, and given the effects of our compounds on mitochondrial metabolism, in vivo conditions were required to demonstrate the effect of our compounds. Gene array analysis revealed few genes commonly upregulated by both compounds, including succinate dehydrogenase b (SDHB), which is involved in respiration and is a known tumor suppressor gene. Given that we have recently found that honokiol is an activator of Sirt3, this data suggests that honokiol derivatives are acting at least in part through induction of mitochondrial fusion, consistent with decreased phosphorylation of DRP-1 (S616) [41, 42].
Treatment of LM36R with honokiol derivatives leads to an increase in mitochondrial reactive oxygen species. One of the major mechanisms of detoxification of mitochondrially derived ROS is through manganese superoxide dismutase (MnSOD) [43]. A375, which is sensitive to honokiol derivatives, has been demonstrated to have extremely low levels of MnSOD, and is moderately resistant to vemurafenib [44, 45]. We compared expression of MnSOD in less aggressive LM36 and more aggressive LM36R and found that expression of MnSOD is nearly absent in LM36R compared with LM36 (Figure 7). Tumors that have low levels of MnSOD are known to be highly aggressive and absence of MnSOD may be a novel mechanism of resistance to vemurafenib. In addition, the absence of MnSOD may be a biomarker of sensitivity to honokiol derivatives. The resistance of the less aggressive LM36 to honokiol DCA is intriguing. The LM36 appear to have little response to 20 mM honokiol DCA in terms of phosphorylation of DRP1 compared to the more aggressive LM36R and A375 cells. The combination of lack of effect on DRP1 phosphorylation and thus mitochondrial fusion may allow the less aggressive cells to survive reactive oxygen stressors better than tumor cells which both lack the detoxification enzyme MnSOD and have increased mitochondrial fusion. Finally, honokiol derivatives may prove to be useful in killing cells with defective detoxification of mitochondrial ROS by increasing the biogenesis of these mitochondria and thus selectively killing tumors with defective elimination of ROS. Thus the context of the tumor cell may determine the role of whether a drug is a pro or anti-oxidant. In tumor cells that contain defective mitochondria or detoxification systems, activation of mitochondrial fusion could potentially amplify mitochondrial induced reactive oxygen, while in cells with normal mitochondria, honokiol could stimulate an antioxidant activity by stimulating normal mitochondrial biogenesis [46]. Treatment with honokiol derivatives may selectively target tumor cells with defective mitochondria, which often are the most resistant to current therapies.
MATERIALS AND METHODS
Synthesis
Honokiol bis-dichloroacetate (Honokiol DCA)
Honokiol DCA was synthesized according to a procedure previously described by us [47]. Briefly, honokiol (1.0 g, 3.7 mmol) was dissolved in dry dichloromethane (200 mL), followed by addition of 4-dimethylaminopyridine (200 mg) from Sigma-Aldrich (St. Louis, MO). The reaction was heated to 40° C while stirring and dichloroacetylchloride (1.45 mL, 15 mmol) was added dropwise over 10 min. Next, the reaction mixture was refluxed for 5 h, after which all starting material was consumed according to TLC. After cooling the solution was washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (ethyl acetate/hexanes 1:9), which resulted in the wanted product (1.6 g, 76%). The obtained NMR was in accordance with our previous publication [47].
NMR spectra were recorded in deuterated chloroform (CDCl3) with a Varian INOVA 400 MHz instrument, calibrated using residual undeuterated chloroform (1H: δ = 7.24 ppm) as internal standard. The following abbreviations, or a combination thereof, are used to explain the multiplicities: s = singlet, d = doublet, t = triplet. High resolution mass spectrometry (HRMS) analysis was performed with a Thermo Scientific LTQ FT Ultra Hybrid mass spectrometer set on positive ionization.
ortho-Allyl hexafluorobisphenol A (hexafluoro)
Hexafluorobisphenol A (1.0 g, 3.0 mmol) and anhydrous K2CO3 (2.07 g, 15 mmol) were dissolved in acetone (20 mL). The mixture was heated to reflux while stirring and allylbromide (1.0 mL, 11.5 mmol) was added dropwise. After 4h at reflux the solvent was removed in vacuum, and the remaining residue was taken up in ethyl acetate and washed with water and brine. The organic layer was dried over MgSO4, filtered, and concentrated in vacuum. The crude hexafluorobisallyloxyphenyl A (4,4’-(perfluoropropane-2,2-diyl)bis((allyloxy)benzene)) was used without further purification.
