
INTRODUCTION
Tumor responses to radiotherapy are influenced by many factors, including those of genetic, pathological, and tumor micro-environmental. For example, intra-tumoral hypoxia is one of the factors that contribute to radioresistance [1]. In recent years, efforts have been focused to identify molecular elements in regulating tumor radiosensitivity, and gene products that are involved in the DNA damage response (DDR) have been found to be associated with radiosensitivity [2]. However, it is still not clear how these DDR elements contribute to radioresistance in the tumor setting, and there is clearly a lack of clinical significance of these factors.
As a class of critical elements in the DDR, microRNAs (miRNAs) have been implicated in regulation of radiosensitivity [3, 4]. miRNAs are small non-coding RNAs that are involved in biological processes, including development, proliferation, cell-cycling and DNA damage repair [5]. These 20–25 nucleotide RNAs are processed from larger nucleotide fold-back structures (pri-RNA) into hairpin pre-miRNAs before finally being converted into mature miRNAs. The number of unique, mature human miRNAs reported is 2,588 (according to the June 2014 release of miRBase 21). The regulation of miRNA expression can be governed by a number of internal and external factors, and these factors uniquely regulate the maturation of specific miRNAs [6].
The differential expression of miRNAs in human tumors has led to the idea that these miRNAs can contribute to oncogenesis by acting as tumor suppressors or oncogenes. These changes in expression can promote genetic instability by negatively regulating DDR proteins, resulting in a decrease in the DNA repair capacity of cancerous cells [7, 8]. Studies have implicated a hypoxic tumor micro-environment and the increased expression of HIF-1α in the up-regulation of two specific miRNAs, miR-210 and miR-373. The expression of these two miRNAs was found to specifically alter the nucleotide excision repair and homology-dependent repair pathways [9]. Similarly, exposure to ionizing radiation (IR) results in an altered miRNA expression profile [10]. Alterations in expression become critical in terms of radiosensitivity, because activation of the DDR, in an Ataxia-Telangiectasia mutated (ATM) kinase-dependent manner, provide regulatory targets for miRNAs expressed in the presence of IR at each stage of the checkpoint [10]. Internal regulation and maturation of specific miRNAs induced by DNA damage can be linked to activation of ATM, and the subsequent phosphorylation of the KH-type splicing regulatory protein (KSRP) [11].
The determination and characterization of miRNA expression profiles, particularly in cancer subtypes, can play an important role in understanding disease progression and treatment responses. Examination of miRNA expression profiles in both laboratory and clinical settings might establish miRNA “fingerprints” that correlate to, among other parameters, cancer type, aggression, and response to treatment. In Head and Neck Squamous cell carcinomas (HNSCC) for example, a comparative analysis of normal and cancerous tissues revealed that a handful of miRNAs were differentially regulated in the cancerous tissues with one in particular, Mir-21 as a putative oncogenic miRNA [12]. Most recent studies underscoring the robustness of The Cancer Genome Atlas (TCGA) database utilized the database to annotate and analyze somatic variants in HNSCC patients [13]. Here we focus specifically on the effects of ionizing radiation on miRNA expression levels, and the correlation of the expression to radiosensitivity to identify a miRNA signature for the predication of radiosensitivity.
