
INTRODUCTION
Breast cancer has been shown to be a highly heterogeneous disease through genomic analysis of both human and the mouse tumor samples [1–7]. Classification based on gene expression profiles of tumor samples resulted in a detailed analysis of human breast cancer, allowing for a better understanding of the disease at both molecular and clinical levels. However, breast cancer is still lethal, predominately due to metastatic progression [8]. Breast cancer metastasis is a complex multistep process that involves detachment of tumor cells from the original site, intravasation into the blood vessel, extravasation out of the blood vessel, and colonization of distant organs such as the bone, brain, lung, and liver [9–11].
To study breast cancer development and progression, one approach that has been used is the generation of genetically engineered mouse model systems. One well studied model is the highly metastatic MMTV-PyMT strain, where metastasis to the lung is observed in virtually all mice reaching tumor endpoint [12, 13]. Conversely, the MMTV-Myc model [14] is poorly metastatic [15–17]. This suggests that MMTV-Myc tumors would require additional genetic events in order to model metastasis found in c-Myc associated human breast cancer metastasis.
In mouse models of breast cancer, the E2F transcription factors have been predicted to be activated in many tumor models [7]. Traditionally, E2F transcription factors have been well described to regulate cell cycle [18–20]. In cancer, E2Fs have been implicated in tumor development, progression [21] and angiogenesis [22]. Specifically in breast cancer, E2F1 expression has been shown to be reduced in primary and metastatic breast carcinoma [23, 24] and deletion of the E2F2 chromosomal region was also observed [25]. In the analysis of mouse mammary tumors from MMTV-Myc mice, an enrichment of E2F bound genes in the EMT/squamous subset of tumor samples was noted [26]. Recently, perturbation of individual E2Fs in the MMTV-PyMT model was shown to affect latency, histology, vasculature, and importantly, the metastatic capability of these tumors [13]. Specifically, E2F1 and E2F2 loss was shown to vastly reduce metastasis. Other studies have implicated the E2Fs in the tumor process, including an examination of Neu and Myc initiated tumors in the absence of various E2Fs with impacts on latency and metastasis that varied by model [27–29]. In the metastatic process, E2Fs have been shown to mediate metastasis by regulating matrix metalloproteinase [30] and that expression of E2F1 increased extravasation of circulating cancer cells from the endothelium [31].
We have previously shown that the perturbation of activator E2Fs levels in the MMTV-Myc mouse model of breast cancer affected incidence and latency [28]. Here we demonstrate that loss of E2F2 in Myc induced tumors dramatically increased breast cancer metastasis. To define the role of E2F2 in the metastasis process, we generated gene expression data with and without E2F2 in Myc induced tumors. Candidate genes regulating metastasis were identified and tested for roles in metastasis. This analysis demonstrated that a tumor suppressor gene, PTPRD [32–34], may act in conjunction with E2F2 to mediate metastasis.
RESULTS
E2F2 loss induces metastasis in MMTV-Myc driven tumors
Our previous research had predicted and demonstrated a role for E2F pathway activation in MMTV-Myc tumors [28]. In that work, a Kaplan-Meier survival plot revealed a significant acceleration in tumor onset when E2F1 was lost and a delay in tumor onset when E2F2 or E2F3 was lost. Indeed, we saw that loss of E2F2 increased time to tumor onset by an average of 160 days (Figure 1A; p = 0.0057). Here we observed that the loss of any E2F increased metastases in MMTV-Myc intiated tumors which are normally not highly metastatic (13%). However, only the loss of E2F2 significantly increased the percentage of tumor bearing mice with metastasis to the lung to 67% (Figure 1B; p = 0.0361).
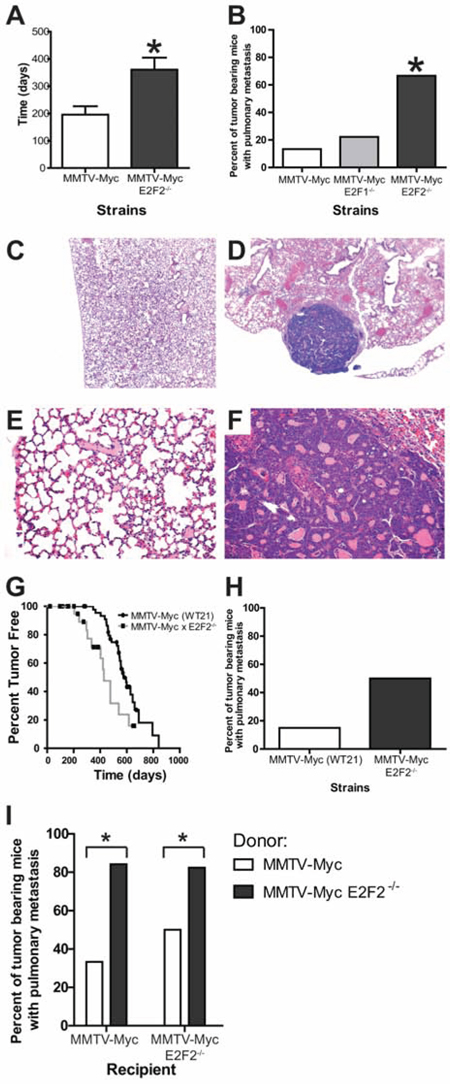
Figure 1: E2F2 loss induces metastasis in Myc driven tumors. MMTV-Myc transgenic mice were interbred with E2F2−/− mice and tumor latency was examined. Myc tumors developing in the absence of E2F2 had a significantly increased time to tumor onset (A. p = 0.0057). Metastasis is rarely observed in MMTV-Myc mice with only 13% of tumor bearing mice having lung metastasis (B. n = 2/15). Metastatic incidence is increased to 67% when Myc tumors develop in the E2F2 knockout background (n = 6/9; p-value = 0.0361). Histology of a MMTV-Myc mouse lung showing the absence of lung metastasis at 4X C. compared with the metastases observed in the MMTV-Myc E2F2 null strain D. Increased magnification (20X) of these sections revealed secondary structure within the metastatic lesion E and F. To ensure that the metastatic effect of E2F2 loss was not a strain specific effect, MMTV-Myc WT21 mice were interbred with E2F2−/− mice. Loss of E2F2 in the MMTV-Myc WT21 background resulted in decreased latency G. and trend towards increased percentage of metastasis bearing mice H. Transplantation of MMTV-Myc WT21 E2F2−/− tumors into MMTV-Myc WT21 or MMTV-Myc WT21 E2F2−/− backgrounds produced striking metastases, suggesting that loss of E2F2 affected metastasis in a cell autonomous manner I.