A procedure was adapted from the literature as follows: BCl3 (1M in CH2Cl2, 12 mL, 12 mmol) was added to a stirred solution of hexafluorobisallyloxyphenyl A (1.25 g, 3.0 mmol) in CH2Cl2 (10 mL) at –78 °C under argon. The reaction mixture was allowed to reach room temperature followed by stirring under argon for 4h. The reaction was quenched by addition of H2O (20 mL), and the mixture was extracted with CHCl3 (3 × 20 mL). The organic layers were combined, washed with brine, dried over MgSO4, filtered, and concentrated under vacuum. The crude product was purified by column chromatography on silica gel (ethyl acetate/hexanes 1:9), which afforded hexafluoro as a yellow solid (1.15 g, 92%). 1H-NMR (CDCl3): δ 7.13 (d, J = 8.8 Hz, 2H), 7.08 (s, 2H), 6.77 (d, J = 8.8 Hz, 2H), 5.99 – 5.89 (m, 2H), 5.12 – 5.03 (m, 4H), 3.35 (d, J = 6.0 Hz, 4H). HRMS calculated for C21H19F6O2 417.12838, found 417.12877.
Proliferation studies
Proliferation Studies were done using a Beckman Z1 Coulter cell counter. Cells were seeded 5 × 104 cells per well in a 24-well plate. Cells were then treated the next day with 20 μM of honokiol, honokiol DCA, or hexafluoro from 10mM stock solutions in DMSO. Cells were incubated at 37 °C with 5% CO2. Each compound was treated in quadruplicate wells. After 24 hours, cells were washed twice with PBS and trypsined. Cells were then counted in an isotonic solution.
MTT assays
MTT assays were used to evaluate the effect of 72h drug treatment on LM36/LM36R cell growth as described previously [17].
DHE assays
A375, LM36 and LM36R cells were treated with control, honokiol DCA, hexafluoro or vehicle for 24 hours. Cells were washed with PBS, digested with 0.05 % trypsin/0.53 mM EDTA and pelleted at 600g for 5 min. Cells were resuspended in 10 μM dihydroethidium (DHE) and incubated while gently shaking in the dark for 10 min and place on ice in the dark. Analysis of DHE fluorescence was performed on a Becton Dickinson FACScan flow cytometer, 10,000 cells were counted and analyzed by FlowJo 7.6.4. Mean values of the DHE fluorescence intensity were compared. Error bars represent the standard error of the mean of triplicate data points.
MitoSOX assay
A375, LM36 and LM36R cells were treated with control, honokiol DCA, hexafluoro or vehicle for 24 hours. Cells were washed with PBS followed by the addition of 5 μM MitoSOX in phenol red free RPMI 1640. Cells were incubated for 30 mins at 37 C, 5 % CO2. MitoSOX was removed and cells were digested with 0.05 % trypsin/0.53 mM EDTA and pelleted at 600g for 5 min. Cells were resuspended in HANKS and placed on ice. Analysis of DHE fluorescence was performed on a Becton Dickinson FACScan flow cytometer, 10,000 cells were counted and analyzed by FlowJo 7.6.4. Mean values of the DHE fluorescence intensity were compared. Error bars represent the standard error of the mean of triplicate data points.
