
INTRODUCTION
Glioblastoma is the commonest and most devastating primary brain cancer [1]. The disease has a universally fatal prognosis despite aggressive treatment in which over 85% of patients die within two years [2]. While the median age group is middle age to elderly, a smaller numbers of cases are found in young adults and children [2, 3]. The predominant group of middle aged and elderly glioblastomas has been extensively investigated. Molecular classification in glioblastomas based on gene expression profiles classified the heterogeneous disease into proneural, neural, classical and mesenchymal subtypes, with each subtype carried distinct genomic aberrations [4]. Isocitrate dehydrogenase-1 (IDH1) mutation or platelet-derived growth factor receptor alpha (PDGFRA) amplification, epidermal growth factor receptor (EGFR) amplification and neurofibromin 1 (NF1) mutations were associated with proneural, classical and mesenchymal glioblastomas, respectively [4]. Genome-wide methylation study further identified a subset of adult glioblastoma with glioma-CpG island methylation phenotype (G-CIMP) which was enriched in proneural subgroup, tightly associated with IDH1 mutation and exhibiting favorable prognosis [5]. For the younger group, only the children’s glioblastomas has been extensively studied [6–9]. Distinct genomic aberrations including PDGFRA alterations and hotspot mutations in histone 3.3 (H3F3A) at codons 27 (K27) and 34 (G34) as well as histone 3.1 (HIST1H3B) at codon 27 (K27) were frequently found in glioblastomas in children [6–8]. Genomic study in combined series of pediatric and adult glioblastomas further identified age-specific biological subgroups which can be defined by driver events including H3F3A-K27M, H3F3A-G34R/V and IDH1 mutations, strongly indicating that glioblastomas are different diseases in different age groups [10, 11]. Recent study also reported the activating mutation BRAF-V600E identified a distinct clinical subgroup of pediatric high grade gliomas [12]. While current literature has been only focused in glioblastomas of either children or older patients and in particular, vast majority of adult glioblastomas studied were above 35 years as a result of the skewed distribution towards older age (median age 64 years) [2], the young adult age group was known to have better prognosis [3, 13] but was understudied in the literature. In this regards, we investigate a set of subgroup-defining molecular biomarkers in young adult glioblastomas aged from 17 to 35 years and evaluate the prognostic impact of the biomarkers. Our study reveals that BRAF, H3F3A and IDH1 mutations are associated with distinct clinical features and can stratify young adult glioblastomas into prognostic subgroups, which have important clinical implications in refining the prognostic classification of glioblastomas in young adults.
RESULTS
Cohort characteristics
Clinical and molecular data of the cohort was summarized in Table 1 and Figure 1. The age of the young adult glioblastoma cohort ranged from 17 years to 35 years. The mean and median ages were 25 years, respectively. The male-to-female ratio was 1:1.61. Eighty-eight tumors (82.2%) were located in cerebral hemisphere. There were 51 tumors (47.7%) involving frontal lobe, 18 tumors (16.8%) involving parietal lobe, 30 tumors (28%) involving temporal lobe and seven tumors (6.5%) involving occipital lobe, with 18 cases (16.8%) affected two cerebral lobes and five frontal tumors also affected corpus callosum (four cases) and lateral ventricle (one case). Twenty-four tumors (22.4%) affected midline structures including one case (0.9%) in basal ganglia, seven cases (6.5%) in thalamus, six cases (5.6%) in ventricular system, five cases (4.7%) in corpus callosum, one case (0.9%) in cerebellum and four cases (3.7%) in spinal cord. Treatment data in operation and chemo-radiotherapy was available in 87 patients (81.3%) and 80 patients (74.8%), respectively. Fifty-eight of 87 patients (67%) received total resection. Sixty-one of 80 patients (76.3%) received radiotherapy and 64 of 80 patients (80%) received chemotherapy, with 52 of 80 patients (65%) received concomitant chemo-radiotherapy. Survival data was available in 94 patients (87.9%). The median overall survival and median follow-up were 14.7 months and 31.6 months, respectively.
Table 1: Clinical and molecular data of the young adult glioblastoma cohort
n = 107 |
|
|---|---|
Gender (Male/Female) |
66/41 |
Age (Mean/median/range) |
25.0/25/(17–35) |
Tumor location |
|
Cerebral hemisphere |
65 (60.7%) |
Cerebellum |
1 (0.9%) |
Midline structures |
18 (16.8%) |
More than one location affected |
23 (21.5%) |
Operation |
|
Total resection |
58 (54.2%) |
Non-total resection |
29 (27.1%) |
Not available |
20 (18.7%) |
Adjuvant therapy |
|
Radiotherapy + chemotherapy |
52 (48.6%) |
Radiotherapy only |
9 (8.4%) |
Chemotherapy only |
12 (11.2%) |
No adjuvant therapy |
7 (6.5%) |
Not available |
27 (25.2%) |
BRAF |
|
Mutant |
16 (15%) |
Wild-type |
91 (85%) |
IDH1 |
|
Mutant |
18 (16.8%) |
Wild-type |
89 (83.2%) |
H3F3A |
|
K27M |
17 (15.9%) |
G34R/V |
3 (2.8%) |
wild-type |
87 (81.3%) |
HIST1H3B |
|
Mutant |
0 (0%) |
wild-type |
107 (100%) |
EGFR |
|
Amplified |
4 (3.7%) |
Non-amplified |
103 (96.3%) |
TERTp |
|
C228T mutant |
6 (5.6%) |
C250T mutant |
3 (2.8%) |
Wild-type |
95 (88.8%) |
Not available |
3 (2.8%) |
CDKN2A homozygous deletion |
|
Yes |
31 (29%) |
No |
56 (52.3%) |
Not available |
20 (18.7%) |
PDGFRA expression |
|
Positive |
33 (30.8%) |
Negative |
74 (69.2%) |
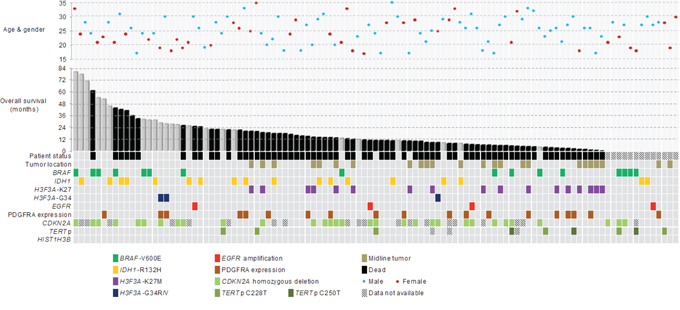
Figure 1: Clinical, genetic and molecular characteristics of 107 young adult glioblastomas aging from 17 years to 35 years. Mutation rates of BRAF, H3F3A and IDH1 were 15% (16/107), 18.7% (20/107) and 16.8% (18/107), respectively. No HIST1H3B mutation was detected. BRAF-V600E mutation was associated with CDKN2A deletion (p = 0.0002) and younger age (p = 0.013). H3F3A-K27M mutation was associated with midline tumor location (p < 0.00001). Positive PDGFRA expression co-occurred in 50% of H3F3A mutated tumors. IDH1-R132H mutation was associated with older age (p = 0.012). BRAF, IDH1, H3F3A-G34R/V mutations and EGFR amplification predominantly developed in hemispheric locations.