Other published work noted that E2F2 loss in Wap-Myc mice decreased time to tumor onset and did not describe a metastatic phenotype [29]. Given the differences in latency effects between the two published MMTV-Myc E2F reports, we sought to ensure that the metastatic effects were not a strain specific artifact. To do this, we interbred a separate MMTV-Myc transgenic, MMTV-Myc WT21 [26] with the E2F2 knockouts. With the loss of E2F2 in this strain, tumors developed 147 days earlier (Figure 1G). Importantly, despite the differences in latency, loss of E2F2 in this strain also increased the metastatic frequency by 35% (Figure 1H), demonstrating the role of E2F2 in MMTV-Myc mediated metastasis was not a strain dependent artifact. Furthermore, transplantation of MMTV-Myc WT21 E2F2−/− tumors into MMTV-Myc WT21 or MMTV-Myc WT21 E2F2−/− background revealed that loss of E2F2 increased metastasis in a cell autonomous manner (Figure 1I; p = 0.0177 and 0.0382, respectively) which further confirmed our finding of the role of E2F2 in MMTV-Myc mediated metastasis.
Histology of pulmonary sections for the MMTV-Myc strains typically resulted in sections lacking metastases (Figure 1C) in 87% of tumor bearing mice. Conversely, metastatic lesions were readily visible in the MMTV-Myc mice lacking E2F2 (Figure 1D) in 67% of tumor bearing mice. At high power, normal lung morphology was observed in the MMTV-Myc strain (Figure 1E) while secondary structure in the lung metastases was observed (Figure 1F). Lung metastases at necropsy were occasionally large enough (2–3 mm in diameter) that flash frozen samples were isolated for gene expression studies.
Gene expression alterations associated with lung metastasis
To begin to determine the mechanism by which loss of E2F2 function increased metastasis in Myc induced tumors, we examined gene expression by microarray in MMTV-Myc and MMTV-Myc E2F2−/− tumors. We included tumors from MMTV-Myc E2F1−/− and MMTV-Myc E2F3+/− as controls for tumors with loss of activator E2Fs without the presence of metastases. In addition to 20 primary tumors from each genotype, we assayed 6 E2F mutant lung metastasis samples. Unsupervised hierarchical clustering divided these primary samples based on their histological type, rather than by their genotype. Interestingly, the lung metastasis samples clustered together and were most closely related to the papillary subtype. (Figure 2A). Focusing on metastasis in a clustering analysis, groups of genes that were upregulated uniquely in each cluster were identified. For instance, in Cluster D, 108 genes defined the 6 lung metastases samples and clearly differentiated between these samples and other samples. The transcriptional control of these groups was examined through an over-representation analysis using predicted transcription factor binding sites. This revealed an enrichment in genes with predicted E2F binding sites in genes upregulated in lung metastasis (gene cluster D; Figure 2B). Given that epithelial-mesenchymal transition (EMT) has been linked to the propensity of cells to metastasize [35], we compared EMT and lung metastasis clusters using GSEA. As expected, enrichment of genes regulated by SMAD2, SMAD3, and SMAD4 were enriched in the EMT cluster (Figure 2C; p = 0.03). Interestingly, this experiment demonstrated an enrichment of an invasive ovarian epithelial cancer geneset in the lung metastastic samples, suggesting that similar gene expression patterns could be shared (Figure 2D, p = 0.016).
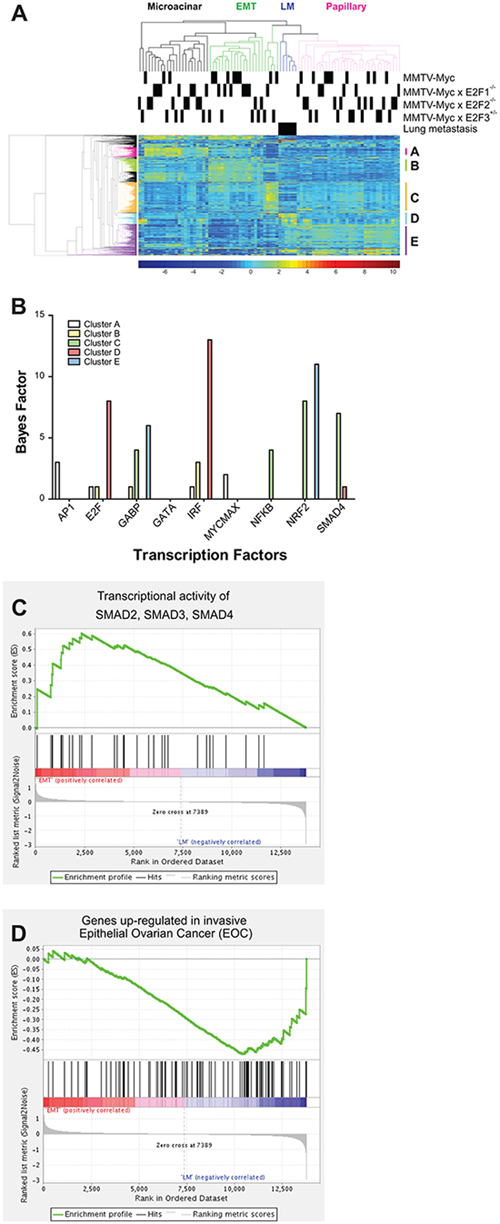
Figure 2: Gene expression alterations associated with lung metastasis. Gene expression analysis of MMTV-Myc tumors in E2F the WT, E2F1−/−, E2F2−/− and E2F3+/− backgrounds and 6 sample of lung metastasis were analyzed by microarray. Unsupervised hierarchical clustering revealed that samples were clustered based on their histological type rather than their genotype with lung metastasis samples being clustered together A. The clustering of the metastatic samples in one group, closely related to the papillary subtype was noted. We examined the indicated sub-clusters of genes for predicted transcription factor binding demonstrating that genes upregulated in metastasis, represented in cluster D, were enriched for E2F binding motifs B. Comparison of EMT and lung metastasis samples by GSEA demonstrated enrichment of genes regulated by SMAD2, SMAD3, and SMAD4 was elevated in EMT relative to lung metastases (C. p = 0.03). Enrichment of genes up-regulated in invasive ovarian epithelial cancer was noted in the metastasis samples (D. p = 0.016).
