
INTRODUCTION
Osteosarcoma is the most common primary malignant tumor in children, adolescents, and young adults [1]. It comprises a group of mesenchymal bone-forming tumors with multiple histologic subtypes, including osteoblastic, chondroblastic, fibroblastic, and unconventional. The biology of osteosarcoma is complex, and treatment options have remained essentially unchanged for several decades [2]. Since the 1980s, front-line treatment for primary osteosarcoma has consisted of induction chemotherapy followed by surgery for patients with non-metastatic disease. Adjuvant chemotherapy is also given. Options are even more limited for patients with relapsed osteosarcoma. A recent Phase II study of sorafenib and everolimus represents the first positive study for relapsed osteosarcoma in decades, but even this study did not reach statistical significance for the primary outcome [3]. Radium 223 di-chloride, an alpha particle, may be another potential treatment option for patients with osteoblastic metastases [4, 5].
In the era of precision medicine, the molecular profile of a patient's tumor can be determined readily and can be matched to a targeted therapy in real time. This presents an opportunity to systematically analyze an individual patient's tumor and offer them personalized therapy [6]. With few advancements in treatment and the dismal prognosis for patients with relapsed, refractory disease, osteosarcoma patients may benefit from deep molecular, genomic sequencing and comprehensive molecular profiling [2, 6]. As a demonstration of this approach, we performed comprehensive molecular profiling of two patients with metastatic osteosarcoma and matched their profiles to molecularly targeted therapy. Here we report the results of this profiling and discuss the implications and limitations of this approach.
RESULTS
The patients' clinical histories, molecular profiling results, and targeted therapy are summarized here.
Patient #1
This 21-year-old male was found to have chrondroblastic osteosarcoma of the right distal femur in 2011. Upon diagnosis, he began treatment with standard first-line chemotherapy comprising methotrexate, doxorubicin, and cisplatin (MAP). He experienced immediate relief from pain with the initial cisplatin and doxorubicin infusions. He then underwent limb salvage surgery, and pathologic review showed his resected tumor to have 80% necrosis. He received postoperative chemotherapy because of his high-risk status and was closely monitored with periodic computed tomographic (CT) scans. Nine months after completion of adjuvant chemotherapy, a routine CT scan showed a 3.5 × 3.5–cm nodule in the left upper lobe of the lung and a 2.0 × 2.5–cm nodule in the right middle lobe, representing distant relapse of the osteosarcoma (Figure 1). Despite treatment with high-dose ifosfamide (1 cycle at 14 g/m2), the disease progressed and the patient subsequently developed a pneumothorax. He received another course of cisplatin and doxorubicin, and although the pneumothorax resolved, there was evidence of continued progression of his intrathoracic metastases. He subsequently underwent thoracotomy of the left lung with upper lobe resection and metastectomy. The final pathology report revealed at least 40 foci of tumor in the resected lung tissue. This tumor tissue was sent for comprehensive molecular profiling to identify potential targeted therapy options. The patient was referred to the molecular treatment planning group.
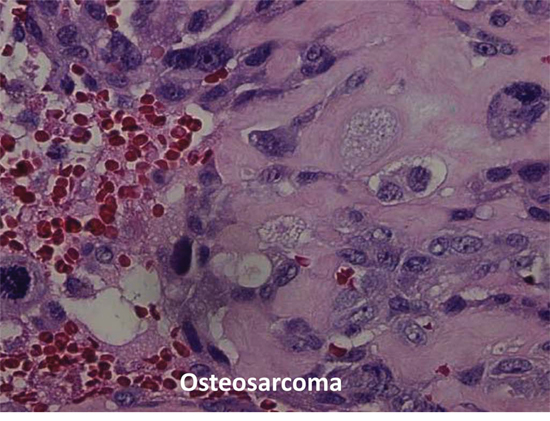
Figure 1: Histopathology figure of osteosarcoma from patient # 1.
Patient #1: next-generation exome sequencing
Next-generation sequencing by FoundationOne identified four genomic aberrations: MET amplification, PIK3CA V344G mutation, CCNE1 amplification, and a loss of function mutation in PTPRD (S1845f*). Our institutional 46-gene panel confirmed the PIK3CA V344G mutation. Caris Target One gene profiling revealed positivity for TLE3 (2+, 80%), androgen receptor (1+, 60%), TS, RRM1, and PTEN. Further testing was negative for ERCC1, MGMT, TOPO1, ER/PR, and SPARC. FISH analysis revealed no amplification in Her2/neu or TOP2A. P1K3CA and BRAF were wild type by sequencing for hotspot alterations. Genetic testing results for Patient #1 are summarized in Table 1 and Table 2.
Table 1: Summary of molecular aberrations in genes, receptors, and pathways by various CLIA-certified methods for osteosarcoma Patient #1 and their targeted agents
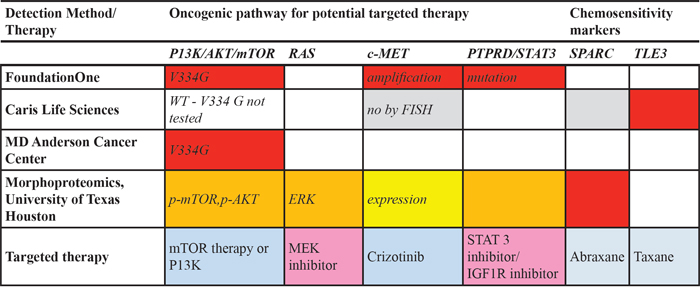
Table 2: Complete molecular profiling analysis by various CLIA-certified methods for osteosarcoma Patient #1
Marker |
Result |
Details |
Comments |
|---|---|---|---|
Topoisomerase II alpha |
Positive |
~70–90% of tumor nuclei (higher percentage of nuclear expression in cellular foci and less in chondroid regions); 26 mitotic figures/10 hpf in cellular regions |
Facilitates S to G2 and M phase transitions |
Bcl-2 |
Positive |
2+ in cytoplasm of tumor cells |
Anti-apoptotic protein |
Fatty acid synthase (FASN) |
Positive |
Highly expressed in cytoplasm (up to 3+) |
Tumorigenic protein; correlates with propensity for pulmonary metastasis [62] |
P38 mitogen-activated protein kinase (MAPK) |
Positive |
Expressed to varying degrees in tumor nuclei |