Young adult glioblastomas show recurrent BRAF, H3F3A and IDH1 mutations but infrequent TERTp mutation and EGFR amplification
Mutational status of BRAF, H3F3A, HIST1H3B and IDH1 were examined in all 107 young adult glioblastomas. Over half of the cases exhibited mutations in BRAF, IDH or H3F3A. Mutation rates of BRAF, H3F3A and IDH1 across the cohort were 15% (16/107), 18.7% (20/107) and 16.8% (18/107), respectively. None of the tumors showed mutation in HIST1H3B. Among the H3F3A mutated tumors, 17 cases harbored K27M mutation, two cases harbored G34R mutation and one case harbored G34V mutation. All IDH1 mutated tumors showed IDH1-R132H mutation and all BRAF mutated tumors showed BRAF-V600E mutation. TERTp mutation was only identified in 8.7% (9/104) of young adult glioblastomas, including six cases of C228T mutation and three cases of C250T mutation. EGFR amplification was detectable in only 3.7% (4/107) of young adult glioblastomas (Table 1 and Supplementary Figure S1). Notably, with the exception of three BRAF mutated glioblastomas concurrently harboring TERTp mutation, BRAF, H3F3A, IDH1, TERTp mutations and EGFR amplification were mutually exclusive (Figure 1).
BRAF mutated glioblastomas show frequent CDKN2A homozygous deletion and younger patient age
CDKN2A homozygous deletion was detected in 35.6% (31/87) of cases with analyzable data (Table 1 and Supplementary Figure S1). Data was non-analyzable in 20 cases due to either weak hybridization signals or strong background fluorescence. Correlating with other molecular biomarkers examined, CDKN2A homozygous deletion was associated with BRAF mutation. Eighty percent (12/15) of BRAF mutated glioblastomas concurrently harbored CDKN2A homozygous deletion, compared with 26.4% (19/72) of BRAF wild-type glioblastomas (p = 0.0002) (Figure 2a). Comparing the age between BRAF mutated and BRAF wild-type glioblastomas, patients with mutated BRAF were younger than those with wild-type. The mean ages of BRAF mutated glioblastomas and BRAF wild-type glioblastomas were 22.3 years and 25.5 years, respectively (p = 0.013) (Figure 2b). Further comparison of patient age between the mutually exclusive molecular subgroups also revealed that BRAF mutated glioblastomas were younger than IDH1 mutated glioblastomas (mean age 22.3 years vs 27.9) (p = 0.0008, One-way ANOVA).
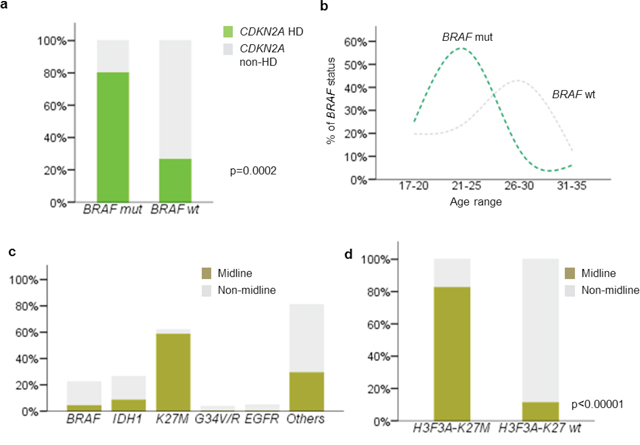
Figure 2: Correlation between clinicopathological and molecular variables of young adult glioblastoma. a. 80% of BRAF-V600E mutated glioblastomas showed concurrent CDKN2A deletion. BRAF-V600E mutation was closely associated with CDKN2A deletion in young adult glioblastoma (p = 0.0002). b. Patients with mutant BRAF-V600E glioblastomas are older than those wild type tumors (p = 0.013). c. H3F3A-K27M mutation was closely associated with midline structures (p < 0.00001), while the other mutations, IDH1-R132H, BRAF-V600E, H3F3A-G34R/V and EGFR amplification were mainly identified in tumors originated from hemispheric locations. d. Only 3.6% of hemispheric glioblastomas harbored H3F3A-K27M mutation (p < 0.00001). Mut, mutated; wt, wild type; hd, homozygous deletion.