RESULTS
Ionizing radiation-induced miRNA changes in cell lines with proficient or deficient of ATM
In order to assess the relationship of miRNA changes with radiosensitivity, we selected a pair of human lymphoblastoid cell lines derived from a patient (proband) of Ataxia-Telangiectasia and a direct relative (normal). These two cell lines have been extensively used to study the functional role of the ATM kinase in the DDR previously [14–16]. We conducted a full microRNA array in cells treated with or without IR. To analyze the data, we first established the threshold fluorescence intensity for each of the four experimental conditions (GM0536 mock or IR and GM1526 mock or IR):
Table 1: miRNA targets identified from microArray screening
miRNA |
Fold change (after IR) ATM+/+ |
± SD |
Fold Change (After IR) ATM−/− |
± SD |
|---|---|---|---|---|
hsa-let-7e |
0.89194 |
0.0388 |
1.42886 |
0.0335 |
hsa-let-7f |
1.03497 |
0.1200 |
1.62202 |
0.0341 |
hsa-let-7g |
1.20312 |
0.0359 |
1.07888 |
0.0470 |
hsa-mir-016 |
0.88144 |
0.1411 |
2.24896 |
0.1462 |
hsa-mir-019a |
0.89458 |
0.0141 |
0.91168 |
0.0298 |
hsa-mir-019b |
0.88981 |
0.0490 |
0.9242 |
0.0610 |
hsa-mir-020a |
1.19119 |
0.1565 |
0.82158 |
0.1914 |
hsa-mir-027a |
0.97308 |
0.0415 |
1.40207 |
0.0356 |
hsa-mir-029b |
0.78003 |
0.0145 |
1.56938 |
0.1775 |
hsa-mir-029c |
1.07076 |
0.1039 |
1.20673 |
0.1883 |
hsa-mir-030c |
0.55318 |
0.0699 |
1.60287 |
0.1257 |
hsa-mir-030e 3p |
1.05512 |
0.0711 |
0 |
0 |
hsa-mir-101 |
0.92582 |
0.0323 |
0.73823 |
0.0517 |
hsa-mir-106a |
0.80357 |
0.0579 |
1.92872 |
0.0678 |
hsa-miR-1248 |
2.2929 |
0.1513 |
0 |
0 |
hsa-miR-1254 |
0.75457 |
0.0154 |
0 |
0 |
hsa-miR-1308 |
0.85653 |
0.0533 |
1.06383 |
0.0970 |
hsa-mir-142 3p |
0.79681 |
0.0180 |
0.83335 |
0.1186 |
hsa-mir-150 |
1.14678 |
0.0265 |
0 |
0 |
hsa-miR-1826 |
1.22729 |
0.0339 |
3.11047 |
0.0832 |
hsa-miR-18b |
0.80097 |
0.0894 |
0.86415 |
0.0261 |
hsa-miR-320c |
1.18737 |
0.0490 |
0 |
0.0368 |
hsa-mir-565-Pre |
1.37948 |
0.0744 |
1.02796 |
0.0482 |
hsa-mir-566-Pre |
0.59372 |
0.0217 |
1.29855 |
0.1471 |
hsa-mir-595 |
0 |
0 |
0 |
0 |
hsa-miR-768-3p |
1.16511 |
0.0261 |
0 |
0 |
hsa-miR-768-5p |
1.28797 |
0.0771 |
0 |
0 |
hsa-miR-886-5p |
1.08908 |
0.0675 |
0.76209 |
0.0305 |
Validation of the miRNA hits using the TCGA data base
To assess the clinical relevance of the 28 miRNAs hits, we accessed the TCGA database and focused on available cases of Head and Neck Squamous Cell Carcinomas. We compared the levels of each miRNA expression in patient samples. Of the 523 available patient cases in the database, 435 patients were monitored by clinical follow-up. These patients were monitored for either complete remission or additional tumor event, and were categorized as having radiation or chemotherapy. The clinical data for a majority of the patients was often incomplete, so we focused our analysis on those patients with the most complete set of criteria in terms of tumor status and course of treatment. Specifically, we chose a subset of cancer patients in which the primary course of treatment was radiotherapy. An analysis of three groups of patient miRNAs was performed, with each group comprised of 10 patients representative of their respective groups. The groups analyzed were as follows: radiated with a complete response (defined as radiosensitive), radiated with tumor progression (defined as radioresistant), and not irradiated. Highlighting the expression of those 28 miRNAs identified in the microArray screen, a ratio of the average expression levels in the patients receiving radiotherapy vs. those not receiving radiotherapy was calculated (Figure 1A). The ratios were then compared to the fold change values from Table 1. From the correlation of the patient data with the in vitro data, a subset of 5 miRNAs emerges as a signature for predicting radiation response in the clinical setting (Figure 1B). In this pattern, upregulation of miR-016, miR-29b, miR-150 and miR-1254, and down regulation of let-7e after radiation correlated with a complete response. On the contrary, an inverse expression pattern was seen to be associated with additional tumor events in the samples.