Western blot analysis
Cells were plated in T25 flasks and treated when 80% confluent for 24 hours with 20 μM of each compound (honokiol, honokiol DCA, and hexafluoro. Blots were probed with antibodies for p-akt, p-mapk 42/44, p-p38, p-DRP1, total akt, total ERK, FOXD3, MDMX, p-Rac1b, MnSOD or beta actin were added at a concentration of 1:1000 in 5% non-fat dry milk in TBS and allowed to shake overnight in the coldroom (4°C) [48]. The next day, the blots were probed with anti-rabbit or anti-mouse HRP linked antibodies 1:10,000 (Cell Signaling) 5% non-fat dry milk in TBS with anti-rabbit or anti-mouse HRP linked antibodies (Cell Signaling). Super-Substrate from Thermofisher was used to activate the HRP linked secondary antibodies for development. The membranes were then developed using a Bio-rad docking station and camera. The software used was Bio-Rad Image Lab version 4.0.
Gene chip analysis
Tumors from animals treated with control vehicle, honokiol, honokiol DCA, and hexafluoro were harvested and snapped-frozen in liquid nitrogen until RNA Extraction. RNA extraction was performed according to the Qiagen RNeasy kit. RNA samples were then submitted to Emory University’s Intergrated Genomics Core for RNA quality analysis and gene expression assay. Gene expression analysis was performed using an Illumina HumaHT-12 v3 Expression Bead Chip and Gene Expression Module of Illumina’s GenomeStudio Software package (v2011.1, Illumina).
qRT-PCR
Gene chip analysis results were confirmed via qRT-PCR using the Applied Biosystems 7500 FAST Real Time PCR System. Briefly, cDNA was generated from RNA extracts using SuperScript VILO cDNA Synthesis Kit (Invitrogen) and the Eppendorf Mastercycler gradient. Taqman primers for SDHB, AKR1B10, PIAS4, TRAF2, and S18 (endogenous control) were used with TaqMan Fast Universal PCR Mastermix (2X) (Applied Biosystems).
In vivo studies
A375, LM36, and LM36R cells were injected 1 × 106 cells per mouse, in groups of four. The cells were allowed two days of incubation before treatment. Honokiol DCA, hexafluoro were injected 5 times per week at 140 mg/kg in groups of 4 mice per compound. The injection cocktail was made by dissolving 16 mg of compound into 100 μl of absolute ethanol. The ethanol-compound solutions were then added to 1 ml of 20% soy-fat Intralipid (Frensenius Kabi) and vortexed vigorously [13]. Mice were injected with 0.25cc per mouse via intraperitoneal injections. The animals’ weights and their tumors’ lengths and widths were measured weekly. Tumor volumes were calculated via the tumor volume formula: (L × W2) × 0.52, with the smallest dimension being assigned the width and squared [49].
Statistical analysis
Genechip analysis
Probe level intensity values were extracted and quantile normalization was performed using the Gene Expression Module of Illumina’s GenomeStudio Software package (v2011.1, Illumina). Principle Component Analysis (PCA) and unsupervised hierarchical clustering were used to assess the variation of the samples’ expression profiles. To derive a refined list of genes most affected by honokiol DCA and hexaxfluoro compared to Control, the Significance Analysis of Microarrays (SAM; http://statweb.stanford.edu/~tibs/SAM/) was used.
qRT-PCR
The statistical analysis was performed in triplicate using Applied Biosystems Sequence Detection Software (SDS Version 1.3.1). Using the software, relative quantification (RQ) values were obtain using the cycle time (CT) values which were first normalized to an endogenous control (S18) and then to experimental control and evaluated; p < 0.05.
In vivo studies
The statistical analysis for tumor volumes (defined by (L × W2) × 0.52, with the smallest dimension being assigned the width and squared) in the animal studies was performed on groups of 4, using Microsoft Office Excel 2007. P-values were determined using a two-tailed, two-sample equal variance (homoscedastic) student t test; p < 0.05.
ACKNOWLEDGMENTS
Research reported in this publication was supported in part by the Emory Integrated Genomics Core (EIGC) Shared Resource of Winship Cancer Institute of Emory University and NIH/NCI under award number P30CA138292. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
CONFLICTS OF INTEREST
JLA is the inventor on US PATENT#: 8,586,627 on Honokiol DCA and Hexafluoro.
