
INTRODUCTION
The poly (ADP-ribose) polymerases (PARPs) are a family of enzymes comprising 18 members, and they play a vital role in maintaining the stability of the genome [1]. Among them, PARP-1 and PARP-2 are best known for their contribution to DNA damage repair. PARP-1, which was first found ~50 years ago, is activated by DNA damage and plays a crucial role in the repair of single-strand breaks [2]. Another family member, PARP-2, has 69% structural similarity to PARP-1, and some of their functions overlap [3]. Evidence demonstrates that PARP is significantly increased in some cancer types, compared with adjacent non-tumorous tissues [4, 5]. This suggests that inhibition of PARP may provide a novel strategy for cancer therapy.
Genomic instability accompanied with elevated DNA damage response is one of the most common features of human cancers [6], while Double strand breaks (DSBs) are the most severe type of DNA damage. DSBs are repaired mainly through non-homologous end-joining (NHEJ) and homologous recombination repair (HRR) pathways, which two play complementary roles [7]. PARP1-dependent end-joining (PARP-EJ) is a backup NHEJ repair pathway; when NHEJ is defective, PARP1-EJ pathway is activated [8, 9]. PARP inhibitors (such as Olaparib, Iniparib, Veliparib, Rucaparib, and Niraparib), a class of small-molecule drugs inhibiting PARP enzymes, can induce synthetic lethality in HRR deficiency cancer cells [10]. HRR deficiency is common in tumor cells, for example with BRCA gene mutations. In vitro experiments have established that cells with defective HRR are killed by PARP inhibitors [11]. Since their first clinical trial in 2003 [12], PARP inhibitors have shown benefit in the treatment of HRR-deficient tumors. Many kinds of PARP inhibitors have been designed since then; for example, Olaparib has been clinically approved for use in human testing by the Food and Drug Administration (FDA) in the USA.
However, on the other hand, serious adverse events have been reported in the PARP inhibitor arms of some clinical trials, and the therapeutic effect also seemed to be unsatisfactory [13, 14]. Since their appearance, PARP inhibitors have attracted controversy as to whether they are effective and safe anti-tumor agents. Thus, we set out to make a systematic review and meta-analysis of randomized controlled trials (RCTs) to gain insight relative risks and benefits of PARP inhibitors in patients with cancer.
RESULTS
Literature search
We initially identified 2180 potentially eligible studies by title and abstract screening. However, 2054 were excluded as they were not relevant to our analysis, leaving 126 articles for full review. After assessing the full texts of these potentially relevant studies, 115 were excluded for the following reasons: 14 contained no relative outcomes; 88 were phase I or single-arm phase II trials; 12 were duplicate publication; and 1 used PARP inhibitors in both the experimental and control groups. Ultimately, 11 eligible RCTs [15–25] involving a total of 2274 patients were included for analyses. A flow diagram of the trial selection process is shown in Figure 1. One article (OZA, 2013) [25] from EMBASE was partly overlapped with a previous publication (OZA 2015) [15], but it provided elaborated progression-free survival (PFS) data on the BRCA status, which was not mentioned in previous article, so it was also included.
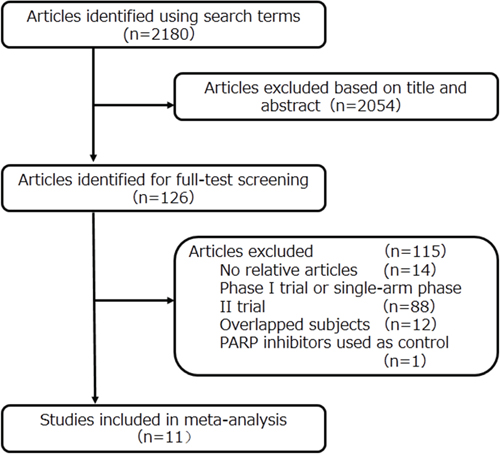
Figure 1: Flow diagram of the literature search and trial selection process.