Pipeline for identification of genes regulating E2F2−/− breast cancer metastasis
In order to identify the genetic mechanism altered with the loss of E2F2 that resulted in an increase of lung metastasis in Myc induced tumors we examined gene expression data from tumors and metastases. The goal of this analysis was to identify genes that were regulated directly or indirectly by E2F2. Importantly, we sought to examine genes that also affected human metastatic outcome. The data analysis pipeline (Figure 3A), began by examining fold change from MMTV-Myc and MMTV-Myc E2F2−/− tumors as well as lung metastasis samples. 451 genes that are homologous to human were differentially expressed (Figure 3B and 3C). These putative target genes were then stratified based on their correlation to human distant metastasis survival, resulting in 61 genes with expression that correlated to human distant metastasis survival (Supplementary Table 1). A cox-hazard ratio analysis further narrowed this list to 28 genes (Supplementary Table 2). To then identify direct E2F target genes, we examined these genes for predicted E2F binding sites. This analysis predicted that 21 genes had E2F binding sites in a loose prediction and 3 genes in more stringent motif predictions (Supplementary Table 3). Combining these various criteria, we identified 7 genes that had potential to mediate metastasis by E2F2 loss in Myc induced breast cancer. Of those 7 genes, PTPRD was the strongest candidate but appeared to be an indirect E2F2 target. Interestingly, PTPRD was recently identified in the TCGA project [32] as being significantly mutated in breast cancer.
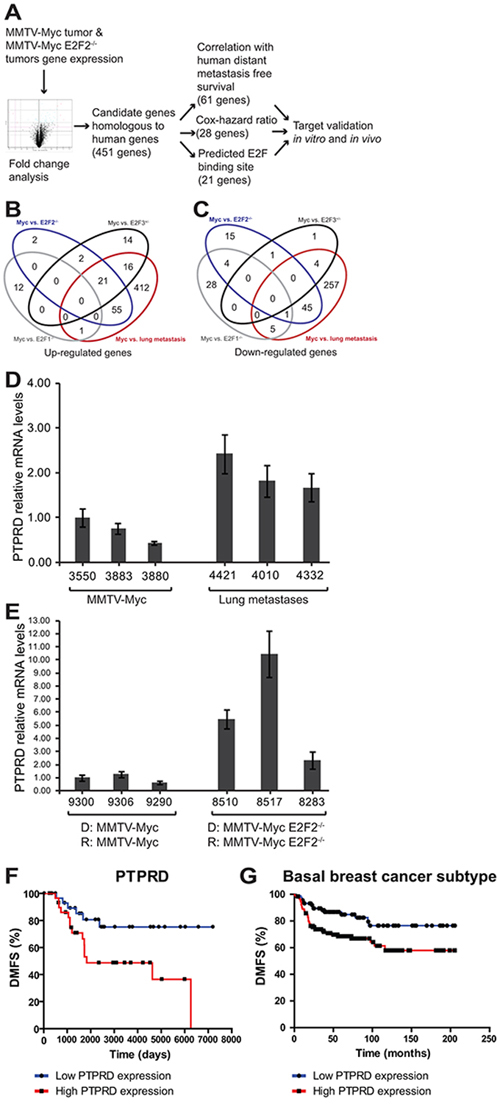
Figure 3: Pipeline for identification of genes regulating E2F2−/− breast cancer metastasis. To examine the regulatory mechanisms involved in E2F2-mediated human breast cancer metastasis, we established a pipeline for analysis. Genes that were differentially expressed by MMTV-Myc, MMTV-Myc x E2F2−/− and lung metastasis samples A–C. were identified. Mouse gene expression data for significantly expressed genes was clustered together with human datasets, revealing 451 candidate genes that are homologous to human genes. We ranked these potential target genes based on their correlation with human distant metastasis free survival, their cox-hazard ratio, and the existence of E2F motifs proximal to the transcriptional start site. Comparison between MMTV-Myc tumors and lung metastases revealed elevated PTPRD expression in the lung metastases samples D. Comparison of MMTV-Myc WT21 non-metastatic tumors and MMTV-Myc WT21 E2F2−/− metastatic tumors passaged in their respective genotype revealed increased PTPRD expression in the MMTV-Myc WT21 E2F2−/− tumors (E. D = donor, R = recipient). Elevated levels of PTPRD were found to be correlated with human distant metastasis free survival (F. p = 0.0153) and is associated with basal breast cancer subtype (G. p = 0.0085).
Microarray analyses revealed that PTPRD was upregulated in lung metastases samples compared to tumor samples. This finding was confirmed by qRT-PCR (Figure 3D). Furthermore, examination of MMTV-Myc WT21 tumors and MMTV-Myc WT21 E2F2−/− tumors that were passaged in their respective background showed relatively high PTPRD expression in the metastatic MMTV-Myc WT21 E2F2−/− tumors compared to the MMTV-Myc WT21 tumors, further confirming our initial findings (Figure 3E). The metastatic importance of PTPRD in human breast cancer was observed in the examination of the distant metastasis free survival (DMFS) curve where high levels of PTPRD were associated with poor DMFS outcomes (Figure 3F; p = 0.0153) relative to low levels of PTPRD and was associated with basal subtype of human breast cancer (Figure 3G; p = 0.0085, hazard ratio = 2.3 (1.2–3.78)).
E2F2 knockdown in human breast cancer increases migration and lung colonization
In order to establish a system where we could assess the effects of PTPRD knockdown, we began by examining whether the mouse effects of E2F2 translated to human breast cancer. Using an shRNA approach we knocked down E2F2 levels in MDA-MB-231 cells. The knockdown of E2F2 was validated in stable clones through immunoblotting for E2F2 in relation to the Grb2 loading control (Figure 4A). No alterations to proliferation were noted (Supplementary Figures 1 and 2). Parental cells and a scrambled shRNA construct were used as controls when we assayed for migratory ability in a transwell assay. The transwell migration assay revealed that knockdown of E2F2 resulted in increased cell migration through the transwell insert membrane relative to controls (Figure 4B–4D, p < 0.0001; Supplementary Figures 1 and 2; p < 0.05). This increased propensity to migrate was also demonstrated through a wound healing assay (data not shown).