GRANT SUPPORT
JLA was supported by the grant RO1 AR47901and P30 AR42687 Emory Skin Disease Research Core Center Grant from the National Institutes of Health, a Veterans Administration Hospital Merit Award, as well as funds from the Margolis Foundation, Rabinowitch-Davis Foundation for Melanoma Research and the Betty Minsk Foundation for Melanoma Research. EV is recipient of FIRC/AIRC fellowship “Luciana Selce”.
REFERENCES
1. Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM, Moses TY, Hostetter G, Wagner U, Kakareka J, Salem G, Pohida T, Heenan P, Duray P, Kallioniemi O, Hayward NK, et al. High frequency of BRAF mutations in nevi. Nature genetics. 2003; 33:19-20.
2. Flaherty KT. Targeting metastatic melanoma. Annual review of medicine. 2012; 63:171-183.
3. Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, Chiarion Sileni V, Lebbe C, Mandala M, Millward M, Arance A, Bondarenko I, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. The New England journal of medicine. 2014; 371:1877-1888.
4. Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, Salton M, Dahlman KB, Tadi M, Wargo JA, Flaherty KT, Kelley MC, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011; 480:387-390.
5. Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, Caponigro G, Hieronymus H, Murray RR, Salehi-Ashtiani K, Hill DE, Vidal M, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010; 468:968-972.
6. Whittaker SR, Theurillat JP, Van Allen E, Wagle N, Hsiao J, Cowley GS, Schadendorf D, Root DE and Garraway LA. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer discovery. 2013; 3:350-362.
7. Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, Kelley MC, Kefford RF, Chmielowski B, Glaspy JA, Sosman JA, van Baren N, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer discovery. 2014; 4:80-93.
8. Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G, Sosman JA, Ribas A and Lo RS. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010; 468:973-977.
9. Villanueva J, Infante JR, Krepler C, Reyes-Uribe P, Samanta M, Chen HY, Li B, Swoboda RK, Wilson M, Vultur A, Fukunaba-Kalabis M, Wubbenhorst B, Chen TY, Liu Q, Sproesser K, DeMarini DJ, et al. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep. 2013; 4:1090-1099.
10. Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, Rosenberg M, Goetz EM, Sullivan RJ, Farlow DN, Friedrich DC, Anderka K, Perrin D, Johannessen CM, McKenna A, Cibulskis K, et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer discovery. 2014; 4:61-68.
11. Cohen C, Zavala-Pompa A, Sequeira JH, Shoji M, Sexton DG, Cotsonis G, Cerimele F, Govindarajan B, Macaron N and Arbiser JL. Mitogen-actived protein kinase activation is an early event in melanoma progression. Clinical cancer research. 2002; 8:3728-3733.
12. Govindarajan B, Bai X, Cohen C, Zhong H, Kilroy S, Louis G, Moses M and Arbiser JL. Malignant transformation of melanocytes to melanoma by constitutive activation of mitogen-activated protein kinase kinase (MAPKK) signaling. The Journal of biological chemistry. 2003; 278:9790-9795.
13. Bai X, Cerimele F, Ushio-Fukai M, Waqas M, Campbell PM, Govindarajan B, Der CJ, Battle T, Frank DA, Ye K, Murad E, Dubiel W, Soff G and Arbiser JL. Honokiol, a small molecular weight natural product, inhibits angiogenesis in vitro and tumor growth in vivo. J Biol Chem. 2003; 278:35501-35507.
14. Raja SM, Chen S, Yue P, Acker TM, Lefkove B, Arbiser JL, Khuri FR and Sun SY. The natural product honokiol preferentially inhibits cellular FLICE-inhibitory protein and augments death receptor-induced apoptosis. Molecular cancer therapeutics. 2008; 7:2212-2223.
15. Garcia A, Zheng Y, Zhao C, Toschi A, Fan J, Shraibman N, Brown HA, Bar-Sagi D, Foster DA and Arbiser JL. Honokiol suppresses survival signals mediated by Ras-dependent phospholipase D activity in human cancer cells. Clinical cancer research. 2008; 14:4267-4274.
16. Pillai VB, Samant S, Sundaresan NR, Raghuraman H, Kim G, Bonner MY, Arbiser JL, Walker DI, Jones DP, Gius D and Gupta MP. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat Commun. 2015; 6:6656.