Chemoresistance factor; phosphorylated on threonine180/tyrosine182 |
Excision repair cross complementation group1 (ERCC1) |
Positive |
Expressed to varying degrees in tumor nuclei |
Chemoresistance factor; downstream effector of MAPK |
Sirt1 (silent mating type information regulator 2 homolog), NAD+histone deacetylase |
Positive |
Expressed in 2–3+ in majority of tumor nuclei |
Associated with upregulation in response to chemotherapy, mechanism of cancer resistance in human osteosarcoma [63] |
Nuclear factor (NF)-kappaB |
Positive |
0–1/1+, nuclear/cytoplasm |
Prosurvival protein, constitutively activated by virtue of expression with nuclear translocation of p-NF-kappaBp65(Ser536) |
Signal transducer and activator of transcription (STAT)-3 |
Positive |
0–3+ (majority positive), nuclear |
Activated with nuclear expression of p-STAT3(Tyr705) [64, 65] |
Cyclooxygenase (COX)-2 |
Positive |
1–3+, cytoplasm |
Correlates inversely with survival in patients with osteosarcoma and lung metastasis; highly expressed in cytoplasm of tumor cells, especially more cellular regions [66] |
WNT/B-catenin signaling pathway. |
Positive |
Strong cytoplasmic and nuclear (up to 3+) expression in minor component of tumor cells (cellular foci) |
Coincides with nuclear expression of c-Myc protein, whose expression can be upregulated by WNT/B-catenin signaling in ~1/3 of tumor cells [67]; cytoplasmic and/or nuclear staining of Beta-catenin associated in preclinical study and in primary osteosarcomas with lung metastasis [68] |
Hypoxia-inducible factor (HIF)-1 alpha |
Present |
2+ cytoplasmic compartment, without apparent nuclear translocation |
Hypoxia-associated protein analyte |
HIF-2 alpha, VEGF-A |
Present |
0–1+ in cytoplasmic compartment |
Ischemic-type coagulative necrosis not apparent in the biopsy specimen |
Nestin |
Present |
Strong expression; up to 3+; cytoplasmic aspect |
High expression level in osteosarcoma predicted a worse clinical outcome in one study [69] |
CD44 |
Present |
Cytoplasmic/plasmalemmal aspect of tumor cells |
|
Glioma-associated oncogene protein (Gli2) |
Present |
Variable expression (0–3+) in majority of nuclei |
Reflection of either sonic hedgehog pathway signaling or TGF-beta [Smad3] activation of Gli2 [70–72] |
Secreted protein acidic and rich in cysteine (SPARC; osteonectin) |
Present |
Up to 3+ cytoplasmic expression |
Predicts poorer relapse and event-free survival rates as well as therapeutic option [73] |
Patient #1: immunohistochemistry based morphoproteomics analysis and therapy
Immunohistochemistry-based morphoproteomic analysis showed constitutive activation of the c-MET pathway in the tumor cells to a mild degree, as evidenced by the expression of phosphorylated (p)-c-Met (Tyr1234/1235) in the cytoplasmic compartment (0 to 1+ with a rare 2+ chromogenic signal on a scale of 0 to 3+) (Figure 2). The insulin-like growth factor (IGF) pathway was expressed with 1+ to 3+ signal in the cytoplasmic compartment of total IGF-1R (Tyr1165/1166). Furthermore, there was moderate expression of PKC-alpha (up to 3+ in both the cytoplasmic and plasmalemmal compartments). The ras/Raf kinase extracellular signal-regulated kinase (ERK) pathway was constitutively activated in the form of phosphorylated p-ERK 1/2 (Thr202/Tyr204). Also present was constitutive activation of the mammalian target of rapamycin C2 (mTORC2) pathway as evidenced by the expression, with nuclear subcellular compartmentalization, of p-mTOR [Ser2448] with concomitant expression of its upstream effector p-Akt (Ser473). The activation of mTORC2 coincides with both downstream signaling from the IGF-1R pathway and strong PKC-alpha expression [7, 8].
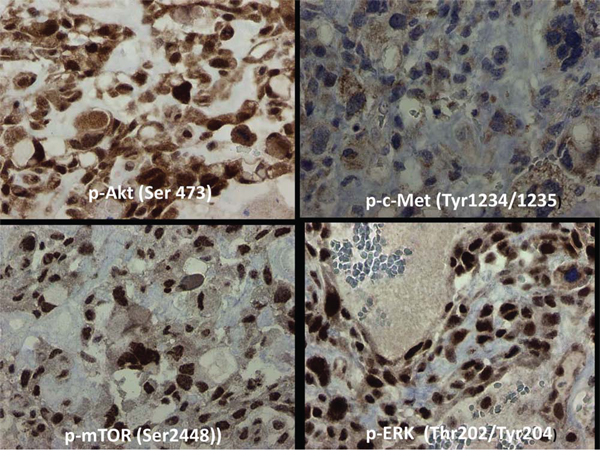
Figure 2: Immunohistochemistry based morphoproteomic studies of osteosarcoma Patient #1. In the tumor cells of Patient #1, c-Met was expressed at 0–1+ (rarely 2+) in the cytoplasm, mTOR at 3+ in nucleus and cytoplasm, ERK 1/2 at 3+ in the nucleus, and Akt at 0–3+ in both nucleus and cytoplasm. Representative sections are shown.
The biological pathway networks evoked by IPA from the molecular profiles were reviewed for potential targeted therapy options. Of the six biological pathway networks evoked, only one differentiated the patient from the control. In the patient, there were losses of control pathway interactions with insulin and follicle stimulating hormone, and there were gains of interactions with MYC, a key transcription regulator in cancer. Of particular interest was the potentially strong influence of PROP1 on the patient's network, possibly to lower the concentration of beta-catenin (CTNNB1). PROP1 encodes a transcription factor in the pituitary gland. Seven drugs were considered for therapy: 1-alpha25 (OH)2-vitamin D3, celecoxib, gemcitabine, lovastatin, melatonin, metformin, and nab-paclitaxel. As can been seen in (Figure 3), these drugs provided coverage of the patient's pathway network.
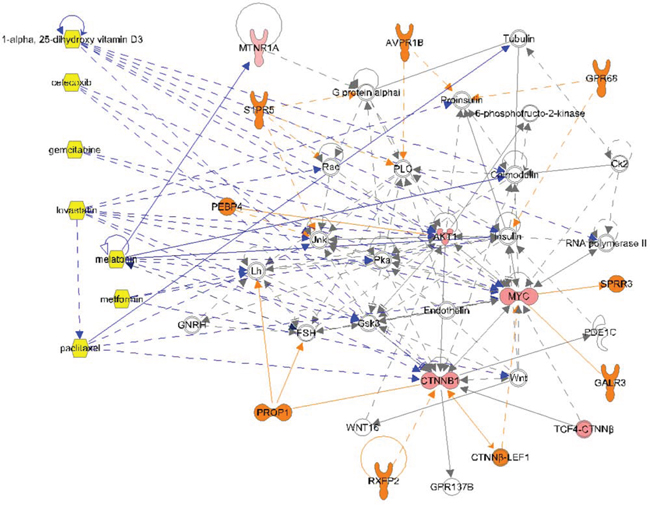
Figure 3: Complex molecular signaling network for osteosarcoma Patient #1. The patient's molecular profiling results were subjected to Ingenuity® Pathway Analysis to identify therapeutic targets. ORANGE: only in disease. PINK: measured analytes (lighter color is lower score). YELLOW: Drugs. DARK BLUE lines: drug interactions.