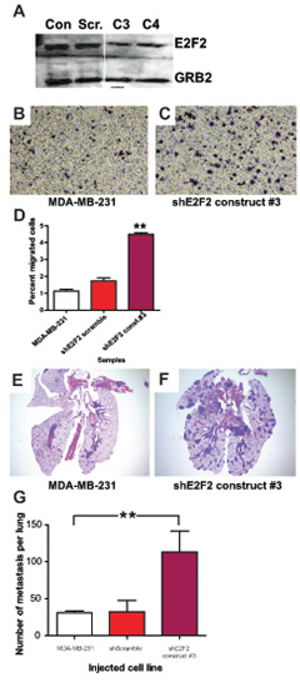
Figure 4: E2F2 knockdown in human breast cancer increases migration and lung colonization. E2F2 knockdown in human breast cancer was achieved by transfection of MDA-MB-231 cells with shE2F2. Efficacy of E2F2 knockdown was assayed by western blotting A. with untransfected MDA-MB-231 (Con.), MDA-MB-231 transfected with shScramble (Scr.), shE2F2 construct #3 (C3), shE2F2 construct #4 (C4). Migration of MDA-MB-231 control cells B. and with E2F2 knockdown C. in transwell migration assays revealed that the percentage of cells that migrated across the membrane increased when the level of E2F2 was decreased (D. p < 0.0001). In colonization assays with and without the knockdown, lesions were found in the lungs of mice injected with MDA-MB-231 E. and greatly increased with transfection of shE2F2 F. Quantification of the numbers of metastatic lesions revealed an increased number of metastatic lesions in mice injected with MDA-MB-231 transfected with shE2F2 (G. p = 0.0184).
Given that the in vitro experiments only examine a portion of the functions necessary for metastasis, we examined the ability of the cells to colonize the lungs by injecting the cells into the bloodstream. As expected, in control cell lines this resulted in localized discreet colonization (Figure 4E). Strikingly, knockdown of E2F2 resulted in a significant increase in metastatic lesions (Figure 4F). Quantitation of these effects revealed a large increase in number of metastatic lesions in a section of the lung by more than four folds (Figure 4G, p = 0.0184). Taken together our results in vitro and in vivo showed that knockdown of E2F2 increased the metastatic capability, correlating well with the increased metastasis in the MMTV-Myc E2F2−/− mouse model.
PTPRD knockdown in human breast cancer decreases migration and lung colonization
After establishing an experimental system whereby E2F2 knockdown increased metastatic potential, we then examined if loss of PTPRD would reduce metastasis in the same system. To this end, we used an shRNA approach to knockdown levels of PTPRD to 40% of wild type levels (Figure 5A, p = 0.01). No effects on proliferation were noted (Supplementary Figure 2B).
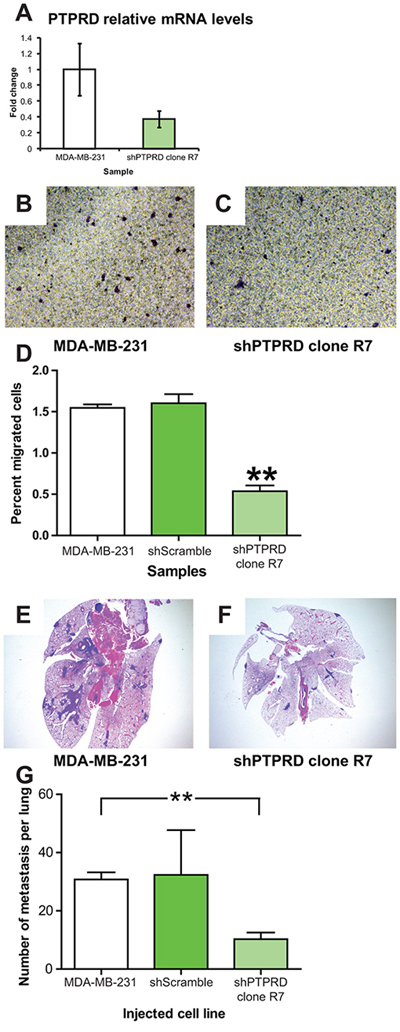
Figure 5: PTPRD knockdown in human breast cancer decreases migration and lung colonization. PTPRD knockdown in human breast cancer was achieved by transfection of MDA-MB-231 cells with shPTPRD. Efficacy of PTPRD knockdown was assayed by qRT-PCR (A. p = 0.01). Migration of MDA-MB-231 control cells B. and with PTPRD knockdown C. in transwell migration assays revealed that the percentage of cells that migrated across the membrane decreased when the level of PTPRD was decreased (D. p < 0.0001). In colonization assays with and without the knockdown, lesions were found in the lungs of mice injected with MDA-MB-231 E. and decreased with injection of MDA-MB-231 transfected with shPTPRD F. Quantification of the numbers of metastatic lesions revealed a decreased number of lesion in mice injected with MDA-MB-231 transfected with shPTPRD (G. p = 0.0009).
Effects of PTPRD knockdown were asssayed in vitro by a transwell migration assay. This demonstrated that knockdown of PTPRD resulted in a significant decrease of the percentage of cells that migrated through the transwell insert membrane (Figure 5B–5D; p < 0.0001; Supplementary Figures 3 and 4; p < 0.01 and p < 0.05, respectively). In vivo, knockdown of PTPRD resulted in fewer metastatic lesions in the lung relative to controls after cells were injected retro-orbitally (Figure 5E–5G; p = 0.0009). This result reflected our gene expression finding in the MMTV-Myc mouse model where PTPRD was found to be upregulated by 2.83 fold in lung metastasis compared to MMTV-Myc tumor samples. This fold change difference was further validated by using qRT-PCR which showed that PTPRD RNA expression was increased by 2 fold in lung metastases samples compared to MMTV-Myc samples (Figure 3D). Taken together, these data indicated that PTPRD knockdown significantly decreased metastatic capability.