Tumor location differ between H3F3A-K27M mutated glioblastomas and other glioblastoma subgroups
Distribution of tumor location according to molecular biomarkers was shown in Figure 2c. H3F3A-K27 mutation was found in 58.3% (14/24) of glioblastomas affecting midline structures, including six thalamic tumors, four ventricular tumors and four cervical/thoracic spinal cord tumors. In contrast, only 3.6% (3/83) of hemispheric glioblastomas harbored the mutation (p < 0.00001) (Figure 2d). These three K27M mutated hemispheric glioblastomas were all located in temporal/frontal lobe. Young adult glioblastomas with BRAF, IDH1, H3F3A-G34R/V mutations and EGFR amplification predominantly developed in hemispheric locations without affecting midline structure, with the exception of one BRAF mutated glioblastoma in corpus callosum and three IDH1 mutated glioblastomas in basal ganglia (one case) and corpus callosum (two cases).
Frequent PDGFRA expression in H3F3A mutated glioblastomas
Positive PDGFRA immunohistochemical expression was detected in 30.8% (33/107) of cases. Comparing PDGFRA expression across BRAF, IDH1 and H3F3A mutated glioblastomas, positive expression co-occurred in 50% (10/10) of H3F3A mutated tumors, 27.8% (5/13) of IDH1 mutated tumors and 12.5% (2/16) of BRAF mutated tumors (p = 0.05). Among H3F3A mutated tumors, 100% (3/3) of G34V/R mutated tumors and 42.2% (7/17) of K27M mutated tumors demonstrated PDGFRA expression.
Prognostication of young adult glioblastoma by molecular biomarkers
We investigated the survival data of the young adult glioblastoma cohort according to the clinical parameters and molecular biomarkers by univariate analysis as shown in Table 2. Patients receiving total resection (p = 0.002) and chemo-radiotherapy (p < 0.0001) were associated with better prognosis and midline tumor location was associated with poor outcome (p < 0.0001) (Supplementary Figure S2). Among the molecular biomarkers evaluated, BRAF, H3F3A-K27M, IDH1 and PDGFRA demonstrated prognostic relevance in young adult glioblastomas. BRAF mutated glioblastomas exhibited better prognosis than those with wild-type BRAF (p = 0.032). The median overall survival was 43.2 months for BRAF mutated glioblastomas and 13.6 months for BRAF wild-type glioblastomas. IDH1 mutated tumors, as expected, also exhibited favorable prognosis comparing to IDH1 wild-type tumors, with median overall survival of 24.2 months in the mutant group and 13.5 months in the wild-type group (p = 0.034). H3F3A-K27M mutation was associated with poor prognosis across the cohort. The median overall survival was 6 months in K27M mutated tumors, compared to 17.6 months in the wild-type counterparts (p < 0.0001). Tumors with positive PDGFRA expression showed shorter survival (8.6 months) than those with negative expression (17.4 months) (p = 0.03). Co-evaluation of BRAF, IDH1 and H3F3A-K27M status stratified young adult glioblastomas into four prognostic groups across the cohort (p < 0.00001). BRAF mutated tumors (p = 0.038) and IDH1 mutated tumors (p = 0.028) had better prognosis than BRAF/IDH1/K27 wild-type tumors, which in turn showed better survival than K27M mutated tumors (p = 0.002) (Figure 3a to 3e). Subset analysis demonstrated that positive PDGFRA expression was associated with poor outcome within the K27M mutated glioblastoma subgroup (p = 0.03) (Supplementary Figure S2). Multivariate analysis was performed by including molecular biomarkers showing prognostic relevance in univariate analysis and adjusted for patient age, tumor location, operation and adjuvant treatment. Older patient age (HR = 1.085, 95% CI = 1.015 to 1.159, p = 0.016) and tumors involving midline structure (HR = 3.86, 95% CI = 1.509 to 9.877, p = 0.005) independent poor prognostic factors. Tumors treated by concomitant chemo-radiotherapy (HR = 0.196, 95% CI = 0.072 to 0.531, p = 0.001) and IDH1 mutation (HR = 0.389, 95% CI = 0.172 to 0.878, p = 0.023) were independent favorable prognostic factors.