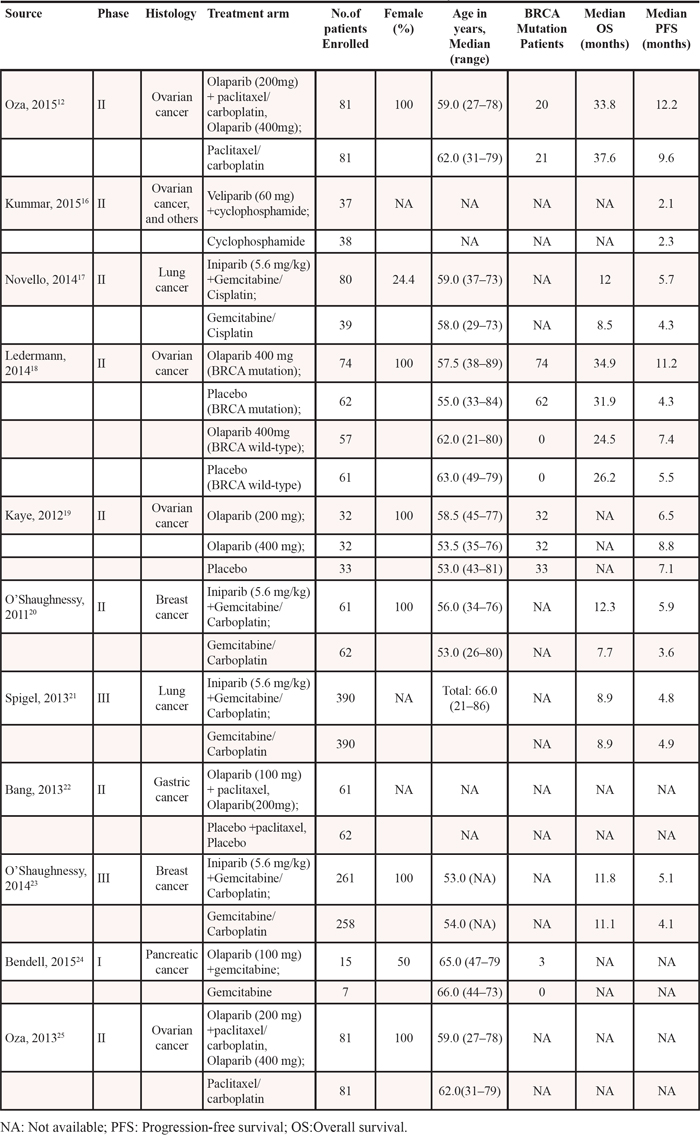
Of all 11 RCTs, 4 were BRCA mutation-correlated; 6 used the PARP inhibitor Olaparib, 4 used Iniparib, and 1 used Veliparib; 4 were about ovarian cancer, 2 about lung cancer, 2 about breast cancer, 1 about gastric cancer, 1 about pancreatic cancer, and 1 about ovarian, peritoneal, and fallopian tube cancers. The characteristics of 11 included trials are presented in Table 1.
Table 1: Characteristics of the trials included in the meta-analysis
Outcomes
Progression-free survival
7 trials reported PFS of the overall population. Overall, PFS was significantly longer in the PARP inhibitors group than in the control group [Hazard ratio (HR), 0.67; 95% confidence interval (CI), 0.50–0.90] (Figure 2). BRCA mutation status was known in 3 trials, and pooled results showed a HR of 0.32 (95% CI, 0.11–0.94); 6 trials on BRCA status unknown or non-mutation subgroup, the HR was 0.78 (95% CI, 0.65–0.95) (Table 2). 4 trials provided data on Iniparib and 3 trials on Olaparib. For Iniparib, no significant difference was observed between experiment group and control group (HR, 0.83; 95%CI, 0.68–1.02) (Table 2). For Olaparib, a significant improvement in PFS was recorded in the Olaparib group compared with the control group (HR, 0.50; 95% CI, 0.32–0.80) (Table 2). In the subgroup analysis by cancer type, 2 trials concerning lung cancer, and they gave a HR of 0.98 (95% CI, 0.83–1.15); 3 trials on ovarian cancer, the HR of PFS was 0.50 (95%CI, 0.32–0.80); 2 trials about breast cancer, and they gave a HR of 0.72 (95% CI, 0.56–0.94) (Table 2).
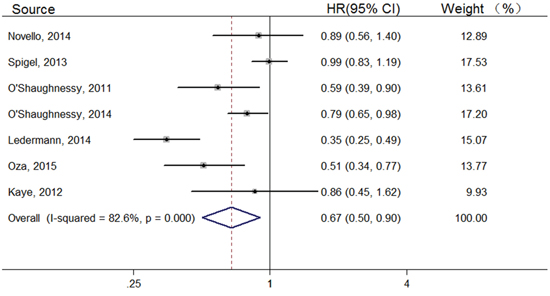
Figure 2: Forest plots of the pooled HRs for PFS by overall population.
Table 2: Summary results of the pooled HRs for PFS and OS by subgroup analysis
Pooled PFS |
Pooled OS |
||||||||
|---|---|---|---|---|---|---|---|---|---|
No.of trials |
HR (95%CI) |
I2 |
P |
No.of trials |
HR (95%CI) |
I2 |
P |
||
BRCA status |
|||||||||
BRCA status unknown or non-mutation |
617,18,20,21,23,25 |
0.78(0.65,0.95) |
49.90% |
0.076 |
517,18,20,21,23 |
0.89(0.73,1.09) |
50.80% |
0.087 |
|
BRCA mutation |
315,18,19 |
0.32(0.11,0.94) |
85.60% |
0.001 |
315,18,19 |
0.83(0.57,1.23) |
0.00% |
0.604 |
|
Drug type |
|||||||||
Iniparib |
417,20,21,23 |
0.83(0.68,1.02) |
51.10% |
0.105 |
417,20,21,23 |
0.86(0.67,1.10) |
63.00% |
0.044 |
|
Olaparib |
315,18,19 |
0.50(0.32,0.80) |
68.90% |
0.040 |
315,18,19 |
0.99(0.78,1.25) |
0.00% |
0.542 |
|
Cancer type |
|||||||||
Ovarican cancer |
315,18,19 |
0.50(0.32,0.80) |
68.90% |
0.040 |
315,18,19 |
0.99(0.78,1.25) |
0.00% |
0.542 |
|
Breast cancer |
220,23 |
0.72(0.56,0.94) |
33.70% |
0.219 |
220,23 |
0.74(0.49,1.12) |
62.90% |
0.100 |
|
Lung cancer |
217,21 |
0.98(0.83,1.15) |
0.00% |
0.672 |
217,21 |
1.00(0.76,1.31) |
35.10% |
0.214 |
|
PFS: Progression-free survival; OS:Overall survival; P: P-value of Q-test for heterogeneity test.