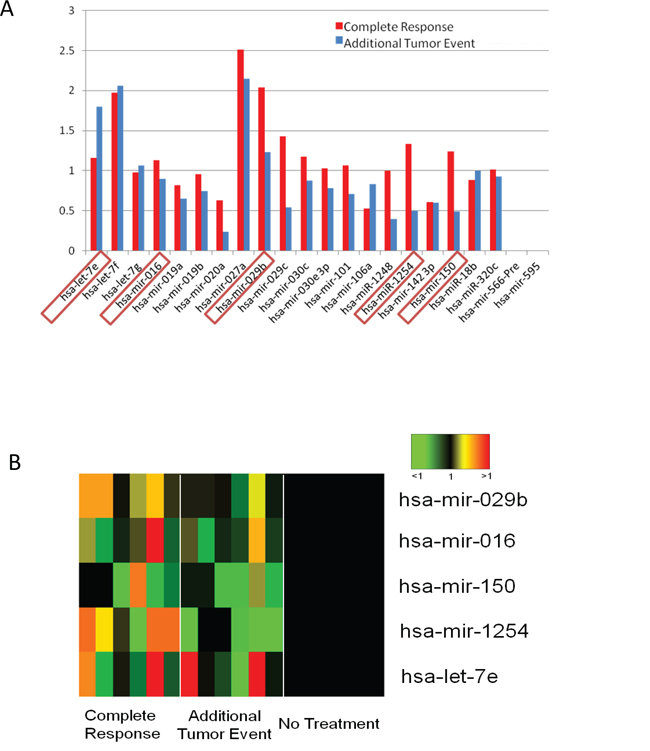
Figure 1: Differences in miRNA expression patterns between patients with complete response and those with additional tumor events. A. Relative miRNA expression of the 22 miRNAs identified in the in vitro microarray was plotted. Fold change was cross referenced with the in vitro fold change. The boxed miRNAs positively correlate with in vitro data. Significance was determined by Student's t-test; *p ≤ 0.05, **p ≤ 0.005, ***p ≤ 0.0005 B. A heat map of the levels of the 5 correlating miRNAs in patients with complete response or additional tumor event relative to patients receiving no treatment indicates an increase in the event of a complete response.
ATM expression level in HNSCC is associated with the radiation response
As a central DNA damage response protein, ATM-deficiency leads to radiosensitivity [17]. ATM-mediated radiosensitivity is regulated by its phosphorylation of downstream targets such as SMC1 and Snail [18, 19]. To determine whether the various radiation responses in the HNSCC patients were linked to different ATM expression, we looked at the ATM expression level in the same patient population in the TCGA database. Protein expression for the TCGA HNSC samples was determined using MDA Reverse Phase Protein Array. The average levels of ATM expression from the radio-sensitive samples and radio-resistant samples were calculated. Interestingly, we found that ATM expression levels were significantly lower in the radio-sensitive patients when compared to the resistant patient levels (Figure 2).
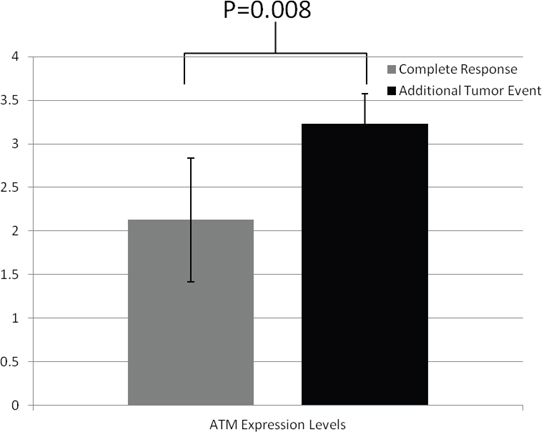
Figure 2: ATM expression levels are higher in patients with additional tumor events following radiation treatment. RPPA data from the patients cataloged in the TCGA database shows that there is an increase in ATM protein levels in patients with additional tumor events.