Table 2: Univariate analysis of clinical parameters and molecular markers
n |
HR |
[95% CI] |
Median OS (months) |
p |
|
|---|---|---|---|---|---|
Age |
|||||
≤ 25 years |
54 |
1 |
17.6 |
0.094 |
|
> 25 years |
53 |
1.5 |
[0.93 – 2.4] |
12.3 |
|
Tumor location |
|||||
Midline |
16 |
3.503 |
[1.974 – 6.216] |
8.4 |
<0.0001 |
Non-midline |
78 |
1 |
18.2 |
||
Operation |
|||||
Total resection |
58 |
0.456 |
[0.271 – 0.767] |
19.8 |
0.002 |
Non-total resection |
29 |
1 |
11.1 |
||
Adjuvant therapy |
|||||
Chemotherapy + Radiotherapy |
52 |
0.089 |
[0.036 – 0.221] |
19.8 |
<0.0001 |
Chemotherapy only |
12 |
0.176 |
[0.063 – 0.494] |
7.6 |
|
Radiotherapy only |
9 |
0.289 |
[0.098 – 0.848] |
10.3 |
|
No adjuvant treatment |
7 |
1 |
4.5 |
||
BRAF |
|||||
Mutant |
12 |
0.405 |
[0.173 – 0.951] |
43.2 |
0.032 |
Wild-type |
82 |
1 |
13.6 |
||
IDH1 |
|||||
Mutant |
16 |
0.476 |
[0.236 – 0.962] |
24.2 |
0.034 |
Wild-type |
78 |
1 |
13.5 |
||
H3F3A-K27 |
|||||
Mutant |
17 |
3.448 |
[1.91 – 6.225] |
6 |
<0.0001 |
Wild-type |
77 |
1 |
17.6 |
||
H3F3A-G34 |
|||||
Mutant |
3 |
0.331 |
[0.046 – 2.389] |
NR |
0.248 |
Wild-type |
91 |
1 |
14.7 |
||
TERTp |
|||||
Mutant |
6 |
1.492 |
[0.539 – 4.134] |
5.1 |
0.438 |
Wild-type |
85 |
1 |
15.6 |
||
EGFR |
|||||
Amplified |
3 |
1.521 |
[0.476 – 4.857] |
10.7 |
0.476 |
Non-amplified |
91 |
1 |
15 |
||
CDKN2A |
|||||
Homozygous deletion |
28 |
0.616 |
[0.35 – 1.086] |
13.9 |
0.091 |
No homozygous deletion |
47 |
1 |
15 |
||
PDGFRA expression |
|||||
Positive |
30 |
1.732 |
[1.048 – 2.862] |
8.6 |
0.03 |
Negative |
64 |
1 |
17.4 |
HR, hazard ratio; 95% CI, 95% confidence interval
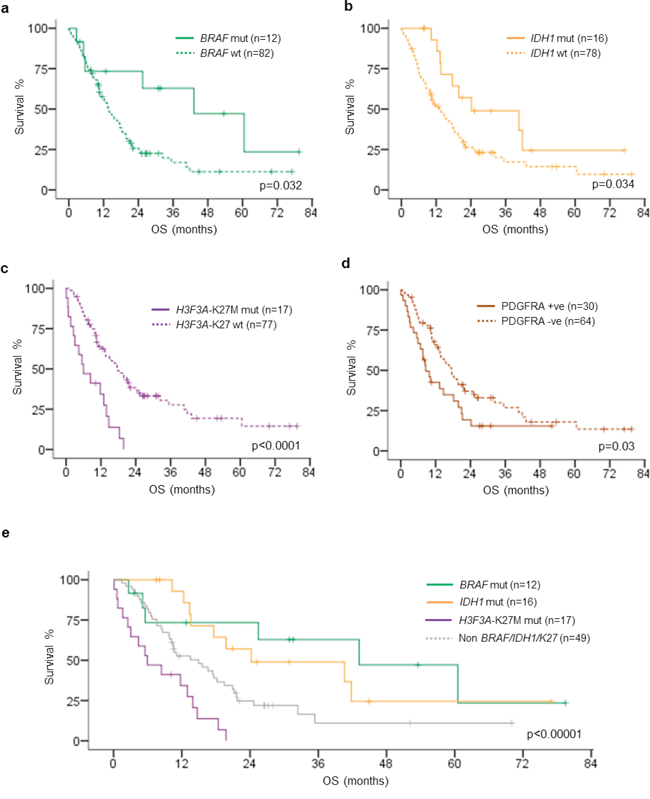
Figure 3: Kaplan–Meier survival analysis of BRAF mutation, IDH1 mutation, H3F3A-K27M mutation, PDGFRA immunohistochemistry positivity and subgroups defined by BRAF, IDH1, H3F3A-K27M mutations. a. BRAF-V600E mutation was associated with longer OS comparing to BRAF wild type (p = 0.032). b. IDH1-R132H mutation was associated with longer OS comparing to IDH1 wild type (p = 0.034). c. H3F3A-K27M mutation was associated with shorter OS comparing to H3F3A wild type (p < 0.0001). d. PDGFRA immunohistochemistry positivity was associated with OS comparing to PDGFRA immunohistochemistry negativity (p = 0.03). e. BRAF-V600E mutated tumors exhibited the longest OS followed by IDH1-R132H mutated tumors, while H3F3A-K27M mutated tumors showed the worst OS (p < 0.00001). OS, overall survival; mut, mutated; wt, wild type.
DISCUSSION
While the predominant middle-aged and elderly glioblastomas as well as the pediatric glioblastomas have been extensively investigated, there is lack of focusing study in the young adult age group. In this study, we showed that prognostication and possibly classification of young adult glioblastoma can be biomarker-based and demonstrated that a significant portion of young adult glioblastomas could be genetically defined by mutually exclusive BRAF-V600E mutation (15%), H3F3A-K27M mutation (15.9%), H3F3A-G34R/V mutation (2.8%) and IDH1-R132H mutation (16.8%). BRAF-V600E mutation was frequently identified in pediatric low-grade gliomas including pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma [14, 15]. In contrast to these pediatric low-grade glioma subtypes, mutation frequency was much lower in adult diffuse gliomas [15–19]. Knobbe and co-workers investigated 94 glioblastomas and identified three cases (3.2%) harboring BRAF mutation [16]. In the study by Schindler and colleagues examining 1,320 nervous system tumors, BRAF mutation was detected in less than 2% of adult glioblastomas investigated [15]. Dahiya and colleagues evaluated the BRAF status in 39 adult glioblastomas and identified 7.7% (three cases) possessing the mutation [19]. The mutation was reported in up to 54% of epithelioid glioblastoma, an uncommon histologic variant of glioblastoma in young adults [17]. In our current study focusing on young adult patients aged from 17 to 35 years, 15% of patients had BRAF mutation but no BRAF mutation was identified in seven cases of secondary young adult glioblastomas. And patients had BRAF mutation were significantly younger than those without the mutation. Mutated cases had mean age of 22.3 years and median age of 21 years, compared to mean age of 25.5 years and median age of 26 years in wild-type cases (p = 0.013). Additionally, BRAF mutation identified a subset of patients with favorable prognosis in our cohort. In univariate analysis, the median overall survival of patients with BRAF mutated glioblastomas was 43.2 months and those with wild-type BRAF was 13.6 months (p = 0.032). Notably, among the 16 BRAF mutated glioblastomas, 11 cases had operation record and total resection could be achieved in all cases (p = 0.013), suggesting BRAF mutated glioblastomas were more amendable to surgical resection. Seven cases received concomitant chemotherapy and