Overall survival rates
Our meta-analysis showed no significant difference in overall survival rates between the PARP inhibitor and placebo arms in the overall population (HR, 0.92; 95% CI, 0.79–1.08) (Figure 3). In the BRCA mutation group, the HR was 0.83 (95% CI, 0.57–1.23); and in the BRCA status unknown or non-mutation subgroup, the HR was 0.89 (95% CI, 0.73–1.09) (Table 2). Olaparib showed no statistical difference in overall survival rates, with an HR of 0.99 (95% CI, 0.78–1.25), compared with controls group; Similar results was observed in the Iniparib group (HR, 0.86; 95% CI, 0.67–1.10) (Table 2). In addition, compared with control group, PARP inhibitors showed no statistically significant difference in improving the overall survival (OS) of patients with ovarian (HR, 0.99; 95% CI, 0.78–1.25), breast (HR, 0.74; 95% CI, 0.49–1.12), or lung cancer (HR, 1.00; 95% CI, 0.76–1.31) (Table 2).
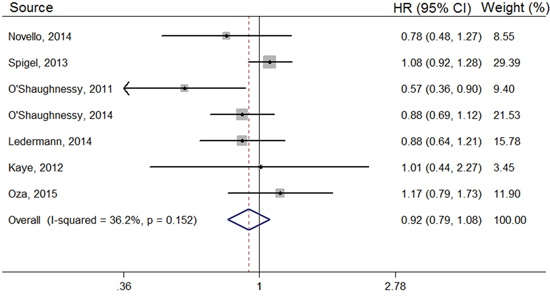
Figure 3: Forest plots of the pooled HRs for OS by overall population.
Safety
To evaluate the safety of PARP inhibitors, we analyzed the risk factor of any side-effects in the overall population with Grade 3 (G3) or more serious adverse events (AEs). Furthermore, since two PARP inhibitors were mainly used in the RCTs, we also evaluated the safety of the Iniparib and Olaparib subgroups separately.
Analysis of patients in the overall population showed that, compared with the control arms, PARP inhibitors were associated with a decreased risk of asthenia (RR, 0.34; 95%CI, 0.14–0.82) but increased risk of neutropenia (RR, 1.14; 95%CI, 1.01–1.29). In our study, we did not find any association between PARP inhibitors and other AEs (Table 3). In addtion, there were no significant differences in the AEs in the Olaparib or Iniparib subgroup (Supplementary Tables 1 and 2).
Table 3: Relative risks with 95% confidence intervals for common adverse events (Grade ≥ 3)
Adverse event |
No.of Trials |
Subjects |
RR [95% CI] |
P |
I2 (%) |
Pb |
|---|---|---|---|---|---|---|
Abdominal pain |
5 |
350/265 |
0.48[0.18,1.25] |
0.13 |
0.00 |
0.86 |
Anaemia |
8 |
703/609 |
1.45[0.77,2.75] |
0.25 |
49.00 |
0.06 |
Anorexia |
2 |
72/66 |
0.67[0.11,4.17] |
0.67 |
0.00 |
0.61 |
Arthralgia |
2 |
193/187 |
2.96[0.31,28.17] |
0.34 |
0.00 |
0.97 |
Asthenia |
3 |
278/199 |
0.34[0.14,0.82] |
0.02 |
2.00 |
0.36 |
Constipation |
4 |
454/374 |
1.63[0.46,5.81] |
0.45 |
0.00 |
0.98 |
Cough |
2 |
312/303 |
1.32[0.25,6.97] |
0.74 |
0.00 |
0.54 |
Dehydration |
2 |
94/97 |
3.09[0.33,29.18] |
0.32 |
0.00 |
1.00 |
Diarrhoea |
5 |
590/502 |
1.90[0.84,4.29] |
0.12 |
0.00 |
0.80 |
Dyspnea |
4 |
405/349 |
1.14[0.53,2.46] |
0.74 |
16.00 |
0.31 |
Fatigue |
5 |
548/450 |
1.34[0.82,2.19] |
0.24 |
0.00 |
0.62 |
Headache |
2 |
391/372 |
1.45[0.41,5.20] |
0.57 |
8.00 |
0.30 |
Increased ALT |
3 |
327/310 |
0.95[0.25,3.62] |
0.94 |
42.00 |
0.18 |
Increased AST |
2 |
312/303 |
1.10[0.56,2.17] |
0.78 |
0.00 |
0.52 |
Leukopenia |
4 |
427/380 |
0.99[0.71,1.38] |
0.95 |
0.00 |
0.44 |
Nausea |
6 |
605/509 |
1.17[0.51,2.66] |
0.71 |
10.00 |
0.35 |
Neutropenia |
7 |
639/577 |
1.14[1.01,1.29] |
0.03 |
0.00 |
0.46 |
Peripheral edema |
3 |
327/310 |
1.10[0.28,4.42] |
0.89 |
0.00 |
0.73 |
Pulmonary embolism |
2 |
93/46 |
1.02[0.29,3.54] |
0.98 |
0.00 |
0.50 |
Thrombocytopenia |
4 |
442/387 |
1.26[0.99,1.60] |
0.06 |
0.00 |
0.88 |
Vomiting |
6 |
605/509 |
1.43[0.66,3.09] |
0.36 |
0.00 |
0.89 |
RR: Relative risk; CI: Confidence interval; Pb: P-value of Q-test for heterogeneity test.