Verification of miRNA signature by qRT-PCR
In order to determine the validity of the identified miRNAs as a signature, we tested a head and neck cancer cell line Cal27, a tongue Squamous cell carcinoma line. The miRNA expression profile of the Cal27 before and after radiation, with and without ATM inhibition is consistent with both the initial miRNA screen and, more importantly, the TCGA clinical data (Figure 3A). As with the TCGA data and the initial miRNA screen, in the absence of ATM function, miR-16, -29b, -1254, and -150 were up-regulated, while Let-7e was down-regulated. To confirm the qRT-PCR, the amplified products were run on an agarose gel (Figure 3B). The results indicate that this identified signature represents radiosensitivity in HNSCC. A comparison of the basal miRNA levels with and without ATM inhibition indicates that ATM plays a minimal role in the basal regulation of these miRNAs (Supplemental Figure 1).
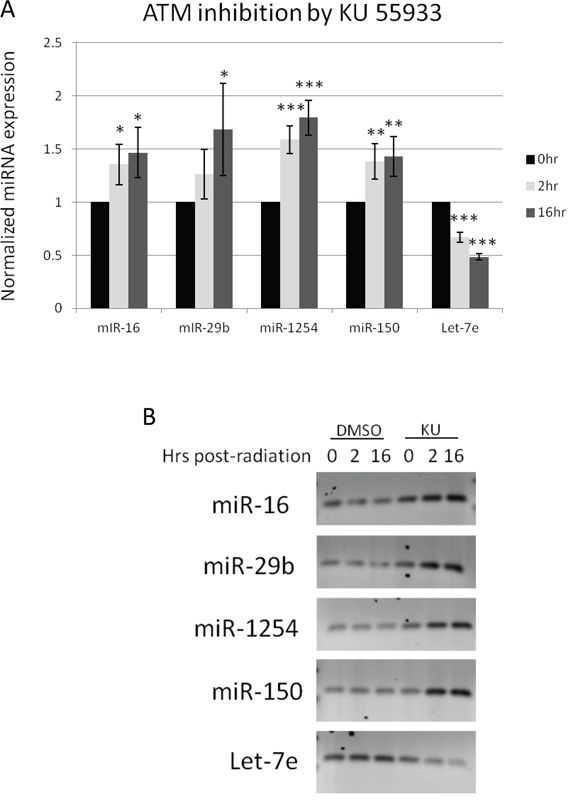
Figure 3: qRT-PCR of miRNA signature in HNSCC cells. A–B. Cal27 cells were subjected to ionizing radiation in the presence or absence of the ATM kinase inhibitor KU-55933. The miRNA levels were quantified by qRT-PCR, the expression levels of each miRNA were normalized according to the Cq values. In the KU-55933 treated cells, there were significant changes in all 5 miRNAs probed as determined by Student's t-test for variance. P values are reported as *p ≤ 0.05, **p ≤ 0.005, ***p ≤ 0.0005.
A pathway analysis of the miRNA signature that predict radiation responsiveness
A pathway analysis of the targets regulated by these 5 signature miRNAs was determined using the String database (STRING 9.1) (Figure 4). Of the multiple gene targets of these miRNAs, a handful is implicated in the DDR. Interestingly, we found that SMC1A, a direct target of ATM in radiosensitivity [18], is regulated by let-7e by the pathway analysis. Conversely, BCL-2 and MCL1 are regulated by miR-16 and miR-29b respectively [20, 21], which we have observed to be up-regulated in radiosensitive patients with complete regression of the disease. The direct connection of the targets of the miRNAs to key proteins in the DDR, specifically ATM, MDM2 and TP53, indicated that the expression patterns of these miRNAs present a viable biological fingerprint for radiosensitivity phenotypes.