Given that the PIK3CA V344G mutation was an activating one, Patient #1 was started on metformin and rapamycin therapy, and the response was characterized as stable disease. The c-MET amplification seen in the exome sequencing was confirmed by IHC, and thus the patient also received crizotinib, a c-MET/ALK and ROS1 inhibitor. Unfortunately, his disease progressed on the crizotinib, rapamycin, and metformin combination. He was evaluated for another specific c-MET inhibitor study, but that study required a washout period of 4 weeks. Given the aggressive clinical course of his disease, he was started on gemcitabine and nab-paclitaxel therapy on the basis of the SPARC expression identified on morphoproteomic profiling. Celecoxib was added because of the COX2 expression in the IHC specimen. Unfortunately, his disease continued to progress and he eventually died of his disease.
Patient #2
This 16-year-old male was found to have a tumor in the left proximal tibia with anterior cruciate ligament tear. After biopsy of the tumor, osteoblastic osteosarcoma was diagnosed. The patient received neoadjuvant standard MAP chemotherapy according to the protocol AOST0331 regimen, but he was not officially enrolled in the trial. The patient underwent rotationplasty with surgery that revealed tumor necrosis greater than 95%, with negative margins. He then continued the MAP chemotherapy and developed bilateral hip and groin pain that did not respond to conservative management. An MRI of the left hip suggested a tumor focus, and biopsy showed high-grade metastatic osteosarcoma. The patient began therapy with liposomal doxorubicin and high-dose methotrexate with zoledronic acid. A follow-up positron emission tomographic–CT scan showed progression of disease in the left femur and right hip. He was referred to MD Anderson for treatment on a clinical trial of the alpha emitter radium 223 (http://ClinicalTrials.gov identifier: NCT01833520) [4] and was enrolled on the protocol. After one dose his disease progressed. He then received two cycles of ifosfamide, but his disease continued to progress. The patient underwent left leg amputation due to progressive disease. The patient was presented at treatment planning meeting after he was found to have progression on ifosfamide.
Patient #2: next-generation sequencing and therapy
Patient #2 had a comprehensive genomic profile that showed loss of NF2 exons 2–16, PDGFRA amplification, C17orf39 amplification, RB1 loss, and TP53 loss. The patient's Variant of Unknown Significance report showed the following aberrations: CUX1 L509V, GPR124 A1090P, RAD50 V842A, FANCD2 amplification, HGF R328H, VHL amplification, FGF14 rearrangement, IL7R E392K, FLCN amplification, and PCLO P1027T. The PDGFR amplification was confirmed by IHC, showing 90% PDGFRα positive (3+ expression in 90% of tumor cells).
Patient #2 was presented at the molecular treatment planning meeting at MD Anderson. On the basis of his molecular profile, the patient was enrolled in a Phase I clinical trial (http://ClinicalTrials.gov Identifier: NCT01187199) of bevacizumab and temsirolimus in combination with sorafenib for the treatment of advanced cancer [9]. The patient tolerated the therapy reasonably well, with the exception of grade 2 mucositis and grade 1 fatigue. Re-staging scans revealed multiple new bilateral lung opacities with minimal FDG activity, suggesting metastases. There also were new FDG-avid right hilar and left bronchial nodes and a new focus of FDG activity within the left scapula, also suggesting metastasis. Because of progressive disease, he was taken off the clinical trial. The patient elected to receive best supportive care.
DISCUSSION
Here we report the clinical course, comprehensive molecular profiling, and molecularly matched therapy for two patients with relapsed osteosarcoma. This in-depth analysis of two patients' cases offers insight into the complex biology of high risk osteosarcoma.
In Patient #1, c-MET amplification was shown by exome sequencing, and there was evidence that downstream activation of related pathways contributed to the growth of this patient's tumor. MET overexpression has been demonstrated to convert primary human osteoblasts into osteosarcoma [10]. The Met/HGF receptor has been shown to be overexpressed in human osteosarcomas and is activated by either a paracrine or an autocrine circuit, and therefore might play a role in aggressive osteosarcomas [11]. HGF activates both the mitogen and “motogen” machinery in human osteosarcoma cell lines, and this is related to activation of the ERK and Akt pathways [12]. P-ERK 1/2 (Thr202/Tyr204) and p-Akt (Ser473) were expressed in this patient's tumor (Figure 2). A small-molecule Met inhibitor, PF2362376, inhibited phosphorylation of Met, Erk, and Akt while inhibiting the proliferation of canine osteosarcoma cell lines and induced cell death at biologically achievable concentrations, and further was found to block activities associated with Met signaling, including migration, invasion, branching morphogenesis, and colony formation in soft agar [13].
In the morphoproteomic laboratory, the phospho-specific probe for tyrosine 1234/1235 was used to assess the state of phosphorylative activation of c-Met in Patient #1′s tumor cells; it detected 0 to 1+ (rare 2+) staining intensity on a scale of 0 to 3+. This is a relatively low level of expression of the constitutively activated c-Met pathway. The Caris profile, furthermore, did not show c-MET amplification by FISH. One of the reasons could be tumor heterogeneity.
The PIK3CA missense mutation V344G was detected in Patient #1′s tumor by exome sequencing by two different laboratories. This may represent an activating mutation and thus contribute to PI3K/Akt/mTOR pathway signaling. This was further reflected in the constitutive activation of the mTOR signal transduction pathway in the tumor. The PI3K/Akt/mTOR pathway has been reported to contribute to osteosarcoma progression [14]. The confirmed presence of MET amplification and p-ERK and p-AKT by IHC prompted the patient's treatment with crizotinib and rapamycin, but unfortunately he did not have a significant clinical response to this combination.
The CCNE1 amplification identified in Patient #1′s tumor by FoundationOne next-generation sequencing is important in that it encodes the cyclin E1 protein (which regulates G1 to S phase transition) via binding to and activating cyclin-dependent protein kinase 2 (cdk2). This protein complex has a direct role in initiation of replication and overall maintenance of genomic instability [15]. This particular amplification was reported in 27% of osteosarcomas (6/22) in one study, and was directly correlated with increased cyclin E1 expression, but it does not correspond with chemotherapy response rates [16]. There are no approved therapies that target the CCNE1 amplification, but clinical trials of cdk2 protein inhibitors are underway.