Analysis of publication bias
We used Funnel plot and Egger's regression asymmetry test to access the publication bias of literatures. Arrangement of data points did not reveal any evidence of obvious asymmetry. This was further confirmed by Egger's linear regression asymmetry test for each outcome and the results still did not show any evidence of publication bias (PFS: t = −1.24, P = 0.271; OS: t = −1.30, P = 0.251) (Supplementary Figures 1 and 2).
DISCUSSION
To our best knowledge, this study is the first systematic review and meta-analysis to evaluate the efficiency and safety of the novel antitumor PARP inhibitors. In our study, all RCTs included were published from 2011 to 2015, which reflects the popularity of PARP inhibitors in the past few years. Of all the 2274 patients, several types of cancers were reported, such as ovarian, lung, breast, and gastric cancers. Besides these, many trials [26–32] in different kinds of cancers not eligible for inclusion are still under way. Although the results have not come out, it is possible that PARP inhibitors may work in patients against some certain types of tumors. Various inhibitors suppressing PARP enzymes were involved in this study, including Olaparib, Iniparib, and Veliparib. It is worth noting that Olaparib was recently approved for use in human testing by the FDA in 2014 [33]. Will PARP inhibitors be a powerful and safe strategy for personalized cancer treatment in the future? According to our study, PARP inhibitors do well in prolonging the PFS of cancer patients, despite of some reported adverse events.
In our study, PARP inhibitors significantly improved the PFS in the overall population (HR, 0.67; 95% CI, 0.50–0.90), while the overall HR of the BRCA status unknown or non-mutation group was a little higher (HR, 0.78; 95% CI, 0.65–0.95). However, the difference was greater in the BRCA mutation group (HR, 0.32; 95% CI, 0.11–0.94). There is no doubt that BRCA mutation made a major contribution to the improved results. Individuals with BRCA mutation are at an increased risk of developing breast, ovarian, and other cancers [34]. More than 1 million women develop breast or ovarian cancer every year worldwide, and about 10% of them have a BRCA mutation [35–37], moreover that cancer patients with BRCA mutation have better outcomes than non-BRCA carriers [38, 39]. And a recent study [40] suggested that BRCA mutation should be taken into account when devising therapeutic strategies. BRCA plays a vital role in DNA damage repair by the HRR process, while PARP enzymes are involved in crucial complementary repair process [41]. With BRCA mutation, cancer cells are unable to perform HRR efficiently [42], then PARP in turn plays a major role in repairing damaged DNA to maintain cell survival. Thus, PARP could be targeted for treating BRCA-mutant tumors using a synthetic lethal approach. In this analysis, PARP inhibitors appeared to be efficient in killing BRCA-deficient cancer cells and prolonging the PFS of patients, mainly due to suppression of PARP enzyme activity [43].
Since PARP inhibitors have emerged as promising antitumor drugs, many efforts have been made to develop compounds such as Olaparib, Iniparib, Veliparib, and others as antineoplastic agents [44]. In our study, the main PARP inhibitors included were Olaparib and Iniparib. Olaparib has already shown benefit in treating patients with BRCA mutation, and our results confirmed that again, with an overall HR of 0.50 (95% CI, 0.32–0.80) between the Olaparib and control arms. Although Iniparib had a tendency to improve the PFS, the difference did not reach a statistically significant. The mechanism of action of Iniparib seems not to be closely correlated to PARP enzymes, it was reported that Iniparib carries a carboxyl group swiveled around an amino bond, and this may weakens its ability to bind PARP [45]. Although in the four trials with Iniparib it was combined with gemcitabine, cisplatin, or carboplatin, data [46] have already revealed that Iniparib fails to sensitize cells to cisplatin, gemcitabine, or paclitaxel. We suppose that one possibility of the difference, caused between the Iniparib and Olaparib subgroups, may be different criteria of patient enrolment. Patients enrolled in the Olaparib trials mostly had BRCA mutations, while those in the Iniparib subgroup mainly had non-mutation BRCA status. It should also be noted that an initial submission of Olaparib to FDA was rejected. The application was only accepted following addition data provided to support that benefit was restricted to the BRCA mutated patients.