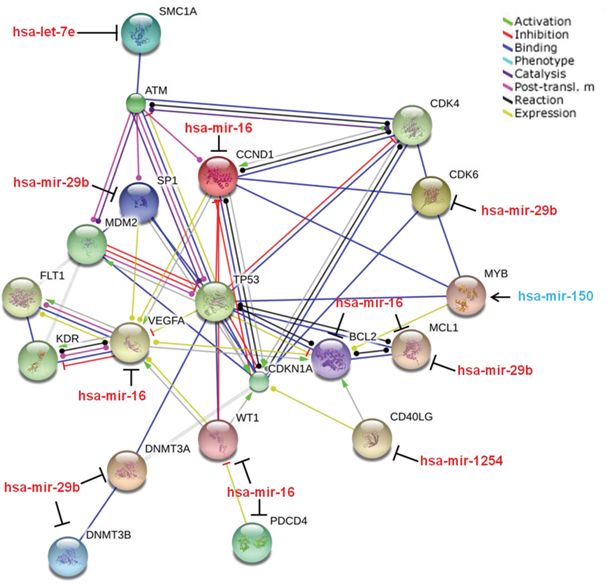
Figure 4: Pathway analysis of proteins regulated by the 5 “fingerprint” miRNAs. String pathway analysis of the proteins regulated by the identified miRNA fingerprint links elevated expression of the 5 miRNAs with cell survival, cell cycle, and DNA damage pathways.
DISCUSSION
Identification of a miRNA signature to predict tumor response to radiotherapy can provide a more comprehensive picture as to how miRNAs are involved in the radiation response. With the correlation of this signature in the TCGA database, we demonstrate a clinically relevant biomarker tool that might provide useful information to help make a treatment decision. For example, if the radiation-induced miRNA signature change can be obtained after the first week of radiation treatment, and analyzing the data might determine whether a booster dose of radiation is needed by the end of radiotherapy course.
The ability to link in vitro observations to clinical relevance relies on the curation of a patient database. The TCGA network provides a large scale view of numerous parameters for cancers in nearly a dozen tissue types. The public availability of this data allows for the reconciliation of non-clinical data with information gathered from patients in order to more effectively translate results from the bench into practical clinical application. Recently, it was shown that the patient repository of the TCGA database can be utilized in a disease specific manner to highlight parity in somatic mutations that can be leveraged into advances in treatment of HNSCC patients [13]. The database parameters examined here focused on miRNA and protein expression levels, and disease progression of patients with HNSCC. The radiosensitivity miRNA signature identified was validated by the clinical data, indicating that miRNAs can be used as biomarker to predict clinical responsiveness to radiotherapy.
Previous mechanistic studies have provided clues on how these 5 miRNA signature proteins are involved in regulating radiosensitivity. Specifically, pathway analysis shows that identified signature miRNAs regulate mRNA levels of proteins involved in pro-survival pathways and DNA repair pathways. Using differential miRNA expression as a means for establishing a fingerprint or biomarker has the potential to be a reliable prognostic tool, particularly if the absence of a functional ATM results in an elevated basal expression of these miRNAs. The miRNAs that were identified in this study have been implicated in disease progression in a number of cancer types; however their expression has not be examined in the context of radiation treatment.
By considering the 5 miRNAs as a signature as well as the correlation of ATM expression with radiation resistance we can begin to validate two means for predictive biomarkers: 1) Prior to radiation, ATM expression can be assessed. Based on the TCGA data as well as the initial microarray screen, the direct correlation between ATM levels and radio-resistance can be used to determine the effectiveness of radiotherapy. 2) A post-radiation assessment of our target miRNA signature expression early in the radiation course can be predictive of the sensitivity of the tumor to the treatment. If the signature indicates a resistance, as determined by the relative expression levels defined above, then a booster radiation dose may be required for a complete tumor response.