The PTPRD S1845fs*2 mutation detected in Patient #1′s tumor is an altered tumor suppressor gene that dephosphorylates the oncoprotein Stat3 [17], thus inactivating it. There are no approved treatments or clinical trials focused on PTPRD. However, the increased Stat3 activity induced by this mutation is a potential target, as clinical trials of inhibitors of Stat3 are underway. Interestingly, somatic and germline PTPRD mutations have been noted in Ewing sarcoma, another bone sarcoma that occurs in children and young adults. In a small study that sequenced the tumors of a few patients, it was intriguing that PTPRD germline aberrations conferred clinical benefit to patients with Ewing sarcoma who received therapy directed at the IGF1R and mTOR pathway [18].
After failure of crizotinib and rapamycin in Patient #1, additional morphoproteomic analysis was done that showed SPARC and COX2 expression. The patient then was started on a combination of gemcitabine, nab-paclitaxel, and celecoxib on the basis of these findings. Nab-paclitaxel binds to tubulin and stabilizes it, leading to mitotic catastrophe and apoptosis [19, 20]. By stabilizing beta-tubulin, the taxanes sequester Smad3 and reduce TGF-beta signaling [20]. Nab-paclitaxel's uptake is facilitated by SPARC [21–23], which was highly expressed in the patient's tumor, suggesting that the tumor may have been sensitive to this cytotoxic agent. In a xenograft model of human osteosarcoma with high SPARC expression, nab-paclitaxel showed a tumor inhibition rate of 98.8%, which was greater than those of doxorubicin (46.1%) or paclitaxel (40.8%) [24].
The sensitivity of tumor cells to taxanes can be enhanced by metformin via downregulation of p38MAPK-dependent ERCC1. Metformin inhibits both the mTORC1 pathway and components of the mTORC2 pathway (expressed in this patient's tumor) by activating AMPK [25]. It also inhibits the tumorigenic FASN pathway [25, 26], which was expressed in Patient #1′s tumor and has previously been associated with aggressive lung metastasis in osteosarcoma [27]. In vitro, metformin inhibits the mevalonate pathway and leads to increased cytotoxicity of anticancer drugs in an additive manner against human osteosarcoma cells [28]. It decreases the capacity of advanced glycation end products to upregulate the expression of RAGE in osteoblastic cells [29] and thereby could contribute to downregulation of the tumorigenic ERK pathway [30]. This is relevant to the observation that S100A7 promotes the migration and invasion of osteosarcoma cells via the receptor for advanced glycation end products [31].
In spite of the interrelated targetable pathways in Patient #1′s tumor, his cancer continued to progress. The lack of clinical benefit of the targeted therapy combinations may be attributable to the identified aberrations not being the driver aberrations or to the drugs chosen off-label not being the best ones to target the aberrations. Perhaps a specific P13K inhibitor or a more potent c-MET inhibitor could have yielded a more favorable response. These suggestions are speculative, but they reflect the complex biology of relapsed osteosarcoma biology, which may entail activation of multiple drug resistance pathways.
In Patient #2, the potential actionable aberrations included NF2 loss and PDGFRA aberration. Merlin, encoded by NF2, functions by coordinating signaling of receptor tyrosine kinases, such as the epidermal growth factor receptor (EGFR), with cell contact; the inactivation of Merlin in cancer disrupts this mechanism and leads to unrestrained receptor tyrosine kinase signaling despite cell contact [32]. Any alteration that results in the partial or complete loss of the FERM domain (amino acids 22–311) or the NF2 C-terminal moesin domain (aa 532–595) is predicted to cause a loss of function [33, 34]. Therefore, the alteration observed in this tumor is expected to cause a loss of function. The NF2 mutation was not reported in 14 osteosarcomas analyzed (COSMIC, Aug 2014). Mice heterozygous for NF2 develop a variety of cancers, including osteosarcomas [35], although neurofibromatosis 2 patients do not normally develop osteosarcomas, nor are mutations in NF2 frequently found in human osteosarcoma samples [36]. At present, there are no approved therapies that directly target NF2 loss. However, preclinical studies in models of NF2 loss have suggested that the mTOR inhibitors may be a relevant approach [37]. The mTOR inhibitors everolimus and temsirolimus have been approved by the U.S. Food and Drug Administration (FDA) for other cancer types and are under clinical investigation in solid tumors [38].
PDGFRA encodes platelet-derived growth factor receptor alpha (PDGFR-alpha), a tyrosine kinase receptor that, upon binding of cognate ligands (PDGFA or PDGFB), activates several signaling pathways, including PI3K and MAPK11. PDGFR aberrations, including point mutations, translocations, amplification, and/or overexpression, have been associated with various malignancies, leading to consideration of their protein products as oncoproteins [39]. PDGFRA alterations have not been reported in any of the 54 osteosarcoma cases analyzed (COSMIC, Nov 2014). PDGFR-alpha expression was reported in 4–90% of osteosarcomas, but its expression was not correlated with overall or disease-free survival [40, 41]. Several tyrosine kinase inhibitors that target the PDGFR-alpha and Beta proteins, as well as other kinases, are FDA-approved for other indications. These agents, including imatinib, sunitinib, sorafenib, dasatinib, nilotinib, regorafenib, ponatinib, and pazopanib, are currently in clinical trials for patients with various solid tumor types.
PDGFRA activation leads to activation of the PI3K/Akt and mTOR pathways [42]. Therefore, PI3K and mTOR pathway inhibitors may be relevant in a tumor with PDGFRA amplification. The mTOR inhibitors everolimus and temsirolimus are FDA-approved for other indications and remain under investigation in multiple solid tumor types.
Functional loss of the tumor suppressor p53, which is encoded by the TP53 gene, is common in aggressive advanced cancers [43]. Germline mutations in TP53 are associated with the very rare Li-Fraumeni syndrome and the early onset of many cancers, including osteosarcoma [44]. TP53 mutations have been reported in 27% of osteosarcomas (COSMIC, October 2014). Interestingly, it has been shown that, among patients with advanced cancer, those whose tumor had a TP53 mutation had a longer progression-free survival interval on a bevacizumab-containing regimen than those whose tumor had a wild-type TP53 [45].
These aberrations directed us to choose for Patient #2 a clinical trial of the combination of sorafenib, bevacizumab, and temsirolimus. Even this targeting of multiple pathways with a cocktail of drugs, however, was not able to overcome the complex biology of osteosarcoma. mTOR inhibitors were used in combination with other targeted therapies. It has been shown that mTOR inhibitors can overcome primary and/or acquired resistance to various tyrosine kinase inhibitors and monoclonal antibodies in various malignancies [9, 18, 46, 47].