Soon after their development, PARP inhibitors were used in hundreds of clinical trials, and many different kinds of tumors with or without BRCA mutation were involved [47]. For example, Olaparib was reported to be effective against several tumor types including ovarian, breast, pancreatic, and prostate cancers [48]. The main tumor types included in our study were ovarian, breast, and lung cancers. According to our results, there was significant statistical heterogeneity in the PFS of the ovarian cancer subgroup (HR, 0.50; 95%CI, 0.32–0.80), while in the lung (HR, 0.98; 95%CI, 0.83–1.15) and breast cancer groups (HR, 0.72; 95%CI, 0.56–0.94), there seemed to be no odds difference between the PARP inhibitor and control arms. As far as we know, various PARP inhibitors were designed to suppress tumors with BRCA mutation, no matter what kind of cancer. Thus, the reason may be that most of the patients in the ovarian cancer group had BRCA mutation, while those in lung and breast cancer subgroups did not. The results would be better if the patients in lung and breast cancer group were also with BRCA mutation as those in ovarian cancer group.
Consistent with previous hypothesis, our study show that cancer patients with BRCA mutation may increase sensitivity to PARP inhibitors [49]. PARP inhibitors can induce synthetic lethality in HRR deficient cancer cells, such as BRCA dysfunction [50]. BRCA mutation was at a high prevalence among breast cancer patients. About 20% of breast cancer patients were BRCA mutations carriers [51, 52]. Beyond breast cancer, several other malignancies, including ovarian cancer, pancreatic cancer, melanoma, and prostate cancer, were also correlated to BRCA mutations [53]. On top of BRCA, mutations of other DNA repair genes, such as RAD51, ATM, PALB2, CHEK2, may also increase sensitivity to PARP inhibitors [54]. It is therefore important to perform genetic testing and in prior to this therapy.
However, PAPR inhibitors failed to improve the OS of cancer patients in this analysis. Apart from the overall level, we also analyzed the OS from the perspective of cancer type, PARP inhibitor category, and BRCA status. According to our analysis, other than the OS of the Iniparib subgroup, none of the results showed significant differences between the PARP inhibitor and control arms. And even the result in the Iniparib subgroup was not satisfactory, with an HR of 0.86 (95%CI, 0.67–1.10). Although PARP inhibitors did not statistically improve the OS, in some individual trials, they were reported to clearly increase it. For example, Novello et al17 reported that the median OS in the PARP inhibitor arm was 12 months compared with 8.5 months in the control arm; and in another trial20, the OS was prolonged from 7.7 months to 12.3 months by PARP inhibitors. Research on PARP inhibitors is still ongoing, and many aspects need further improvement. Also, the inherent relationship between PFS and OS should be taken into account, since PFS is contained within OS. PARP inhibitors may be able to effectively improve the PFS, but this was not strong enough to translate PFS effects into OS improvement.
Today, many traditional anti-cancer drugs are able to kill tumor cells, but their toxicity to normal cells restricts their clinical application. PARP inhibitors suppress DNA repair, and kill cancer cells through “synthetic lethality” [55]. In this way, PARP inhibitors are applicable across various cancers, improving the efficacy and reducing the toxicity of individualized therapies. Our study revealed the benefit of the low toxicity of PARP inhibitors. Compared with the control arms, no treatment-correlated risks were seen in the Iniparib and Olaparib subgroups. Only a slightly decreased risk of asthenia (RR, 0.34; 95% CI, 0.14–0.82) and increased risk of neutropenia (RR, 1.14; 95% CI, 1.01–1.29) were seen in the overall population, suggesting that the PARP inhibitors are well-tolerated.
In conclusion, based on the available observational studies, PARP inhibitors do better in improving PFS with little toxicity, especially in patients with BRCA deficiency. However, they fail to increase the OS.
MATERIALS AND METHODS
Study selection
We carried out a comprehensive search to identify potential articles in PUBMED and EMBASE up to January 2015, using the search terms: “PARP inhibitors” or “Olaparib” or “Iniparib” or “Veliparib” or “Rucaparib” or “Niraparib” or “Talazoparib” and “cancer” or “tumor” or “carcinoma”, limited to clinical trials. There was no limit on the language of publication. In order to ensure the completeness and quality of the results, relevant scientific meetings were retrieved, and unpublished trials were checked in the clinical trial registry (http://www.clinicaltrials.gov).
To be included, studies had to be RCTs and had to report at least one outcome of interest, such as PFS, OS, and AEs. Single-arm trials and trials in which PARP inhibitors were used in both arms were excluded, on account of the absence of control groups. Trials in which PARP inhibitors were used to treat other diseases were also excluded. In all the included RCTs, PARP inhibitors were used alone or combination with other chemotherapeutic agents as the treatment group, while in the control group placebo or other chemotherapeutic agents were used. Two investigators reviewed the articles independently to exclude irrelevant and overlapping studies.
Data extraction
We collected the following information from all the included RCTs: first author's surname, year of publication, number of participants, histology, trial phase, treatment arm, median age, BRCA status, median OS, and median PFS. In addition, the HR of the median OS and median PFS with 95% CIs were extracted from most of the trials to evaluate the curative effect of PARP inhibitors. Information on AEs was also retrieved to calculate the safety of PARP inhibitors.