Regulatory network analysis indirectly connected E2F2 and PTPRD
Examination of the networks linking PTPRD and E2F2 showed that E2F2 is potentially indirectly linked with PTPRD (Figure 6A). Given the role of BCAR1 in mediating breast cancer invasion [36], it appears that there may be a critical link for mediation of metastasis in human breast cancer between E2F2 and PTPRD. In order to compare the mouse model data with human breast cancer, we co-clustered the mouse tumors with human breast cancer samples. Interestingly, unsupervised hierarchical clustering revealed that the lung metastases all clustered together with a subset of human breast cancer samples (Figure 6B, cluster B). We predicted the probability of E2F2 pathway activation in these subsets of human tumors and used it to stratify clinical gene expression samples. A comparison between cluster B and the closest neighboring cluster (Figure 6B, cluster A) for samples with low E2F2 activity revealed low E2F2 pathway activation in cluster B was correlated to decreased time to distant metastasis whereas in cluster A low probability of E2F2 pathway activation was correlated to increased time to distant metastasis free survival (Figure 6C; p < 0.0001). Survival analysis showed that overexpression of PTPRD decreased time to distant metastasis in the basal subtype of human breast cancer (Figure 3G). In addition, examination of mouse gene expression data showed that in PTPRD was highly expressed in cluster B (Supplementary Tables 6 and 7; Supplementary Figure 5). Taken together these findings suggested that E2F2 loss regulated breast cancer metastasis in a subpopulation of human tumors, potentially through a PTPRD signaling axis.
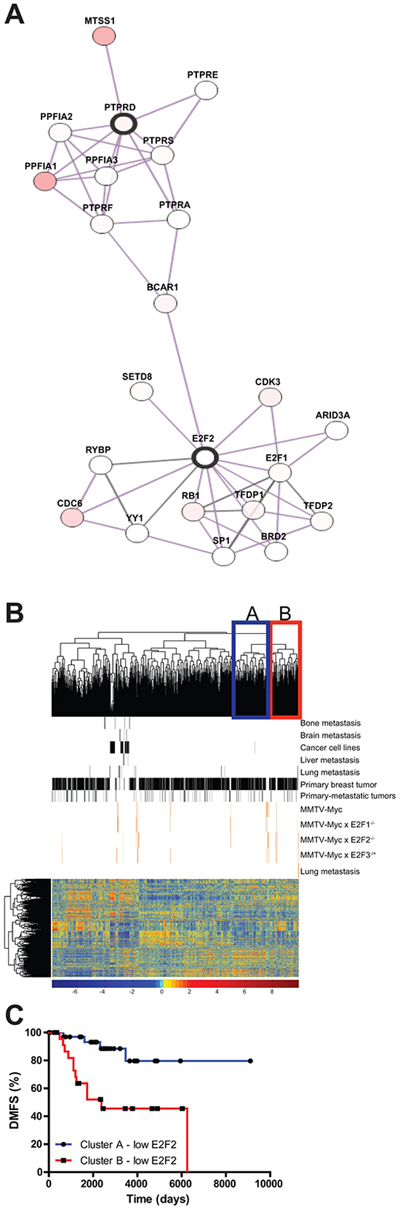
Figure 6: E2F2, PTPRD, and connections to human breast cancer. We examined a protein-protein interaction network and found that E2F2 and PTPRD were connected through MYC and STAT3 A. Unsupervised clustering of human breast tumor datasets and mouse tumor dataset revealed a cluster in which lung metastasis samples and human tumors were clustered B. Stratification of human Distant Metastasis Free Survival data by E2F2 pathway probability values showed differential effect of E2F2 loss in human tumors C. p-value < 0.0001).
DISCUSSION
Amplification of Myc has been associated with poor prognosis and distant metastasis in human breast cancer [37]. However, the MMTV-Myc transgenic mice are normally poorly metastatic, and only become metastatic with additional genetic events [17]. Myc function in vitro is reliant upon the E2Fs, a family of transcription factors essential for Myc mediated cell cycle progression and apoptosis. The E2Fs have been predicted and genetically demonstrated to be critical for Myc induced tumors. Interestingly, the genetic crosses to delete E2F2 in Myc induced tumors have provided different results for tumor latency [28, 29]. These effects are likely due to promoter differences, background, differences in transgene integration and expression as well as developmental timing of transgene activity.
Here we noted that E2F2 loss in Myc induced tumors significantly increased metastasis. This finding was made in two separate Myc transgenic lines and suggests that a critical function of E2F2 in Myc tumors is in regulation of metastasis genes. Strikingly, these results are quite distinct from other mouse models of breast cancer. For instance, loss of E2F2 in MMTV-Neu and MMTV-PyMT transgenics vastly reduced metastatic capacity [13, 27]. Together, these results indicate that there are oncogene specific genetic programs that involve differential signaling through the E2F transcription factors. Importantly, these differences may be regulated by which E2F is activated. Given that other E2Fs can compensate for loss of individual E2Fs [38], it is possible that loss of E2F2 in the MMTV-Myc mouse model increased metastasis by increasing the activity of other E2Fs and thus promotes metastasis by differentially regulating other genes involved in metastatic progression [30, 31]. We noted also that the transplantation of MMTV-Myc WT21 E2F2−/− tumors into the background of MMTV-Myc WT21 produced significantly more metastases compared to MMTV-Myc WT21 tumors transplanted into the same background. This suggested that metastases from MMTV-Myc WT21 E2F2−/− was cell autonomous. We observed increased metastasis when MMTV-Myc WT21 was transplanted into MMTV-Myc WT21 background, a small effect likely due to stromal alteration associated with surgery procedure. Further analysis to explore the mechanisms by which E2Fs influenced metastasis in these models is warranted.
GSEA analyses of EMT and lung metastases gene expression clusters revealed the expected enrichment of SMAD2, SMAD3, and SMAD4 regulated genes in the EMT cluster. Interestingly, when lung metastasis samples were compared to EMT histological type tumors, there was an enrichment of an invasive ovarian tumors geneset in the lung metastases. Given that pulmonary metastasis is frequently observed in ovarian cancer [39], this result suggests that there may potentially be a shared pathway between lung metastases and invasive ovarian tumors.
To elucidate the mechanism by which E2F2 mediated breast cancer metastasis, a gene expression approach for target identification was coupled with human breast cancer distance metastasis free survival times. This identified PTPRD as one of the strongest potential regulators of metastasis in the E2F knockout samples. Both in vitro and in vivo tests revealed that PTPRD activity was required for migration and colonization by human breast cancer cells.
PTPRD is a member of the Protein Tyrosine Phosphatases (PTPs) that is involved in various biological processes in cancer [40]. Specifically, PTPRD has been shown as tumor suppressor in glioma [33, 41], liver, lung, head and neck, colorectal and melanoma [42]. In glioblastoma PTPRD was shown to be deleted through array comparative genomic hybridization and copy number analysis [33]. Further exploration into the mechanistic function of PTPRD in glioblastoma demonstrated that the loss of PTPRD led to the accumulation of active phospho-STAT3 in a p16Ink4A−/− mouse model [34]. In breast cancer, PTPRD was discovered through The Cancer Genome Atlas project to be a novel gene that was frequently mutated in addition to PTPN22, suggesting the emerging roles of protein tyrosine phosphatases in breast cancer associated biological processes [32]. Specifically in breast cancer, PTPRD was found to be hypermethylated in late-stage breast cancer [43]. Our data suggests an additional role for PTPRD in mediating breast cancer metastasis in conjunction with the loss of E2F2.