Cancers like melanoma have frequent aberrations of the BRAF gene in more than 50% of patients and have three US FDA approved drugs targeting the BRAF V600 E aberration. Non-small cell Lung cancer is now a heterogenous disease with EGFR, BRAF, Her2/Neu aberrations or ALK, ROS1, RET or FGFR fusions [48–52]. These cancers are examples of recurrent driver aberrations with tyrosine kinase inhibitors in the clinic that are useful to act upon these genes. Even in these cancers although these genes are considered dominant drivers and frequently have exceptional responses to kinase inhibitors, resistance eventually develops. In fact, resistance mutations have been described with every kinase inhibitor [53]. Resistance mechanisms are complex and can be innate and / or acquired in the same kinase domain or through another mechanism upstream or downstream [18, 54]. Relapsed osteosarcoma is a highly aggressive disease and has an extremely complex biology. The clinical next generation sequencing and the IHC based profiling have their own limitations in terms of the genes or proteins tested. Firstly osteosarcoma is a disease that bears the stamp of chromothripsis arising from a single cellular catastrophic event than accumulation of aberrations over time. The aberrations that arise from this crisis are thus unpredictable other than TP53 which is till date one of the undruggable genes [55]. Secondly, the patients were given targeted therapy after chemotherapy refractory disease when the biology of the tumor and the drug resistance mechanisms are complex. Thirdly, we did not have active drugs in the clinic against several of the proteins tested eg: WNT pathway inhibitor or Glioma-associated oncogene protein (Gli2) pathway inhibitor. Fourthly, there is intra-tumoral and inter-tumoral heterogeneity. The metastatic sites of the disease would probably have a different biology when compared to the primary and secondary sites which was profiled. Lastly, there was a recent study published that showed the lack of efficacy of molecularly targeted agents outside of their approved indications did not improve progression-free survival when compared to physicians choice in advanced cancer patients [56]. So it may be that it may have not been the right drug to the right target at the right time and single kinase inhibitors may work only in rare cancer subsets. Moreover, most of the hallmarks of cancer are controlled by signaling networks at the cellular level that need a strategy combing kinomics, proteomics, drug screening with novel approaches using a comprehensive translational approach in appropriate models including patient derived xenograft models, genetically engineered mouse models and co-culture systems, more than the clinical next generation sequencing and IHC based approaches we have used.
One of the limitations of this study is that the patients were not tested for programmed cell death ligand 1 (PDL1) expression. PDL1 was recently shown to correlate with tumor-infiltrating lymphocytes in a subset of osteosarcoma patients [57, 58]. Given the increasing portfolio of agents targeting PD-1/PDL1, targeting this immune checkpoint may be an option for osteosarcoma.
MATERIALS AND METHODS
We identified two patients with relapsed, metastatic osteosarcoma who presented for targeted therapy options to the Clinical center for targeted therapy at The University of Texas MD Anderson Cancer Center. Their clinical information was obtained through review of their medical records. Treatment, consent and patient treatment on targeted therapy or on the investigational trial and data collection were performed in accordance with the guidelines of The University of Texas MD Anderson Cancer Center Institutional Review Board (IRB).
Molecular profiling of tumor samples
The patients' tumor samples were subjected to molecular profiling by next-generation exome sequencing of 186 cancer genes by the Illumina platform (FoundationOne, Boston, MA); by “Caris Target Now”–based molecular profiling (Caris Life Sciences, Irving, TX) that included immunohistochemistry (IHC), fluorescence in situ hybridization (FISH), and sequencing; by a Clinical Laboratory Improvement Amendments (CLIA)–certified MD Anderson Cancer Center next-generation sequencing 46-gene panel. In addition immunohistochemistry and morphoproteomics analyses were performed. These methods have been previously published [18, 59–61].
Biomedical analytics
The case scales from the morphoproteomic analysis were weighted and normalized to a set of profile scores according to a biomedical analytics algorithm customized for the pathologist. The scores, along with the Uniprot IDs of the measured analytes, were analyzed through the use of QIAGEN's Ingenuity® Pathway Analysis (IPA®, QIAGEN Redwood City, http://www.qiagen.com/ingenuity) to evoke the most likely biological pathways. A “Control” profile, consisting of the same Uniprot IDs without scores, was also created to minimize search bias in the IPA data mining. The patient's evoked pathway networks were then compared with those of the control, and evoked molecules unique to the tumor were identified. The interactions of proposed drugs with the identified disease-related biological pathway network(s) were then highlighted.
CONCLUSION
We report a comprehensive genomic profile and proteomic analysis of two patients with recurrent, metastatic osteosarcoma. Although potential targetable genomic alterations were identified in both tumors, these patients did not derive clinical benefit from the treatments chosen to target their cancer. Osteosarcoma is a heterogenous disease with a high degree of intratumoral variability. Although there is great promise in personalized medicine, these cases highlight the need for alternative, more effective approaches to guiding treatment decisions in this disease given the large volume of data generated by molecular profiling. Future measures exploring the unique biology and molecular pathogenesis of rare tumors may pave the way for the development of more specific and effective targeted therapies.
ACKNOWLEDGMENTS
The authors acknowledge Shanon Wilkes osteosarcoma research funds, High impact clinical research support program (HI-CRSP) funds from The University of Texas MD Anderson Cancer Center for the Radium 223 trial, National Institutes of Health Cancer Center Support Grant CA016672. The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication. We thank the motivated patients, their families for participation in targeted therapy. We also thank the clinical trial co-ordinator Ms. Danxia Ke, the advance practice provider Ms. Jenny Berry, ANP-BC and the clinical pharmacy specialist Allison Bass, PharmD, BCOP for helping to follow up these patients on targeted therapies.
Author contributions
All authors contributed to writing the manuscript. VS and PMA conceived the manuscript. VS, VOL, PMA provided clinical expertise. SW is the primary investigator for the clinical trial and VS is the co-primary investigator for the clinical trial. VS, MJW, MFM, NMS, ED, VOL, SW, SK, REB, PMA analyzed the data. REB provided pathology and morphoproteomics expertise. MFM provided the biomedical analytics expertise. VS, MJW, NMS, REB and PMA wrote the paper. VS and PMA provided sarcoma expertise. VS provided cellular, molecular and targeted therapy expertise. All authors read and approved the final manuscript.
CONFLICTS OF INTERESTS
The authors declare that they have no competing interests.