Statistical analysis
All analyses were done with Stata version 12 (StataCorp, College Station, Texas) and Review Manager (version 5.1, The Cochrane Collaboration, Oxford, UK). A 2-tailed P value of less than 0.05 was judged as statistically significant. HR and 95% CI were used to assess the OS and PFS between PARP inhibitors group and control group. In addition, we extracted dichotomous data form all studies reporting number of patients with adverse events and total participants and pooled them to calculate RR with 95% CI. The degree of heterogeneity was measured by the I2 statistic, with I2 < 25%, 25–75% and > 75% to represent low, moderate and high degree of inconsistency, respectively [56]. Statistical heterogeneity was defined as an I2 statistic value of more than 50% [56]. In analyses, if the heterogeneity was low then we used a fixed-effect model, or else applied the random-effect model. We further performed a subgroup analysis by the status of BRCA, tumor type, and different kinds of PARP inhibitors (Iniparib and Olaparib). Funnel plot and Egger's regression asymmetry test were used to access the publication bias of literatures [57].
ACKNOWLEDGMENTS AND FUNDING
This work was financially supported by National 1000 Talents Program for Young Scholars, National Natural Science Foundation of China (31370901, 81422031), and Zhejiang Provincial Natural Science Foundation of China (LR14H160001).
CONFLICTS OF INTEREST
None.
REFERENCES
1. Ame JC, Spenlehauer C, de Murcia G. The PARP super-family. BioEssays : news and reviews in molecular, cellular and developmental biology. 2004; 26:882–893.
2. Chambon P, Weill J, Mandel P. Nicotinamide mononucleotide activation of a new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochemical and biophysical research communications. 1963; 11:39–43.
3. Amé J-C, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, Muller S, Höger T, Ménissier-de Murcia J, de Murcia G. PARP-2, a novel mammalian DNA damage-dependent poly (ADP-ribose) polymerase. Journal of Biological Chemistry. 1999; 274:17860–17868.
4. Nomura F, Yaguchi M, Togawa A, Miyazaki M, Isobe K, Miyake M, Noda M, Nakai T. Enhancement of poly-adenosine diphosphate-ribosylation in human hepatocellular carcinoma. Journal of gastroenterology and hepatology. 2000; 15:529–535.
5. Ossovskaya V, Koo IC, Kaldjian EP, Alvares C, Sherman BM. Upregulation of Poly (ADP-Ribose) Polymerase-1 (PARP1) in Triple-Negative Breast Cancer and Other Primary Human Tumor Types. Genes cancer. 2010; 1:812–821. doi: 10.1177/1947601910383418.
6. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674.
7. Matsumoto Y. [Smart choice between two DNA double-strand break repair mechanisms]. Igaku butsuri : Nihon Igaku Butsuri Gakkai kikanshi = Japanese journal of medical physics. 2014; 34:57–64.
8. Mansour WY, Rhein T, Dahm-Daphi J. The alternative end-joining pathway for repair of DNA double-strand breaks requires PARP1 but is not dependent upon microhomologies. Nucleic acids research. 2010; 38:6065–6077.
9. Jia Q, den Dulk-Ras A, Shen H, Hooykaas PJ, de Pater S. Poly(ADP-ribose)polymerases are involved in microhomology mediated back-up non-homologous end joining in Arabidopsis thaliana. Plant molecular biology. 2013; 82:339–351.
10. Seimiya H. [Cancer therapy by PARP inhibitors]. Nihon rinsho Japanese journal of clinical medicine. 2015; 73:1330–1335.
11. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly (ADP-ribose) polymerase. Nature. 2005; 434:913–917.
12. Tentori L, Leonetti C, Scarsella M, d’Amati G, Vergati M, Portarena I, Xu W, Kalish V, Zupi G, Zhang J. Systemic administration of GPI 15427, a novel poly (ADP-ribose) polymerase-1 inhibitor, increases the antitumor activity of temozolomide against intracranial melanoma, glioma, lymphoma. Clinical Cancer Research. 2003; 9:5370–5379.
13. Bedikian AY, Papadopoulos NE, Kim KB, Hwu W-J, Homsi J, Glass MR, Cain S, Rudewicz P, Vernillet L, Hwu P. A phase IB trial of intravenous INO-1001 plus oral temozolomide in subjects with unresectable stage-III or IV melanoma. Cancer investigation. 2009; 27:756–763.
14. Khan O, Gore M, Lorigan P, Stone J, Greystoke A, Burke W, Carmichael J, Watson AJ, McGown G, Thorncroft M. A phase I study of the safety and tolerability of olaparib (AZD2281, KU0059436) and dacarbazine in patients with advanced solid tumours. British journal of cancer. 2011; 104:750–755.
15. Oza AM, Cibula D, Benzaquen AO, Poole C, Mathijssen RHJ, Sonke GS, Colombo N, Špaček J, Vuylsteke P, Hirte H, Mahner S, Plante M, Schmalfeldt B, Mackay H, Rowbottom J, Lowe ES, et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. The lancet oncology. 2015; 16:87–97.