17. Vergani E, Vallacchi V, Frigerio S, Deho P, Mondellini P, Perego P, Cassinelli G, Lanzi C, Testi MA, Rivoltini L, Bongarzone I and Rodolfo M. Identification of MET and SRC activation in melanoma cell lines showing primary resistance to PLX4032. Neoplasia. 2011; 13:1132-1142.
18. Govindarajan B, Sligh JE, Vincent BJ, Li M, Canter JA, Nickoloff BJ, Rodenburg RJ, Smeitink JA, Oberley L, Zhang Y, Slingerland J, Arnold RS, Lambeth JD, Cohen C, Hilenski L, Griendling K, et al. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. The Journal of clinical investigation. 2007; 117:719-729.
19. Cimen H, Han MJ, Yang Y, Tong Q, Koc H and Koc EC. Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry. 2010; 49:304-311.
20. Finley LW, Haas W, Desquiret-Dumas V, Wallace DC, Procaccio V, Gygi SP and Haigis MC. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PloS one. 2011; 6:e23295.
21. Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, Hodis E, Rosenberg M, McKenna A, Cibulskis K, Farlow D, Zimmer L, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer discovery. 2014; 4:94-109.
22. Shi H, Moriceau G, Kong X, Lee MK, Lee H, Koya RC, Ng C, Chodon T, Scolyer RA, Dahlman KB, Sosman JA, Kefford RF, Long GV, Nelson SF, Ribas A and Lo RS. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nature communications. 2012; 3:724.
23. Nayak BK, Feliers D, Sudarshan S, Friedrichs WE, Day RT, New DD, Fitzgerald JP, Eid A, Denapoli T, Parekh DJ, Gorin Y and Block K. Stabilization of HIF-2alpha through redox regulation of mTORC2 activation and initiation of mRNA translation. Oncogene. 2013; 32:3147-3155.
24. Yang X, Zhang Y, Hosaka K, Andersson P, Wang J, Tholander F, Cao Z, Morikawa H, Tegner J, Yang Y, Iwamoto H, Lim S and Cao Y. VEGF-B promotes cancer metastasis through a VEGF-A-independent mechanism and serves as a marker of poor prognosis for cancer patients. Proceedings of the National Academy of Sciences of the United States of America. 2015; 112:E2900-2909.
25. Zhang D, Hedlund EM, Lim S, Chen F, Zhang Y, Sun B and Cao Y. Antiangiogenic agents significantly improve survival in tumor-bearing mice by increasing tolerance to chemotherapy-induced toxicity. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108:4117-4122.
26. Zhou Z, Doi M, Wang J, Cao R, Liu B, Chan KM, Kortesmaa J, Sorokin L, Cao Y and Tryggvason K. Deletion of laminin-8 results in increased tumor neovascularization and metastasis in mice. Cancer research. 2004; 64:4059-4063.
27. Tsao H, Atkins MB and Sober AJ. Management of cutaneous melanoma. N Engl J Med. 2004; 351:998-1012.
28. Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Brocker EB, LeBoit PE, Pinkel D and Bastian BC. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005; 353:2135-2147.
29. Fried L and Arbiser JL. The reactive oxygen-driven tumor: relevance to melanoma. Pigment Cell Melanoma Res. 2008; 21:117-122.
30. Harris CA, Derbin KS, Hunte-McDonough B, Krauss MR, Chen KT, Smith DM and Epstein LB. Manganese superoxide dismutase is induced by IFN-gamma in multiple cell types. Synergistic induction by IFN-gamma and tumor necrosis factor or IL-1. J Immunol. 1991; 147:149-154.
31. Fried LE and Arbiser JL. Honokiol, a multifunctional antiangiogenic and antitumor agent. Antioxidants & redox signaling. 2009; 11:1139-1148.
32. Han LL, Xie LP, Li LH, Zhang XW, Zhang RQ and Wang HZ. Reactive oxygen species production and Bax/Bcl-2 regulation in honokiol-induced apoptosis in human hepatocellular carcinoma SMMC-7721 cells. Environ Toxicol Pharmacol. 2009; 28:97-103.