REFERENCES
1. Anderson P, Salazar-Abshire M. Improving outcomes in difficult bone cancers using multimodality therapy, including radiation: physician and nursing perspectives. Current oncology reports. 2006; 8:415–422.
2. Egas-Bejar D, Anderson PM, Agarwal R, Corrales-Medina F, Devarajan E, Huh WW, Brown RE, Subbiah V. Theranostic Profiling for Actionable Aberrations in Advanced High Risk Osteosarcoma with Aggressive Biology Reveals High Molecular Diversity: The Human Fingerprint Hypothesis. Oncoscience. 2014; 1:167–179.
3. Grignani G, Palmerini E, Ferraresi V, D’Ambrosio L, Bertulli R, Asaftei SD, Tamburini A, Pignochino Y, Sangiolo D, Marchesi E, Capozzi F, Biagini R, Gambarotti M, Fagioli F, Casali PG, Picci P, et al. Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: a non-randomised phase 2 clinical trial. The lancet oncology. 2015; 16:98–107.
4. Subbiah V, Anderson P, Rohren E. Alpha Emitter Radium 223 in High-Risk Osteosarcoma: First Clinical Evidence of Response and Blood-Brain Barrier Penetration. JAMA Oncol. 2015; 1:253–255.
5. Anderson PM, Subbiah V, Rohren E. Bone-seeking radiopharmaceuticals as targeted agents of osteosarcoma: samarium-153-EDTMP and radium-223. Advances in experimental medicine and biology. 2014; 804:291–304.
6. Subbiah V. Prospects and pitfalls of personalizing therapies for sarcomas: from children, adolescents, and young adults to the elderly. Current oncology reports. 2014; 16:401.
7. Zakikhani M, Blouin MJ, Piura E, Pollak MN. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast cancer research and treatment. 2010; 123:271–279.
8. Masri J, Bernath A, Martin J, Jo OD, Vartanian R, Funk A, Gera J. mTORC2 activity is elevated in gliomas and promotes growth and cell motility via overexpression of rictor. Cancer research. 2007; 67:11712–11720.
9. Subbiah V, Westin SN, Wang K, Araujo D, Wang WL, Miller VA, Ross JS, Stephens PJ, Palmer GA, Ali SM. Targeted therapy by combined inhibition of the RAF and mTOR kinases in malignant spindle cell neoplasm harboring the KIAA1549-BRAF fusion protein. Journal of hematology & oncology. 2014; 7:8.
10. Patane S, Avnet S, Coltella N, Costa B, Sponza S, Olivero M, Vigna E, Naldini L, Baldini N, Ferracini R, Corso S, Giordano S, Comoglio PM, Di Renzo MF. MET overexpression turns human primary osteoblasts into osteosarcomas. Cancer research. 2006; 66:4750–4757.
11. Ferracini R, Di Renzo MF, Scotlandi K, Baldini N, Olivero M, Lollini P, Cremona O, Campanacci M, Comoglio PM. The Met/HGF receptor is over-expressed in human osteosarcomas and is activated by either a paracrine or an autocrine circuit. Oncogene. 1995; 10:739–749.
12. Coltella N, Manara MC, Cerisano V, Trusolino L, Di Renzo MF, Scotlandi K, Ferracini R. Role of the MET/HGF receptor in proliferation and invasive behavior of osteosarcoma. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2003; 17:1162–1164.
13. Liao AT, McCleese J, Kamerling S, Christensen J, London CA. A novel small molecule Met inhibitor, PF2362376, exhibits biological activity against osteosarcoma. Veterinary and comparative oncology. 2007; 5:177–196.
14. Zhou R, Zhang Z, Zhao L, Jia C, Xu S, Mai Q, Lu M, Huang M, Wang L, Wang X, Jin D, Bai X. Inhibition of mTOR signaling by oleanolic acid contributes to its anti-tumor activity in osteosarcoma cells. Journal of orthopaedic research: official publication of the Orthopaedic Research Society. 2011; 29:846–852.
15. Moroy T, Geisen C. Cyclin, E. The international journal of biochemistry & cell biology. 2004; 36:1424–1439.
16. Lockwood WW, Stack D, Morris T, Grehan D, O’Keane C, Stewart GL, Cumiskey J, Lam WL, Squire JA, Thomas DM, O’Sullivan MJ. Cyclin E1 is amplified and overexpressed in osteosarcoma. The Journal of molecular diagnostics: JMD. 2011; 13:289–296.
17. Chan TA, Heguy A. The protein tyrosine phosphatase receptor D, a broadly inactivated tumor suppressor regulating STAT function. Cell cycle (Georgetown, Tex). 2009; 8:3063–3064.
18. Subbiah V, Naing A, Brown RE, Chen H, Doyle L, LoRusso P, Benjamin R, Anderson P, Kurzrock R. Targeted morphoproteomic profiling of Ewing’s sarcoma treated with insulin-like growth factor 1 receptor (IGF1R) inhibitors: response/resistance signatures. PloS one. 2011; 6:e18424.
19. Xiao H, Verdier-Pinard P, Fernandez-Fuentes N, Burd B, Angeletti R, Fiser A, Horwitz SB, Orr GA. Insights into the mechanism of microtubule stabilization by Taxol. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103:10166–10173.
20. Zhang D, Sun L, Xian W, Liu F, Ling G, Xiao L, Liu Y, Peng Y, Haruna Y, Kanwar YS. Low-dose paclitaxel ameliorates renal fibrosis in rat UUO model by inhibition of TGF-beta/Smad activity. Laboratory investigation; a journal of technical methods and pathology. 2010; 90:436–447.
21. Gradishar WJ. Albumin-bound paclitaxel: a next-generation taxane. Expert opinion on pharmacotherapy. 2006; 7:1041–1053.
22. Desai NP, Trieu V, Hwang LY, Wu R, Soon-Shiong P, Gradishar WJ. Improved effectiveness of nanoparticle albumin-bound (nab) paclitaxel versus polysorbate-based docetaxel in multiple xenografts as a function of HER2 and SPARC status. Anti-cancer drugs. 2008; 19:899–909.
23. Desai N, Trieu V, Damascelli B, Soon-Shiong P. SPARC Expression Correlates with Tumor Response to Albumin-Bound Paclitaxel in Head and Neck Cancer Patients. Translational oncology. 2009; 2:59–64.
24. Yang Y, Niu X, Zhang Q, Hao L, Ding Y, Xu H. The efficacy of abraxane on osteosarcoma xenografts in nude mice and expression of secreted protein, acidic and rich in cysteine. The American journal of the medical sciences. 2012; 344:199–205.
25. Algire C, Amrein L, Zakikhani M, Panasci L, Pollak M. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocrine-related cancer. 2010; 17:351–360.