radiotherapy, out of nine cases with adjuvant treatment data available. Since majority of the BRAF mutated tumors received total resection and concomitant chemo-radiation which could account for the favorable outcome of this subset of patient in our cohort, we tried to analyze the prognostic value of BRAF mutation in patients receiving total resection and concomitant chemoradiation (n = 37). Seven BRAF mutated tumors showed strong trend of better prognosis than 30 BRAF wild-type tumors (Supplementary Figure S2). The potential prognostic value of BRAF-V600E mutation was also recently reported in pediatric high-grade gliomas [9, 12]. Mistry and colleagues described a series of pediatric secondary high-grade gliomas harboring BRAF mutation and CDKN2A deletion, showing longer latency to transformation from low-grade lesion and better clinical outcome [12]. By conducting genome-wide DNA methylation profiling in 202 pediatric glioblastomas, Korshunov and colleagues identified an epigenomic subset of glioblastoma showing methylation pattern similar to pleomorphic xanthoastrocytoma, enriched for BRAF mutation and CDKN2A homozygous deletion, and showed favorable prognosis [9]. Although those studies were on pediatric glioblastomas, our study provided complementary results to theirs and supported the clinical significance of BRAF-V600E testing in glioblastoma of young person [9, 12, 17, 19]. Apart from the potential prognostic value, BRAF-V600E mutation also served as a novel therapeutic target in this subset of glioblastomas. Robinson and colleagues recently reported a case of BRAF mutated pediatric glioblastoma treated by BRAF inhibitor vemurafenib and showed complete response [20]. Given the potential clinical utility in prognostication and treatment selection, BRAF mutational testing, either by direct sequencing or immunohistochemistry [21], should be conducted in glioblastomas of young patients.
Histone H3 mutation was exclusively observed in H3F3A in 18.7% of young adult glioblastomas. HIST1H3B mutation was not found in any of the 107 young adult samples examined, suggesting the mutation was specific to diffuse intrinsic pontine gliomas (DIPG) but not for non-brainstem high grade gliomas in both pediatric and adult patients [7, 22]. K27M and G34R/V mutated tumors showed distinct tumor locations and clinical outcome in our cohort as in pediatric glioblastomas [9, 10]. In our cohort, vast majority of K27M mutated tumors were located in midline structures including thalamus, ventricular system and cervical/thoracic spinal cord. These rare midline glioblastomas, despite of their different anatomical locations, shared the same driver mutation and suggested a closely-related origin [10, 23]. Notably, K27M mutated glioblastomas showed aggressive clinical course in our cohort with median overall survival of 6 months (range 0.1 months to 19.8 months). Within this aggressive subset of glioblastoma, tumors with positive PDGFRA immunohistochemical expression exhibited a significantly shorter survival (median 2.5 months) than tumors with negative expression (median 11.7 months) (p = 0.03). Interestingly, Puget and colleagues previously reported a distinct transcriptional subgroup of DIPG characterized by oligodendroglial differentiation, driven by PDGFRA upregulation and exhibited significantly worse outcome [23]. The association of PDGFRA expression with poor prognosis in K27M mutated glioblastomas in our study was in line with previous observation. The prognostic value of PDGFRA expression/alterations in the K27M mutated tumors warranted further evaluation in a larger cohort.
IDH1 mutation was present in 16.8% of young adult glioblastomas of which the mutation frequency was comparatively higher than the predominant group of adult/elderly glioblastoma as well as pediatric glioblastomas [8, 9, 24–27]. Previous study by Pollack and colleagues reported that IDH1 mutation was common in adolescent malignant gliomas in which 16.3% of high grade gliomas between 3 to 21 years harbored the mutation and was significantly associated with age greater than 14 years [28]. As expected, IDH1 mutated glioblastomas showed favorable prognosis in our cohort which was independent of age, tumor location, operation and adjuvant treatment among the young adult patients. Favorable prognostic value of IDH1 mutation was also recently demonstrated in pediatric glioblastomas [9]. Notably, although IDH1 mutation was identified in nearly 90% of secondary elderly glioblastomas [27], only 43% (3/7) of IDH1 mutation was detected in our young adult glioblastomas, suggesting that IDH1 mutation wasn’t a main contributing marker for young secondary glioblastomas. Collectively, our study provided complementary evidence to previous studies that IDH1 mutated glioblastoma was a glioblastoma subgroup with favorable prognosis prevalent in adolescents and young adults [11].