16. Kummar S, Fleming GF, Oza AM, Sullivan DM, Gandara DR, Naughton M, Villalona-Calero MA, Morgan R, Szabo PM, Youn A, Chen A, Ji JJ, Allen D, Lih CJ, Mehaffey MG, Walsh WD, et al. Randomized trial of oral cyclophosphamide and veliparib in high-grade serous ovarian, primary peritoneal, or fallopian tube cancers, or BRCA-mutant ovarian cancer. Clinical cancer research. 2015; 21:1574–82.
17. Novello S, Besse B, Felip E, Barlesi F, Mazieres J, Zalcman G, von Pawel J, Reck M, Cappuzzo F, Ferry D, Carcereny E, Santoro A, Garcia-Ribas I, Scagliotti G, Soria JC. A phase II randomized study evaluating the addition of iniparib to gemcitabine plus cisplatin as first-line therapy for metastatic non-small-cell lung cancer. Annals of Oncology. 2014; 25:2156–2162.
18. Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott CL, Meier W, Shapira-Frommer R, Safra T, Matei D, Fielding A, Spencer S, Dougherty B, Orr M, Hodgson D, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. The lancet oncology. 2014; 15:852–61.
19. Kaye SB, Lubinski J, Matulonis U, Ang JE, Gourley C, Karlan BY, Amnon A, Bell-McGuinn KM, Chen LM, Friedlander M, Safra T, Vergote I, Wickens M, Lowe ES, Carmichael J, Kaufman B. Phase, II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J Clin Oncol. 2012; 30:372–379.
20. O’Shaughnessy J, Osborne C, Pippen JE, Yoffe M, Patt D, Rocha C, Koo IC, Sherman BM, Bradley C. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. The New England journal of medicine. 2011; 364:205–214.
21. Spigel D, Kim ES, Lynch T, Mccleod M, Waterhouse D, Paz-Ares L, Harper P, Hainsworth J, De Marinis F, Kabbinavar F. (2013). Randomized phase III trial of gemcitabine (G)/carboplatin (C) with or without iniparib (I) in patients (pts) with previously untreated stage IV squamous lung cancer. JOURNAL OF THORACIC ONCOLOGY: LIPPINCOTT WILLIAMS & WILKINS 530 WALNUT, ST, PHILADELPHIA, PA 19106–3621 USA), pp. S193–S194.
22. Bang Y, Im S, Lee K, Cho J, Song E, Lee K. Olaparib plus paclitaxel in patients with recurrent or metastatic gastric cancer: a randomized, double-blind phase II study. J Clin Oncol. 2013; 31:4013.
23. O’Shaughnessy J, Schwartzberg L, Danso MA, Miller KD, Rugo HS, Neubauer M, Robert N, Hellerstedt B, Saleh M, Richards P, Specht JM, Yardley DA, Carlson RW, Finn RS, Charpentier E, Garcia-Ribas I, et al. Phase III Study of Iniparib Plus Gemcitabine and Carboplatin Versus Gemcitabine and Carboplatin in Patients With Metastatic Triple-Negative Breast Cancer. J Clin Oncol. 2014; 32:3840–3847.
24. Bendell J, O’Reilly E, Middleton M, Chau I, Hochster H, Fielding A, Burke W, Burris H. Phase I study of olaparib plus gemcitabine in patients with advanced solid tumours and comparison with gemcitabine alone in patients with locally advanced/metastatic pancreatic cancer. Annals of Oncology. 2015; mdu581.
25. Oza A, Cibula D, Oaknin Benzaquen A, Poole C, Mathijssen R, Sonke G, Mackay H, Lowe E, Read J, Friedler M. (2013). Olaparib plus chemotherapy, followed by maintenance monotherapy, in women with platinum-sensitive recurrent serous ovarian cancer (PSR SOC): BRCA1/2 mutation (BRCAm) and interim overall survival analyses. EUROPEAN JOURNAL OF CANCER: ELSEVIER SCI LTD THE BOULEVARD, KIDLINGTON, OXFORD OX5 1GB, OXON, ENGLAND), pp. S712–S713.
26. Su JM, Thompson P, Adesina A, Li XN, Kilburn L, Onar-Thomas A, Kocak M, Chyla B, McKeegan E, Warren KE, Goldman S, Pollack IF, Fouladi M, Chen A, Giranda V, Boyett J, et al. A phase I trial of veliparib (ABT-888) and temozolomide in children with recurrent CNS tumors: a Pediatric Brain Tumor Consortium reportdagger. Neuro-oncology. 2014; 16:1661–8.
27. Plummer R, Stephens P, Aissat-Daudigny L, Cambois A, Moachon G, Brown PD, Campone M. Phase 1 dose-escalation study of the PARP inhibitor CEP-9722 as monotherapy or in combination with temozolomide in patients with solid tumors. Cancer chemotherapy and pharmacology. 2014; 74:257–265.