MATERIALS AND METHODS
Cell lines and culture
The Epstein-Barr virus transformed human lymphoblastic cell lines GM0536 and GM1526 cells (obtained from the NIGMS Human Mutant Cell Repository, Camden, NJ) were maintained in RPMI 1640 medium supplemented with 15% fetal bovine serum and 50 μg/ml of penicillin/streptomycin. HNSCC CAL 27 cells (CRL-2095, ATCC Manassas, VA 20108 USA) were maintained in DMEM medium supplemented with 10% FBS containing Pen/Strep antibiotics in a humidified 37°C atmosphere. Cell line authentication was originally done by Short Tandem Repeat profiling at NIGMS and ATCC, respectively. After purchase, the cell lines were not authenticated by authors.
Irradiation
Ionizing radiation was delivered by an X-Rad 320 X-ray irradiator (Precision X-Ray Inc. N Branford, CT) at a dose rate of 2Gy/min.
MiRNA Microarray analysis
MiRNA microarray analyses were performed by GenoSensor (Tempe, Arizona). 2 hours after 0Gy or 6Gy irradiation, cells were harvested and the total RNA isolated followed by miRNA microarray. Genes are positioned by array block, row, and column. All genes are in triplicate. Their names and signal intensity of each probe were directly transferred from the raw data sheets. The average of signal intensity on three redundancies and standard deviation were calculated. Signal intensity is fluorescent signal of each chemical. Since the fluorescence dye is detected at either 635 nm or 532 nm channel, the signals are labeled with either F635 or F532. Bacterial sequences, such as gnd, fixB, etc., were served as negative controls and for assay QA/QC analysis, the sequences of which are not homologous with any targets in the reaction. Housekeeping genes are used as positive controls, such as U6 and 5S-rRNAs. Normalization is represented by the ratio of the average of gene signal intensity of a specific gene vs. average of signal intensity of all positive control genes. Fold change is comparison between samples, calculated by the ratio of normalization value.
Quantification of miRNA
Total RNA was isolated (Trizol Reagent, GIBCO BRL) at 0, 2, and 16 hrs of Cal27 cells post radiation (5Gy IR) treated with and without KU 55933 (ATM inhibitor, Catalog number 3544, Tocris, Bristol, BS11 0QLUnited Kingdom), and cDNA was generated using All-in-One™ miRNA qRT-PCR kit (GeneCopoeia, Rockville, MA 20850 USA). The miRNA levels of 5 target miRNAs were measured by qPCR at miRNA expression levels were compared to those of cells treated with KU 55933 (4 μM Reverse transcription was performed on the Arktik thermal cycler (Thermo) according to the manufacturer's protocol. The cDNA was then mixed with respective miRNA primer probes (GeneCopoeia). The plates were run on the BioRad CFX-96 plate reader. Fluorescence was read in the green channel. Cq values were averaged and compared to positive and non-template controls. The quality of the amplified cDNA was determined by a melt curve at the end of the cycle. Amplification was verified by running 10 μl of the PCR products on a 3% agarose gel. Gels were imaged on an AlphaInnotech FluorChemHD2 gel dock. Changes in miRNA expression were analyzed for significance using the Student's T-test for variance. The experiment was repeated twice with samples in triplicate each time.
Statistical analysis
In running a logistic regression model, we found that the independent variable was a perfect predictor for the dependent variable. Hence, it follows that a threshold analysis would be the best course of action. A threshold analysis was performed on the fluorescence values reported from the microRNA array of the two cell lines under the two experimental conditions. The threshold analysis,
A fold change test was conducted on the data set as a whole using an if/then statement to separate successes from failures. Failures were microRNAs with fluorescence values that reported a zero from the fold change test. Successes produced a value greater than zero. Next, the interval for where the true threshold, θ, actually exists was determined. This was done by taking the minimum fluorescence from the successes and the maximum fluorescence from the failures to form an interval of interest. To do this, a new function was established:
Power analysis for the selection of patient data per condition was determined using the following equation:
TCGA database analysis
Archived patient samples of HNSCC patients in The Cancer Genome Atlas (TCGA) database (https://tcga-data.nci.nih.gov) were selected based on patient samples that had been subjected to both miRNA-Seq testing and reverse-phase protein array (RPPA) analysis. The data from 523 patients was available through the database. The miRNA levels of the specific miRNAs identified in the microarray from the in vitro study were examined and averaged in these patients. The ATM protein levels of the patients were averaged from the RPPA data available. Patients were able to be separated according to response to treatment based on the follow-up data also archived within the database.