Hotspot mutations in TERT promoter region were identified in up to 80% of adult/elderly glioblastomas [29–31] but rare in pediatric glioblastomas [32]. The prevalence of TERTp mutation in the young adult age group was not precisely reported. In our study, only 8.7% of young adult glioblastomas harbored TERTp mutation, suggesting that mutation induced telomerase activation might not be a major mechanism in telomere deregulation in that age group. It remained to be investigated if promoter methylation of TERT causing telomerase upregulation or the alternative lengthening of telomeres (ALT) represented the major mechanism of telomere maintenance in young adult glioblastomas [6, 33]. TERTp mutations were associated with poor prognosis in glioblastomas [30, 31, 34, 35] as well as lower-grade gliomas with wild-type IDH [34, 36, 37]. Although not reached statistical significance, the median overall survival of patients with TERTp mutated glioblastomas was only 5.1 months, compared to 15.6 months in those with wild-type TERTp. Notably, three TERTp mutations were overlapped with BRAF-V600E mutation in the cohort and overall survival was 5.1 months in one patient with survival data available. BRAF-V600E and TERTp mutations were recently found cooperatively identifying the most aggressive subset of papillary thyroid cancer with high recurrence rate [38]. TERTp mutations, either C228T or C250T, generated a consensus binding site (5′-TTCC-3′) for E-twenty-six (ETS) transcription factors and upregulated TERT expression [34, 39, 40]. On the other hand, activation of mitogen-activated protein kinase pathway was also shown to cause upregulation of the ETS system [41–43]. The synergistic effect of BRAF-V600E and TERTp mutations in promoting tumorigenesis was therefore biologically explainable [38]. Although only 2.9% (3/104) of patients harbored concurrent BRAF-V600E and TERTp mutation, this subgroup accounted for 18.8% (3/16) of BRAF mutated glioblastomas of young adults. Further study should be conducted to evaluate the clinical value of concurrent BRAF and TERTp mutations in young adult glioblastomas.
In summary, our study demonstrates recurrent BRAF, IDH1 and H3F3A mutations in young adult glioblastomas with clinical impacts. BRAF mutation and IDH1 mutation identify glioblastomas with less aggressive clinical course and H3F3A-K27M mutation defines glioblastomas with dismal prognosis. The biomarker-based stratification has clinical implications and refines the prognostic classification of young adult glioblastomas.
MATERIALS AND METHODS
Patients and tissue samples
A total of 107 tissue samples (all formalin-fixed paraffin-embedded) were obtained from young adult patients (age from 17–35 years) of Department of Anatomical and Cellular Pathology, Prince of Wales Hospital (Hong Kong) and Department of Neurosurgery, Huashan hospital (Shanghai) with a histological diagnosis of “glioblastoma, WHO grade IV”. Seven out of the 107 samples were secondary glioblastomas progressed from low grade gliomas, including oligoastrocytoma and astrocytoma. Other 102 cases were diagnosed as primary glioblastomas. Clinical and survival data of the patients were retrieved from the respective institutional medical record systems. This study was approved by the Ethics Committee of Shanghai Huashan Hospital and the New Territories East Cluster-Chinese University of Hong Kong Ethics Committee.
Molecular analysis
Mutational analysis was performed as described previously [36, 44]. Tissues from representative tumor area with tumor content 70% were scrapped off from dewaxed sections and treated with proteinase K at a final concentration of 2 mg/ml in 10 mM Tris-HCl buffer (pH 8.5) at 55°C for 2–18 hours and then at 98°C for 10 minutes. Crude cell lysate was centrifuged and supernatant was used for subsequent PCR analysis. The forward and reverse primers then were used to amplify gene BRAF, H3F3A, HIST1H3B, IDH1 and TERT. PCR was performed in 10ul reaction mixture for different thermal protocol (Supplementary Table S1, S2). Sequencing was performed using BigDye Terminator Cycle Sequencing kit v1.1 (Life Technologies). The products were resolved in Genetic Analyzer 3130 × l and analyzed by Sequencing Analysis software. Hotspots BRAF-V600E, H3F3A-K27M, H3F3A-G34R/V, HIST1H3B-K27M, IDH1-R132H and TERTp were detected (Supplementary Figure S1). All base changes were confirmed by sequencing of a newly amplified fragment.
Fluorescence in situ hybridization analysis for EGFR amplification and CDKN2A deletion
Dual-probe fluorescence in situ hybridization (FISH) assay was performed on paraffin-embedded sections, with locus-specific probes for EGFR and CDKN2A paired with centromere probes for chromosome 7p12 and chromosome 9p21 (Supplementary Figure S1, Table S4). Deparaffinization of the sections was carried out, followed by dehydration in 100% ethanol, retrieval by 1M sodium thiocyanate at 80°C for 10 minutes, and digestion in 0.04% pepsin at 37°C was applied on each section for 10 minutes. Simultaneous probe per specimen was denatured at 80°C for 10 minutes with subsequent overnight incubation at 37°C. The sections were washed next day in 1.5 M Urea/2X saline sodium citrate at 50°C for 10 minutes twice. After washing, sections were stained with Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (Vector Laboratories) and visualized under a fluorescent microscope (Carl Zeiss Microscopy LLC, NY, USA). Hybridizing signals in at least 100 non-overlapping nuclei were counted. EGFR amplification was considered when 50% of the tumor cells harbored more than five signals per nuclei CEP7 or innumerable tight clusters of signals of the locus probe [45]. CDKN2A deletion was considered if both signals were lost (homozygous deletion) in at least 20% of tumor nuclei [46].
Immunohistochemistry of PDGFRA
FFPE tissue sections of 4 micron thickness were deparaffinized in xylene and rehydrated in graded alcohols. For PDGFRA, antigen retrieval was carried out by treating the sections in 1M Citrate buffer (PH = 6.0) in a microwave oven. After antigen retrieval, all sections were processed by BondMax automade staining systems (Leica BondMax) using validated protocols. Tissue sections were incubated at 37°C for 30 mins with relevant antibodies of PDGFRA (Supplementary Table S3). Antigen detection was performed using Ultra View diamino benzidine chromogen step (BondMax). The presence of cytoplasmic and membrane staining indicated positivity for PDGFRA [47] (Supplementary Figure S1, Table S5).
Statistical analysis
Statistical analysis was performed using IBM SPSS Statistics 20 (IBM Corporation, NY, USA). Correlation between molecular markers and clinical parameters were examined by X2-test. Comparison between two groups was performed by Student’s t-test or Mann–Whitney U-test. Comparison between three or more groups used one-way analysis of variance (ANOVA). Overall survival (OS) was defined as the duration between the diagnosis and death or last follow-up [25]. Survival curves were plotted by Kaplan-Meier method and analyzed by Log-rank test. Multivariate analysis for independent prognostic marker was performed by Cox-proportional hazards model. Tests with a p value below 0.05 were considered significant.