While PTPRD is not predicted to be a direct transcriptional target of E2F2, analysis of the TCGA data revealed that there was potentially connection at the level of protein regulation, through BCAR1. Alternatively, PTPRD may function through STAT3 [34] given the STAT3 and Myc relationship [44]. Indeed, perturbation to STAT3 levels has been shown to alter the expression of the oncogene c-Myc [45–47]. Moreover, given that Myc interacts with Rb/E2F [48, 49] and there is a c-Myc binding site on the promoter of PTPRD, it is plausible that the loss of E2F2 allowed increased expression of PTPRD mediated by c-Myc. This increase of PTPRD could then lead to increased lung metastasis, a role for PTPRD and E2F2 that is unique to a subpopulation of breast cancer. Further analyses revealed that PTPRD overexpression was correlated to decreased time to metastasis in human basal subtype tumors. Taken together these results demonstrated that E2F2 and PTPRD expression in c-Myc tumors could act as additional risk factors associated with metastasis. Finally, although MYC is known to drive proliferation, it can also suppress metastasis [50] and therefore drug treatment directed towards signaling molecules downstream of MYC signaling should avoid affecting E2F2 signaling to maintain metastasis suppression.
MATERIALS AND METHODS
Animal work
Animal use and husbandry was in accordance to institution and federal guidelines. MMTV-Myc [14], MMTV-Myc (WT21) [26] and E2F2−/− [51] mice were bred and genotyped as previously described [28]. MMTV-Myc E2F2−/− females were kept in continuous breeding cycles. Mice were palpated weekly for tumor presence and tumors were measured by calipers weekly until endpoint when primary tumor reached 20 mm in the largest dimension [28]. Primary tumors were harvested for flash frozen samples, histological analysis and frozen viable transplantable tumors. Lungs were analyzed histologically for metastasis. Large metastatic lesions were flash frozen prior to RNA isolation by RNeasy according to the manufacturer's protocol (Qiagen, Valencia, CA). The quality of RNA samples were checked using Agilent Bioanalyzer (Agilent Biotechnologies, Santa Clara, CA) before being run on Affymetrix GeneChip Mouse Genome 430A 2.0 array chip (Affymetrix, Santa Clara, CA). Transplant assays were performed by transplanting 2.00 mm fragments of either MMTV-Myc WT21 or MMTV-Myc WT21 E2F2−/− tumor into MMTV-Myc WT21 and MMTV-Myc WT21 E2F2−/− backgrounds. 20 mice per genotype were used in the transplantation experiment and tumors were monitored as previously mentioned. Primary tumors and lungs were harvested and submitted for histological analyses as previously mentioned.
Colonization assays were performed by retroorbital injection of nude mice at a concentration of 5 × 105 cells in 50 μl as previously described [52]. Mice observed for signs of labored breathing weekly for a minimum 30 days. At end point, mice were euthanized and lungs were harvested for routine staining. Quantification of colonization was achieved by quantifying the number of lesions found in the lungs.
Gene analysis
Publicly available GEO datasets: GSE11121, GSE14020, GSE2034, GSE2603, GSE3494, GSE4922, GSE6532, GSE7390 and gene expression dataset from human breast cancer cell lines E-TABM-157 were obtained as well as their corresponding clinical annotations (Supplementary Tables 4 and 5). These gene expression datasets were pooled and normalized for batch effects by using Bayesian Factor Regression Modeling [53].
Validated training data from previous studies utilizing adenovirus infection of primary human mammary epithelial cells to build pathway signatures was used to identify samples with Myc and E2F2 activation, Myc and E2F2 signatures were applied as previously described to predict the pathway activation of Myc and E2F2 [54–56]. Briefly, the normalized dataset was merged with training data and a binary regression algorithm was used to calculate the probability of pathway activation for Myc and E2F2.
Mouse gene expression dataset GEO24594 was merged with 6 lung metastasis samples (GEO71815). Briefly, CEL files were analyzed by Affymetrix Expression Console software to ensure that expressions were within bounds. Gene expression datasets were normalized using Microarray Suite 5.0 (MAS5) and Robust Multi-array Average (RMA) methods. Unsupervised hierarchical clustering was performed in RMA normalized samples with Cluster 3.0. Clusters characteristics were identified based on the majority of the histological subtypes present.
To identify samples with human relevance, gene expression from previously mentioned human tumors were co-clustered with mouse tumor dataset. Fold change difference (>1.5) between MMTV-Myc, MMTV-Myc x E2F2−/− and lung metastasis samples were analyzed by GeneSpring. A t-Test was used to determine statistical significance for fold change analysis with p-value cut-off of 0.05. Fold change genes were examined for their correlation with human time to distant metastasis [57, 58] and Univariate Cox regression analysis, which allows genes to be ranked by effect size and does not require the normal assumption of proportional hazards to eliminate bias and maintain stability (Supplementary materials and methods) [59]. To determine whether candidate genes were in/direct E2F targets, genes were tested in GATHER [60] and SwissRegulon [61]. GSEA [62] analysis was completed with the Broad Institute GenePattern public server (http://www.broadinstitute.org/gsea/index.jsp). Regulatory network analysis was performed through examination of TCGA database [63, 64] using the genes: E2F2 and PTPRD.
Cell culture, transfection and migration assays
MCF7 and MDA-MB-231 were chosen based on the probability of Myc and E2F2 pathway activation. Cells were cultured in DMEM, 10% Fetal Bovine Serum (FBS) and 2.0 mM L-glutamine.
shRNA constructs targeting E2F2 and PTPRD were purchased from OriGENE (Rockville, MD). Cells were transfected using ExtremeGENE HP transfection reagent (ROCHE, Indianapolis, IN) according to manufacturer's protocol. Cells were selected using 2 μg/ml puroMyc in 48 hours after transfections. Both populations and colonies were tested using western blot to determine the knockdown efficiency. Wound healing assay and transwell migration assay was performed as previously described [65]. Transwell assays were quantified using ImageJ with Cellcounter plugin.
qRT-PCR
RNA samples were isolated using RNeasy Plus mini-kit (Qiagen, Valencia, CA) according to the manufacturer's protocol. The primers for PTPRD (human) were F-TCACCAAGCTGCGTGAAATG and R-CAGCCATGGGATCTACAACAAA and the primers for PTPRD (mouse) were F-GGCTAGCCATCCTCCAATACC and R-TCCTGGGATTCCTCATATTCC (IDT, Coralville, IA). qRT-PCR was performed on 20 ng of total RNA (MDA-MB-231) or 200 ng of total RNA (MCF7) using QuantiTECT Sybr green PCR kit (Qiagen).