33. Dikalov S, Losik T and Arbiser JL. Honokiol is a potent scavenger of superoxide and peroxyl radicals. Biochem Pharmacol. 2008; 76:589-596.
34. Sheu ML, Chiang CK, Tsai KS, Ho FM, Weng TI, Wu HY and Liu SH. Inhibition of NADPH oxidase-related oxidative stress-triggered signaling by honokiol suppresses high glucose-induced human endothelial cell apoptosis. Free Radic Biol Med. 2008; 44:2043-2050.
35. Ishitsuka K, Hideshima T, Hamasaki M, Raje N, Kumar S, Hideshima H, Shiraishi N, Yasui H, Roccaro AM, Richardson P, Podar K, Le Gouill S, Chauhan D, Tamura K, Arbiser J and Anderson KC. Honokiol overcomes conventional drug resistance in human multiple myeloma by induction of caspase-dependent and -independent apoptosis. Blood. 2005; 106:1794-1800.
36. Battle TE, Arbiser J and Frank DA. The natural product honokiol induces caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia (B-CLL) cells. Blood. 2005; 106:690-697.
37. Chen KH, Dasgupta A, Ding J, Indig FE, Ghosh P and Longo DL. Role of mitofusin 2 (Mfn2) in controlling cellular proliferation. FASEB journal. 2014; 28:382-394.
38. de Brito OM and Scorrano L. Mitofusin-2 regulates mitochondrial and endoplasmic reticulum morphology and tethering: the role of Ras. Mitochondrion. 2009; 9:222-226.
39. Serasinghe MN, Wieder SY, Renault TT, Elkholi R, Asciolla JJ, Yao JL, Jabado O, Hoehn K, Kageyama Y, Sesaki H and Chipuk JE. Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Molecular cell. 2015; 57:521-536.
40. Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006; 6:813-823.
41. Tailor D, Hahm ER, Kale RK, Singh SV and Singh RP. Sodium butyrate induces DRP1-mediated mitochondrial fusion and apoptosis in human colorectal cancer cells. Mitochondrion. 2014; 16:55-64.
42. Yamano K and Youle RJ. Coupling mitochondrial and cell division. Nat Cell Biol. 2011; 13:1026-1027.
43. de Keizer PL, Packer LM, Szypowska AA, Riedl-Polderman PE, van den Broek NJ, de Bruin A, Dansen TB, Marais R, Brenkman AB and Burgering BM. Activation of forkhead box O transcription factors by oncogenic BRAF promotes p21cip1-dependent senescence. Cancer research. 2010; 70:8526-8536.
44. Hirose K, Longo DL, Oppenheim JJ and Matsushima K. Overexpression of mitochondrial manganese superoxide dismutase promotes the survival of tumor cells exposed to interleukin-1, tumor necrosis factor, selected anticancer drugs, and ionizing radiation. FASEB J. 1993; 7:361-368.
45. Suresh A, Tung F, Moreb J and Zucali JR. Role of manganese superoxide dismutase in radioprotection using gene transfer studies. Cancer Gene Ther. 1994; 1:85-90.
46. Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A and Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. The Journal of clinical investigation. 2009; 119:2758-2771.
47. Hahm ER, Karlsson AI, Bonner MY, Arbiser JL and Singh SV. Honokiol inhibits androgen receptor activity in prostate cancer cells. The Prostate. 2014; 74:408-420.
48. Arbiser JL, Moses MA, Fernandez CA, Ghiso N, Cao Y, Klauber N, Frank D, Brownlee M, Flynn E, Parangi S, Byers HR and Folkman J. Oncogenic H-ras stimulates tumor angiogenesis by two distinct pathways. Proceedings of the National Academy of Sciences of the United States of America. 1997; 94:861-866.
49. Arbiser JL, Petros J, Klafter R, Govindajaran B, McLaughlin ER, Brown LF, Cohen C, Moses M, Kilroy S, Arnold RS and Lambeth JD. Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proceedings of the National Academy of Sciences of the United States of America. 2002; 99:715-720.