28. Plummer R, Lorigan P, Steven N, Scott L, Middleton MR, Wilson RH, Mulligan E, Curtin N, Wang D, Dewji R, Abbattista A, Gallo J, Calvert H. A phase II study of the potent PARP inhibitor, Rucaparib (PF-01367338, AG014699), with temozolomide in patients with metastatic melanoma demonstrating evidence of chemopotentiation. Cancer chemotherapy and pharmacology. 2013; 71:1191–1199.
29. Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JH, de Bono JS. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. The New England journal of medicine. 2009; 361:123–134.
30. Plummer R, Jones C, Middleton M, Wilson R, Evans J, Olsen A, Curtin N, Boddy A, McHugh P, Newell D, Harris A, Johnson P, Steinfeldt H, Dewji R, Wang D, Robson L, et al. Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG0699, in combination with temozolomide in patients with advanced solid tumors. Clinical cancer research. 2008; 14:7917–7923.
31. Samol J, Ranson M, Scott E, Macpherson E, Carmichael J, Thomas A, Cassidy J. Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: a phase I study. Investigational new drugs. 2012; 30:1493–1500.
32. Dean E, Middleton MR, Pwint T, Swaisland H, Carmichael J, Goodege-Kunwar P, Ranson M. Phase I study to assess the safety and tolerability of olaparib in combination with bevacizumab in patients with advanced solid tumours. British journal of cancer. 2012; 106:468–474.
33. Deeks ED. Olaparib: first global approval. Drugs. 2015; 75:231–240.
34. Orban T, Olah E. Emerging roles of BRCA1 alternative splicing. Molecular Pathology. 2003; 56:191.
35. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA: a cancer journal for clinicians. 2005; 55:74–108.
36. Boyd J. Specific keynote: hereditary ovarian cancer: what we know. Gynecologic oncology. 2003; 88:S8–S10.
37. Brody LC, Biesecker BB. Breast cancer susceptibility genes: BRCA1 and BRCA2. Medicine. 1998; 77:208–226.
38. Alsop K, Fereday S, Meldrum C, Emmanuel C, George J, Dobrovic A, Birrer MJ, Webb PM, Stewart C, Friedler M. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. Journal of Clinical Oncology. 2012; 30:2654–2663.
39. Bolton KL, Chenevix-Trench G, Goh C, Sadetzki S, Ramus SJ, Karlan BY, Lambrechts D, Despierre E, Barrowdale D, McGuffog L. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. Jama. 2012; 307:382–389.
40. Zhong Q, Peng H-L Zhao, X Zhang, L and Hwang W-T. Effects of BRCA1-and BRCA2-related mutations on ovarian and breast cancer survival: a meta-analysis. Clinical Cancer Research. 2015; 21:211–220.
41. Ying S, Hamdy FC, Helleday T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer research. 2012; 72:2814–2821.
42. Venkitaraman AR. Cancer suppression by the chromosome custodians, BRCA1 and BRCA2. Science. 2014; 343:1470–1475.
43. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005; 434:913–917.
44. Penning TD. Small-molecule PARP modulators—current status and future therapeutic potential. Current opinion in drug discovery & development. 2010; 13:577–586.
45. Sinha G. Downfall of Iniparib: A PARP Inhibitor That Doesn’t Inhibit PARP After All. Journal of the National Cancer Institute. 2014; 106:djt447.
46. Patel AG, De Lorenzo SB, Flatten KS, Poirier GG, Kaufmann SH. Failure of iniparib to inhibit poly(ADP-Ribose) polymerase in vitro. Clinical cancer research. 2012; 18:1655–1662.
47. Lupo B, Trusolino L. Inhibition of poly (ADP-ribosyl) ation in cancer: Old and new paradigms revisited. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer. 2014; 1846:201–215.
48. Olaparib effective in four advanced cancers: Cancer discovery. 2015; 5:of 3.
49. Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002; 108:171–182.
50. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005; 434:917–921.
51. Muendlein A, Rohde BH, Gasser K, Haid A, Rauch S, Kinz E, Drexel H, Hofmann W, Schindler V, Kapoor R, Decker T, Lang AH. Evaluation of BRCA1/2 mutational status among German and Austrian women with triple-negative breast cancer. Journal of cancer research and clinical oncology. 2015; 141:2005–12.
52. Gonzalez-Angulo AM, Timms KM, Liu S, Chen H, Litton JK, Potter J, Lanchbury JS, Stemke-Hale K, Hennessy BT, Arun BK, Hortobagyi GN, Do KA, Mills GB, Meric-Bernstam F. Incidence and Outcome of BRCA Mutations in Unselected Patients with Triple Receptor-Negative Breast Cancer. Clinical Cancer Research. 2011; 17:1082–1089.
53. Mersch J, Jackson MA, Park M, Nebgen D, Peterson SK, Singletary C, Arun BK, Litton JK. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer. 2015; 121:269–275.
54. Euhus D. Genetic Testing Today. Annals of Surgical Oncology. 2014; 21:3209–3215.
55. Kaelin WG. The concept of synthetic lethality in the context of anticancer therapy. Nature reviews cancer. 2005; 5:689–698.
56. Higgins J, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. Bmj. 2003; 327:557–560.
57. Egger M, Smith GD, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. Bmj. 1997; 315:629–634.