Western blotting
Primary antibodies for immunoblotting were rabbit anti-E2F2 (clone E-19 Santa Cruz, Dallas, TX, 1:300) or rabbit anti-PTPRD (Abcam, Cambridge, MA, 1:100).
Data analysis
Statistical analysis was performed using GraphPad Prism 5 and GraphPad Quickcalcs. Non-parametric t-Test was performed on the quantification of tumor metastases, percent area reduction, and transwell migration assays. Differences in latency and distant metastasis free survival was examined by plotting Kaplan-Meier survival plot. To examine the percentage of mice with metastasis, Fisher's test with 2 × 2 contingency table was used.
CONCLUSIONS
This study concludes that; 1. E2F2 loss contributes to metastasis in Myc induced tumors; 2. E2F2 may act through PTPRD to increase metastasis in the MMTV-Myc model of breast cancer.
ACKNOWLEDGMENTS AND FUNDING
We thank the members of the Andrechek laboratory for numerous discussions and critical reading of the manuscript and Jordan Honeysett for technical support. Nude mice were generously donated by the laboratory of Dr. Justin McCormick (Michigan State University, East Lansing, MI). This work was supported by the Susan G. Komen Foundation with grant KG1110510 and with NIH R01CA160514 to ERA.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
REFERENCES
1. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000; 406:747–52.
2. Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001; 98:10869–74.
3. Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A. 2003; 100:8418–23.
4. Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010; 12:R68.
5. Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007; 8:R76.
6. Pfefferle AD, Herschkowitz JI, Usary J, Harrell JC, Spike BT, Adams JR, et al. Transcriptomic classification of genetically engineered mouse models of breast cancer identifies human subtype counterparts. Genome Biol. 2013; 14:R125.
7. Hollern DP, Andrechek E. A genomic analysis of mouse models of breast cancer reveals molecular features of mouse models and relationships to human breast cancer. Breast Cancer Res. 2014; 16:R59.
8. Lu J, Steeg PS, Price JE, Krishnamurthy S, Mani SA, Reuben J, et al. Breast cancer metastasis: challenges and opportunities. Cancer Res. 2009; 69:4951–3.
9. Steeg PS, Ouatas T, Halverson D, Palmieri D, Salerno M. Metastasis suppressor genes: basic biology and potential clinical use. Clin Breast Cancer. 2003; 4:51–62.
10. Weigelt B, Peterse JL, van ’t Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer. 2005; 5:591–602.
11. Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009; 9:274–84.
12. Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992; 12:954–61.
13. Hollern DP, Honeysett J, Cardiff RD, Andrechek ER. The E2F transcription factors regulate tumor development and metastasis in a mouse model of metastatic breast cancer. Mol Cell Biol. 2014; 34:3229–43.
14. Stewart TA, Pattengale PK, Leder P. Spontaneous mammary adenocarcinomas in transgenic mice that carry and express MTV/Myc fusion genes. Cell. 1984; 38:627–37.
15. Hundley JE, Koester SK, Troyer DA, Hilsenbeck SG, Barrington RE, Windle JJ. Differential regulation of cell cycle characteristics and apoptosis in MMTV-Myc and MMTV-ras mouse mammary tumors. Cancer Res. 1997; 57:600–3.
16. Amundadottir LT, Johnson MD, Merlino G, Smith GH, Dickson RB. Synergistic interaction of transforming growth factor alpha and c-Myc in mouse mammary and salivary gland tumorigenesis. Cell growth & differentiation: the molecular biology journal of the American Association for Cancer Research. 1995; 6:737–48.
17. Calvo A, Catena R, Noble MS, Carbott D, Gil-Bazo I, Gonzalez-Moreno O, et al. Identification of VEGF-regulated genes associated with increased lung metastatic potential: functional involvement of tenascin-C in tumor growth and lung metastasis. Oncogene. 2008; 27:5373–84.
18. Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol. 2002; 3:11–20.
19. Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet. 2001; 10:699–703.
20. Nevins JR. Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell growth & differentiation: the molecular biology journal of the American Association for Cancer Research. 1998; 9:585–93.
21. Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer. 2009; 9:785–97.
22. Engelmann D, Mayoli-Nussle D, Mayrhofer C, Furst K, Alla V, Stoll A, et al. E2F1 promotes angiogenesis through the VEGF-C/VEGFR-3 axis in a feedback loop for cooperative induction of PDGF-B. J Mol Cell Biol. 2013; 5:391–403.
23. Worku D, Jouhra F, Jiang GW, Patani N, Newbold RF, Mokbel K. Evidence of a tumour suppressive function of E2F1 gene in human breast cancer. Anticancer Res. 2008; 28:2135–9.
24. Ho GH, Calvano JE, Bisogna M, Van Zee KJ. Expression of E2F-1 and E2F-4 is reduced in primary and metastatic breast carcinomas. Breast Cancer Res Treat. 2001; 69:115–22.
25. Bieche I, Khodja A, Lidereau R. Deletion mapping of chromosomal region 1p32-pter in primary breast cancer. Genes Chromosomes Cancer. 1999; 24:255–63.
26. Andrechek ER, Cardiff RD, Chang JT, Gatza ML, Acharya CR, Potti A, et al. Genetic heterogeneity of Myc-induced mammary tumors reflecting diverse phenotypes including metastatic potential. Proc Natl Acad Sci U S A. 2009; 106:16387–92.
27. Andrechek ER. HER2/Neu tumorigenesis and metastasis is regulated by E2F activator transcription factors. Oncogene. 2015; 34:217–25.
28. Fujiwara K, Yuwanita I, Hollern DP, Andrechek ER. Prediction and Genetic Demonstration of a Role for Activator E2Fs in Myc-Induced Tumors. Cancer Res. 2011; 71:1924–32.