Supplementary information is available at Oncotarget’s website.
ACKNOWLEDGMENTS AND FUNDING
This study was supported by the National Basic Research Program of China and 973 Program (grant no. 2015CB755500), the National Science Foundation of China (grant no. 81172412) and Health and Medical Research Fund of Hong Kong (grant no. 02133146).
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta neuropathologica. 2007; 114:97–109.
2. Ostrom QT, Gittleman H, Farah P, Ondracek A, Chen Y, Wolinsky Y, Stroup NE, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro-oncology. 2013; 15:ii1–56.
3. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine. 2005; 352:987–996.
4. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer cell. 2010; 17:98–110.
5. Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer cell. 2010; 17:510–522.
6. Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, Hovestadt V, Albrecht S, Kool M, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012; 482:226–231.
7. Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, Zhang J, Gajjar A, Dyer MA, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nature genetics. 2012; 44:251–253.
8. Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, Bax DA, Coyle B, Barrow J, Hargrave D, Lowe J, Gajjar A, Zhao W, et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. Journal of clinical oncology. 2010; 28:3061–3068.
9. Korshunov A, Ryzhova M, Hovestadt V, Bender S, Sturm D, Capper D, Meyer J, Schrimpf D, Kool M, Northcott PA, Zheludkova O, Milde T, Witt O, et al. Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta neuropathologica. 2015; 129:669–678.
10. Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tonjes M, Sill M, Bender S, Kool M, Zapatka M, Becker N, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer cell. 2012; 22:425–437.
11. Fontebasso AM, Liu XY, Sturm D, Jabado N. Chromatin remodeling defects in pediatric and young adult glioblastoma: a tale of a variant histone 3 tail. Brain pathology. 2013; 23:210–216.
12. Mistry M, Zhukova N, Merico D, Rakopoulos P, Krishnatry R, Shago M, Stavropoulos J, Alon N, Pole JD, Ray PN, Navickiene V, Mangerel J, Remke M, et al. BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. Journal of clinical oncology. 2015; 33:1015–1022.
13. Gorlia T, van den Bent MJ, Hegi ME, Mirimanoff RO, Weller M, Cairncross JG, Eisenhauer E, Belanger K, Brandes AA, Allgeier A, Lacombe D, Stupp R. Nomograms for predicting survival of patients with newly diagnosed glioblastoma: prognostic factor analysis of EORTC and NCIC trial 26981-22981/CE.3. The Lancet Oncology. 2008; 9:29–38.
14. Dougherty MJ, Santi M, Brose MS, Ma C, Resnick AC, Sievert AJ, Storm PB, Biegel JA. Activating mutations in BRAF characterize a spectrum of pediatric low-grade gliomas. Neuro-oncology. 2010; 12:621–630.
15. Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, Schmieder K, Wesseling P, Mawrin C, Hasselblatt M, Louis DN, Korshunov A, Pfister S, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta neuropathologica. 2011; 121:397–405.
16. Knobbe CB, Reifenberger J, Reifenberger G. Mutation analysis of the Ras pathway genes NRAS, HRAS, KRAS and BRAF in glioblastomas. Acta neuropathologica. 2004; 108:467–470.
17. Kleinschmidt-DeMasters BK, Aisner DL, Birks DK, Foreman NK. Epithelioid GBMs show a high percentage of BRAF V600E mutation. The American journal of surgical pathology. 2013; 37:685–698.
18. Chi AS, Batchelor TT, Yang D, Dias-Santagata D, Borger DR, Ellisen LW, Iafrate AJ, Louis DN. BRAF V600E mutation identifies a subset of low-grade diffusely infiltrating gliomas in adults. Journal of clinical oncology. 2013; 31:e233–236.
19. Dahiya S, Emnett RJ, Haydon DH, Leonard JR, Phillips JJ, Perry A, Gutmann DH. BRAF-V600E mutation in pediatric and adult glioblastoma. Neuro-oncology. 2014; 16:318–319.
20. Robinson GW, Orr BA, Gajjar A. Complete clinical regression of a BRAF V600E-mutant pediatric glioblastoma multiforme after BRAF inhibitor therapy. BMC cancer. 2014; 14:258.
21. Capper D, Preusser M, Habel A, Sahm F, Ackermann U, Schindler G, Pusch S, Mechtersheimer G, Zentgraf H, von Deimling A. Assessment of BRAF V600E mutation status by immunohistochemistry with a mutation-specific monoclonal antibody. Acta neuropathologica. 2011; 122:11–19.
22. Aihara K, Mukasa A, Gotoh K, Saito K, Nagae G, Tsuji S, Tatsuno K, Yamamoto S, Takayanagi S, Narita Y, Shibui S, Aburatani H, Saito N. H3F3A K27M mutations in thalamic gliomas from young adult patients. Neuro-oncology. 2014; 16:140–146.
23. Puget S, Philippe C, Bax DA, Job B, Varlet P, Junier MP, Andreiuolo F, Carvalho D, Reis R, Guerrini-Rousseau L, Roujeau T, Dessen P, Richon C, et al. Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PloS one. 2012; 7:e30313.
24. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008; 321:1807–1812.
25. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, et al. IDH1 and IDH2 mutations in gliomas. The New England journal of medicine. 2009; 360:765–773.
26. Ichimura K, Pearson DM, Kocialkowski S, Backlund LM, Chan R, Jones DT, Collins VP. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro-oncology. 2009; 11:341–347.
27. Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta neuropathologica. 2008; 116:597–602.