26. Vazquez-Martin A, Corominas-Faja B, Cufi S, Vellon L, Oliveras-Ferraros C, Menendez OJ, Joven J, Lupu R, Menendez JA. The mitochondrial H(+)-ATP synthase and the lipogenic switch: new core components of metabolic reprogramming in induced pluripotent stem (iPS) cells. Cell cycle (Georgetown, Tex). 2013; 12:207–218.
27. Cui JJ, Tran-Dube M, Shen H, Nambu M, Kung PP, Pairish M, Jia L, Meng J, Funk L, Botrous I, McTigue M, Grodsky N, Ryan K, Padrique E, Alton G, Timofeevski S, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). Journal of medicinal chemistry. 2011; 54:6342–6363.
28. Rocha GZ, Dias MM, Ropelle ER, Osorio-Costa F, Rossato FA, Vercesi AE, Saad MJ, Carvalheira JB. Metformin amplifies chemotherapy-induced AMPK activation and antitumoral growth. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011; 17:3993–4005.
29. Schurman L, McCarthy AD, Sedlinsky C, Gangoiti MV, Arnol V, Bruzzone L, Cortizo AM. Metformin reverts deleterious effects of advanced glycation end-products (AGEs) on osteoblastic cells. Experimental and clinical endocrinology & diabetes: official journal, German Society of Endocrinology [and] German Diabetes Association. 2008; 116:333–340.
30. Cortizo AM, Lettieri MG, Barrio DA, Mercer N, Etcheverry SB, McCarthy AD. Advanced glycation end-products (AGEs) induce concerted changes in the osteoblastic expression of their receptor RAGE and in the activation of extracellular signal-regulated kinases (ERK). Molecular and cellular biochemistry. 2003; 250:1–10.
31. Kataoka K, Ono T, Murata H, Morishita M, Yamamoto KI, Sakaguchi M, Huh NH. S100A7 promotes the migration and invasion of osteosarcoma cells via the receptor for advanced glycation end products. Oncology letters. 2012; 3:1149–1153.
32. Curto M, McClatchey AI. Nf2/Merlin: a coordinator of receptor signalling and intercellular contact. British journal of cancer. 2008; 98:256–262.
33. Agarwal R, Liebe S, Turski ML, Vidwans SJ, Janku F, Garrido-Laguna I, Munoz J, Schwab R, Rodon J, Kurzrock R, Subbiah V. Targeted therapy for hereditary cancer syndromes: neurofibromatosis type 1, neurofibromatosis type 2, and Gorlin syndrome. Discovery medicine. 2014; 18:323–330.
34. Laulajainen M, Melikova M, Muranen T, Carpen O, Gronholm M. Distinct overlapping sequences at the carboxy-terminus of merlin regulate its tumour suppressor and morphogenic activity. Journal of cellular and molecular medicine. 2012; 16:2161–2175.
35. McClatchey AI, Saotome I, Mercer K, Crowley D, Gusella JF, Bronson RT, Jacks T. Mice heterozygous for a mutation at the Nf2 tumor suppressor locus develop a range of highly metastatic tumors. Genes & development. 1998; 12:1121–1133.
36. Stemmer-Rachamimov AO, Nielsen GP, Rosenberg AE, Louis DN, Jones D, Ramesh V, Gusella JF, Jacoby LB. The NF2 gene and merlin protein in human osteosarcomas. Neurogenetics. 1998; 2:73–74.
37. James MF, Han S, Polizzano C, Plotkin SR, Manning BD, Stemmer-Rachamimov AO, Gusella JF, Ramesh V. NF2/merlin is a novel negative regulator of mTOR complex 1, and activation of mTORC1 is associated with meningioma and schwannoma growth. Molecular and cellular biology. 2009; 29:4250–4261.
38. Subbiah V, Slopis J, Hong DS, Ketonen LM, Hamilton J, McCutcheon IE, Kurzrock R. Treatment of patients with advanced neurofibromatosis type 2 with novel molecularly targeted therapies: from bench to bedside. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012; 30:e64–68.
39. Fletcher JA. Role of KIT and platelet-derived growth factor receptors as oncoproteins. Seminars in oncology. 2004; 31:4–11.
40. Kubo T, Piperdi S, Rosenblum J, Antonescu CR, Chen W, Kim HS, Huvos AG, Sowers R, Meyers PA, Healey JH, Gorlick R. Platelet-derived growth factor receptor as a prognostic marker and a therapeutic target for imatinib mesylate therapy in osteosarcoma. Cancer. 2008; 112:2119–2129.
41. Sulzbacher I, Birner P, Dominkus M, Pichlhofer B, Mazal PR. Expression of platelet-derived growth factor-alpha receptor in human osteosarcoma is not a predictor of outcome. Pathology. 2010; 42:664–668.
42. Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes & development. 2008; 22:1276–1312.
43. Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nature reviews Cancer. 2009; 9:862–873.
44. Agarwal R, Liebe S, Turski ML, Vidwans SJ, Janku F, Garrido-Laguna I, Munoz J, Schwab R, Rodon J, Kurzrock R, Subbiah V. Targeted therapy for hereditary cancer syndromes: hereditary breast and ovarian cancer syndrome, Lynch syndrome, familial adenomatous polyposis, and Li-Fraumeni syndrome. Discovery medicine. 2014; 18:331–339.
45. Said R, Hong DS, Warneke CL, Lee JJ, Wheler JJ, Janku F, Naing A, Falchook GS, Fu S, Piha-Paul S, Tsimberidou AM, Kurzrock R. P53 mutations in advanced cancers: clinical characteristics, outcomes, and correlation between progression-free survival and bevacizumab-containing therapy. Oncotarget. 2013; 4:705–714.
46. Subbiah V, Brown RE, McGuire MF, Buryanek J, Janku F, Younes A, Hong D. A novel immunomodulatory molecularly targeted strategy for refractory Hodgkin’s lymphoma. Oncotarget. 2014; 5:95–102.
47. Agarwal R, Koenig K, Rohren E, Subbiah V. Combined Antiangiogenic and Mammalian Target of Rapamycin Inhibitor Targeted Therapy in Metaplastic Breast Cancer Harboring a PIK3CA Mutation. Journal of breast cancer. 2014; 17:287–290.
48. Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, Wolf J, Raje NS, Diamond EL, Hollebecque A, Gervais R, Elez-Fernandez ME, Italiano A, Hofheinz RD, Hidalgo M, Chan E, et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. The New England journal of medicine. 2015; 373:726–736.
49. Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, Riely GJ, Varella-Garcia M, Shapiro GI, Costa DB, Doebele RC, Le LP, Zheng Z, Tan W, Stephenson P, Shreeve SM, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. The New England journal of medicine. 2014; 371:1963–1971.
50. Shaw AT, Engelman JA. ALK in lung cancer: past, present, and future. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013; 31:1105–1111.
51. Subbiah V, Berry J, Roxas M, Guha-Thakurta N, Subbiah IM, Ali SM, McMahon C, Miller V, Cascone T, Pai S, Tang Z, Heymach JV. Systemic and CNS activity of the RET inhibitor vandetanib combined with the mTOR inhibitor everolimus in KIF5B-RET re-arranged non-small cell lung cancer with brain metastases. Lung Cancer. 2015; 89:76–79.
52. Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, Asaka R, Hamanaka W, Ninomiya H, Uehara H, Lim Choi Y, Satoh Y, Okumura S, Nakagawa K, Mano H, Ishikawa Y. RET, ROS1 and ALK fusions in lung cancer. Nature medicine. 2012; 18:378–381.
53. Subbiah IM, Subbiah V. Exceptional responders: in search of the science behind the miracle cancer cures. Future oncology (London, England). 2015; 11:1–4.
54. Subbiah V, Trent JC, Kurzrock R. Resistance to mammalian target of rapamycin inhibitor therapy in perivascular epithelioid cell tumors. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2010; 28:e415.
55. Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, McLaren S, Lin ML, McBride DJ, Varela I, Nik-Zainal S, Leroy C, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011; 144:27–40.
56. Le Tourneau C, Delord JP, Goncalves A, Gavoille C, Dubot C, Isambert N, Campone M, Tredan O, Massiani MA, Mauborgne C, Armanet S, Servant N, Bieche I, Bernard V, Gentien D, Jezequel P, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. The lancet oncology. 2015; 16:1324–1334.
57. Shen JK, Cote GM, Choy E, Yang P, Harmon D, Schwab J, Nielsen GP, Chebib I, Ferrone S, Wang X, Wang Y, Mankin H, Hornicek FJ, Duan Z. Programmed cell death ligand 1 expression in osteosarcoma. Cancer immunology research. 2014; 2:690–698.
58. Shen JK, Cote GM, Choy E, Hornicek FJ, Duan Z. Targeting programmed cell death ligand 1 in osteosarcoma: an auto-commentary on therapeutic potential. Oncoimmunology. 2014; 3:e954467.
59. Brown RE. Morphogenomics and morphoproteomics: a role for anatomic pathology in personalized medicine. Archives of pathology & laboratory medicine. 2009; 133:568–579.
60. Subbiah V, Brown RE, Jiang Y, Buryanek J, Hayes-Jordan A, Kurzrock R, Anderson PM. Morphoproteomic profiling of the mammalian target of rapamycin (mTOR) signaling pathway in desmoplastic small round cell tumor (EWS/WT1), Ewing’s sarcoma (EWS/FLI1) and Wilms’ tumor(WT1). PloS one. 2013; 8:e68985.
61. Subbiah V, Brown RE, Buryanek J, Trent J, Ashkenazi A, Herbst R, Kurzrock R. Targeting the apoptotic pathway in chondrosarcoma using recombinant human Apo2L/TRAIL (dulanermin), a dual proapoptotic receptor (DR4/DR5) agonist. Molecular cancer therapeutics. 2012; 11:2541–2546.
62. Liu ZL, Wang G, Peng AF, Luo QF, Zhou Y, Huang SH. Fatty acid synthase expression in osteosarcoma and its correlation with pulmonary metastasis. Oncology letters. 2012; 4:878–882.
63. Chu F, Chou PM, Zheng X, Mirkin BL, Rebbaa A. Control of multidrug resistance gene mdr1 and cancer resistance to chemotherapy by the longevity gene sirt1. Cancer research. 2005; 65:10183–10187.
64. Tu B, Du L, Fan QM, Tang Z, Tang TT. STAT3 activation by IL-6 from mesenchymal stem cells promotes the proliferation and metastasis of osteosarcoma. Cancer letters. 2012; 325:80–88.
65. Ryu K, Choy E, Yang C, Susa M, Hornicek FJ, Mankin H, Duan Z. Activation of signal transducer and activator of transcription 3 (Stat3) pathway in osteosarcoma cells and overexpression of phosphorylated-Stat3 correlates with poor prognosis. Journal of orthopaedic research: official publication of the Orthopaedic Research Society. 2010; 28:971–978.
66. Rodriguez NI, Hoots WK, Koshkina NV, Morales-Arias JA, Arndt CA, Inwards CY, Hawkins DS, Munsell MF, Kleinerman ES. COX-2 expression correlates with survival in patients with osteosarcoma lung metastases. Journal of pediatric hematology/oncology. 2008; 30:507–512.
67. Li Y, Gao Q, Yin G, Ding X, Hao J. WNT/beta-catenin-signaling pathway stimulates the proliferation of cultured adult human Sertoli cells via upregulation of C-myc expression. Reproductive sciences. 2012; 19:1232–1240.
68. Iwaya K, Ogawa H, Kuroda M, Izumi M, Ishida T, Mukai K. Cytoplasmic and/or nuclear staining of beta-catenin is associated with lung metastasis. Clinical & experimental metastasis. 2003; 20:525–529.
69. Zambo I, Hermanova M, Adamkova Krakorova D, Mudry P, Zitterbart K, Kyr M, Vesely K, Sterba J, Veselska R. Nestin expression in high-grade osteosarcomas and its clinical significance. Oncology reports. 2012; 27:1592–1598.
70. Navid F, Letterio JJ, Yeung CL, Pegtel M, Helman LJ. Autocrine Transforming Growth Factor-beta Growth Pathway in Murine Osteosarcoma Cell Lines Associated with Inability to Affect Phosphorylation of Retinoblastoma Protein. Sarcoma. 2000; 4:93–102.
71. Dennler S, Andre J, Alexaki I, Li A, Magnaldo T, ten Dijke P, Wang XJ, Verrecchia F, Mauviel A. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer research. 2007; 67:6981–6986.
72. Hirotsu M, Setoguchi T, Sasaki H, Matsunoshita Y, Gao H, Nagao H, Kunigou O, Komiya S. Smoothened as a new therapeutic target for human osteosarcoma. Molecular cancer. 2010; 9:5.
73. Dalla-Torre CA, Yoshimoto M, Lee CH, Joshua AM, de Toledo SR, Petrilli AS, Andrade JA, Chilton-MacNeill S, Zielenska M, Squire JA. Effects of THBS3, SPARC and SPP1 expression on biological behavior and survival in patients with osteosarcoma. BMC cancer. 2006; 6:237.