29. Wu L, de Bruin A, Wang H, Simmons T, Cleghorn W, Goldenberg LE, et al. Selective roles of E2Fs for ErbB2- and Myc-mediated mammary tumorigenesis. Oncogene. 2015; 34:119–28.
30. Johnson JL, Pillai S, Pernazza D, Sebti SM, Lawrence NJ, Chellappan SP. Regulation of matrix metalloproteinase genes by E2F transcription factors: Rb-Raf-1 interaction as a novel target for metastatic disease. Cancer Res. 2012; 72:516–26.
31. Meier C, Spitschak A, Abshagen K, Gupta S, Mor JM, Wolkenhauer O, et al. Association of RHAMM with E2F1 promotes tumour cell extravasation by transcriptional up-regulation of fibronectin. J Pathol. 2014; 234:351–64.
32. Network TCGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012; Oct 4;490:61–70.
33. Veeriah S, Brennan C, Meng S, Singh B, Fagin JA, Solit DB, et al. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc Natl Acad Sci U S A. 2009; 106:9435–40.
34. Ortiz B, Fabius AW, Wu WH, Pedraza A, Brennan CW, Schultz N, et al. Loss of the tyrosine phosphatase PTPRD leads to aberrant STAT3 activation and promotes gliomagenesis. Proc Natl Acad Sci U S A. 2014; 111:8149–54.
35. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009; 119:1420–8.
36. Tornillo G, Defilippi P, Cabodi S. Cas proteins: dodgy scaffolding in breast cancer. Breast Cancer Res. 2014; 16:443.
37. Singhi AD, Cimino-Mathews A, Jenkins RB, Lan F, Fink SR, Nassar H, et al. MYC gene amplification is often acquired in lethal distant breast cancer metastases of unamplified primary tumors. Mod Pathol. 2012; 25:378–87.
38. Kong LJ, Chang JT, Bild AH, Nevins JR. Compensation and specificity of function within the E2F family. Oncogene. 2007; 26:321–7.
39. Sood AK, Sorosky JI, Dolan M, Anderson B, Buller RE. Distant metastases in ovarian cancer: association with p53 mutations. Clin Cancer Res. 1999; 5:2485–90.
40. Ostman A, Hellberg C, Bohmer FD. Protein-tyrosine phosphatases and cancer. Nat Rev Cancer. 2006; 6:307–20.
41. Solomon DA, Kim JS, Cronin JC, Sibenaller Z, Ryken T, Rosenberg SA, et al. Mutational inactivation of PTPRD in glioblastoma multiforme and malignant melanoma. Cancer Res. 2008; 68:10300–6.
42. Julien SG, Dube N, Hardy S, Tremblay ML. Inside the human cancer tyrosine phosphatome. Nat Rev Cancer. 2011; 11:35–49.
43. Chan TA, Glockner S, Yi JM, Chen W, Van Neste L, Cope L, et al. Convergence of mutation and epigenetic alterations identifies common genes in cancer that predict for poor prognosis. PLoS medicine. 2008; 5:e114.
44. Kiuchi N, Nakajima K, Ichiba M, Fukada T, Narimatsu M, Mizuno K, et al. STAT3 is required for the gp130-mediated full activation of the c-Myc gene. J Exp Med. 1999; 189:63–73.
45. Ling X, Arlinghaus RB. Knockdown of STAT3 expression by RNA interference inhibits the induction of breast tumors in immunocompetent mice. Cancer Res. 2005; 65:2532–6.
46. Turkson J, Jove R. STAT proteins: novel molecular targets for cancer drug discovery. Oncogene. 2000; 19:6613–26.
47. Bowman T, Broome MA, Sinibaldi D, Wharton W, Pledger WJ, Sedivy JM, et al. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc Natl Acad Sci U S A. 2001; 98:7319–24.
48. Sears RC, Nevins JR. Signaling networks that link cell proliferation and cell fate. J Biol Chem. 2002; 277:11617–20.
49. Leone G, DeGregori J, Sears R, Jakoi L, Nevins JR. Myc and Ras collaborate in inducing accumulation of active cyclin E/Cdk2 and E2F. Nature. 1997; 387:422–6.
50. Liu H, Radisky DC, Yang D, Xu R, Radisky ES, Bissell MJ, et al. MYC suppresses cancer metastasis by direct transcriptional silencing of alphav and beta3 integrin subunits. Nat Cell Biol. 2012; 14:567–74.
51. Murga M, Fernandez-Capetillo O, Field SJ, Moreno B, Borlado LR, Fujiwara Y, et al. Mutation of E2F2 in mice causes enhanced T lymphocyte proliferation, leading to the development of autoimmunity. Immunity. 2001; 15:959–70.
52. Yardeni T, Eckhaus M, Morris HD, Huizing M, Hoogstraten-Miller S. Retro-orbital injections in mice. Lab animal. 2011; 40:155–60.
53. Carvalho CM, Chang J, Lucas JE, Nevins JR, Wang Q, West M. High-Dimensional Sparse Factor Modeling: Applications in Gene Expression Genomics. J Am Stat Assoc. 2008; 103:1438–56.
54. West M, Blanchette C, Dressman H, Huang E, Ishida S, Spang R, et al. Predicting the clinical status of human breast cancer by using gene expression profiles. Proc Natl Acad Sci U S A. 2001; 98:11462–7.
55. Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006; 439:353–7.
56. Gatza ML, Lucas JE, Barry WT, Kim JW, Wang Q, Crawford MD, et al. A pathway-based classification of human breast cancer. Proc Natl Acad Sci U S A. 2010; 107:6994–9.
57. Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010; 123:725–31.
58. Gyorffy B, Surowiak P, Budczies J, Lanczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One. 2013; 8:e82241.
59. Dunkler D, Schemper M, Heinze G. Gene selection in microarray survival studies under possibly non-proportional hazards. Bioinformatics. 2010; 26:784–90.
60. Chang JT, Nevins JR. GATHER: a systems approach to interpreting genomic signatures. Bioinformatics. 2006; 22:2926–33.
61. Pachkov M, Erb I, Molina N, van Nimwegen E. SwissRegulon: a database of genome-wide annotations of regulatory sites. Nucleic Acids Res. 2007; 35:D127–31.
62. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005; 102:15545–50.
63. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013; 6:pl1.
64. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012; 2:401–4.
65. Chen J, Miller EM, Gallo KA. MLK3 is critical for breast cancer cell migration and promotes a malignant phenotype in mammary epithelial cells. Oncogene. 2010; 29:4399–411.