28. Pollack IF, Hamilton RL, Sobol RW, Nikiforova MN, Lyons-Weiler MA, LaFramboise WA, Burger PC, Brat DJ, Rosenblum MK, Holmes EJ, Zhou T, Jakacki RI. IDH1 mutations are common in malignant gliomas arising in adolescents: a report from the Children’s Oncology Group. Child’s nervous system. 2011; 27:87–94.
29. Arita H, Narita Y, Takami H, Fukushima S, Matsushita Y, Yoshida A, Miyakita Y, Ohno M, Shibui S, Ichimura K. TERT promoter mutations rather than methylation are the main mechanism for TERT upregulation in adult gliomas. Acta neuropathologica. 2013; 126:939–941.
30. Nonoguchi N, Ohta T, Oh JE, Kim YH, Kleihues P, Ohgaki H. TERT promoter mutations in primary and secondary glioblastomas. Acta neuropathologica. 2013; 126:931–937.
31. Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr, Friedman AH, Friedman H, Gallia GL, Giovanella BC, Grollman AP, He TC, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013; 110:6021–6026.
32. Koelsche C, Sahm F, Capper D, Reuss D, Sturm D, Jones DT, Kool M, Northcott PA, Wiestler B, Bohmer K, Meyer J, Mawrin C, Hartmann C, et al. Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta neuropathologica. 2013; 126:907–915.
33. Castelo-Branco P, Choufani S, Mack S, Gallagher D, Zhang C, Lipman T, Zhukova N, Walker EJ, Martin D, Merino D, Wasserman JD, Elizabeth C, Alon N, et al. Methylation of the TERT promoter and risk stratification of childhood brain tumours: an integrative genomic and molecular study. The Lancet Oncology. 2013; 14:534–542.
34. Killela PJ, Pirozzi CJ, Healy P, Reitman ZJ, Lipp E, Rasheed BA, Yang R, Diplas BH, Wang Z, Greer PK, Zhu H, Wang CY, Carpenter AB, et al. Mutations in IDH1, IDH2, and in the TERT promoter define clinically distinct subgroups of adult malignant gliomas. Oncotarget. 2014; 5:1515–1525. doi: 10.18632/oncotarget.1765
35. Labussiere M, Boisselier B, Mokhtari K, Di Stefano AL, Rahimian A, Rossetto M, Ciccarino P, Saulnier O, Paterra R, Marie Y, Finocchiaro G, Sanson M. Combined analysis of TERT, EGFR, and IDH status defines distinct prognostic glioblastoma classes. Neurology. 2014; 83:1200–1206.
36. Chan AK, Yao Y, Zhang Z, Chung NY, Liu JS, Li KK, Shi Z, Chan DT, Poon WS, Zhou L, Ng HK. TERT promoter mutations contribute to subset prognostication of lower-grade gliomas. Modern pathology. 2015; 28:177–186.
37. Labussiere M, Di Stefano AL, Gleize V, Boisselier B, Giry M, Mangesius S, Bruno A, Paterra R, Marie Y, Rahimian A, Finocchiaro G, Houlston RS, Hoang-Xuan K, et al. TERT promoter mutations in gliomas, genetic associations and clinico-pathological correlations. British journal of cancer. 2014; 111:2024–2032.
38. Xing M, Alzahrani AS, Carson KA, Shong YK, Kim TY, Viola D, Elisei R, Bendlova B, Yip L, Mian C, Vianello F, Tuttle RM, Robenshtok E, et al. Association between BRAF V600E mutation and recurrence of papillary thyroid cancer. Journal of clinical oncology. 2015; 33:42–50.
39. Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, Schadendorf D, Kumar R. TERT promoter mutations in familial and sporadic melanoma. Science. 2013; 339:959–961.
40. Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013; 339:957–959.
41. Janknecht R, Ernst WH, Nordheim A. SAP1a is a nuclear target of signaling cascades involving ERKs. Oncogene. 1995; 10:1209–1216.
42. Strahl T, Gille H, Shaw PE. Selective response of ternary complex factor Sap1a to different mitogen-activated protein kinase subgroups. Proceedings of the National Academy of Sciences of the United States of America. 1996; 93:11563–11568.
43. Whitmarsh AJ, Shore P, Sharrocks AD, Davis RJ. Integration of MAP kinase signal transduction pathways at the serum response element. Science. 1995; 269:403–407.
44. Yao Y, Chan AK, Qin ZY, Chen LC, Zhang X, Pang JC, Li HM, Wang Y, Mao Y, Ng HK, Zhou LF. Mutation analysis of IDH1 in paired gliomas revealed IDH1 mutation was not associated with malignant progression but predicted longer survival. PloS one. 2013; 8:e67421.
45. Bax DA, Gaspar N, Little SE, Marshall L, Perryman L, Regairaz M, Viana-Pereira M, Vuononvirta R, Sharp SY, Reis-Filho JS, Stavale JN, Al-Sarraj S, Reis RM, et al. EGFRvIII deletion mutations in pediatric high-grade glioma and response to targeted therapy in pediatric glioma cell lines. Clinical cancer research. 2009; 15:5753–5761.
46. Horbinski C, Nikiforova MN, Hagenkord JM, Hamilton RL, Pollack IF. Interplay among BRAF, p16, p53, and MIB1 in pediatric low-grade gliomas. Neuro-oncology. 2012; 14:777–789.
47. Martinho O, Longatto-Filho A, Lambros MB, Martins A, Pinheiro C, Silva A, Pardal F, Amorim J, Mackay A, Milanezi F, Tamber N, Fenwick K, Ashworth A, et al. Expression, mutation and copy number analysis of platelet-derived growth factor receptor A (PDGFRA) and its ligand PDGFA in gliomas. British journal of cancer. 2009; 101:973–982.