Pathway analysis
The miRNAs identified from patient and in vitro screens were then input into the miRecords database (http://mirecords.umn.edu/) to determine validated target mRNAs for these specific miRNAs. The protein hits from miRecords were then collectively input into the STRING functional protein association networks (http://string-db.org/) to determine functional protein interactions.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by in part by NIH grants R01CA133093 and the Alabama Innovation Fund. We thank Drs. Chunying Yang and Qinghua Zeng (Southern Research Institute) for technical assistance.
CONFLICTS OF INTEREST
None.
REFERENCES
1. Chan DA, Giaccia AJ. Hypoxia, gene expression, and metastasis. Cancer Metastasis Rev. 2007; 26:333–339.
2. Raleigh DR, Haas-Kogan DA. Molecular targets and mechanisms of radiosensitization using DNA damage response pathways. Future Oncol. 2013; 9:219–233.
3. Mao A, Liu Y, Zhang H, Di C, Sun C. microRNA expression and biogenesis in cellular response to ionizing radiation. DNA Cell Biol. 2014; 33:667–679.
4. Chaudhry MA. Radiation-induced microRNA: discovery, functional analysis, and cancer radiotherapy. J Cell Biochem. 2014; 115:436–449.
5. Dong H, Lei J, Ding L, Wen Y, Ju H, Zhang X. MicroRNA: function, detection, and bioanalysis. Chemical reviews. 2013; 113:6207–6233.
6. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014; 15:509–524.
7. Bindra RS, Crosby ME, Glazer PM. Regulation of DNA repair in hypoxic cancer cells. Cancer Metastasis Rev. 2007; 26:249–260.
8. Bindra RS, Glazer PM. Co-repression of mismatch repair gene expression by hypoxia in cancer cells: role of the Myc/Max network. Cancer Lett. 2007; 252:93–103.
9. Crosby ME, Kulshreshtha R, Ivan M, Glazer PM. MicroRNA regulation of DNA repair gene expression in hypoxic stress. Cancer Res. 2009; 69:1221–1229.
10. Czochor JR, Glazer PM. microRNAs in cancer cell response to ionizing radiation. Antioxid Redox Signal. 2014; 21:293–312.
11. Zhang X, Wan G, Berger FG, He X, Lu X. The ATM kinase induces microRNA biogenesis in the DNA damage response. Mol Cell. 2011; 41:371–383.
12. Chang SS, Jiang WW, Smith I, Poeta LM, Begum S, Glazer C, Shan S, Westra W, Sidransky D, Califano JA. MicroRNA alterations in head and neck squamous cell carcinoma. Int J Cancer. 2008; 123:2791–2797.
13. The Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015; 517:576–582.
14. Sun M, Guo X, Qian X, Wang H, Yang C, Brinkman KL, Serrano-Gonzalez M, Jope RS, Zhou B, Engler DA, Zhan M, Wong ST, Fu L, Xu B. Activation of the ATM-Snail pathway promotes breast cancer metastasis. J Mol Cell Biol. 2012; 4:304–315.
15. Tang X, Hui ZG, Cui XL, Garg R, Kastan MB, Xu B. A vnovel ATM-dependent pathway regulates protein phosphatase 1 in response to DNA damage. Mol Cell Biol. 2008; 28:2559–2566.
16. Yang C, Hao J, Kong D, Cui X, Zhang W, Wang H, Guo X, Ma S, Liu X, Pu P, Xu B. ATM-mediated Mad1 Serine 214 phosphorylation regulates Mad1 dimerization and the spindle assembly checkpoint. Carcinogenesis. 2014; 35:2007–2013.
17. Boohaker RJ, Xu B. The Versatile Functions of the ATM Kinase. Biomedical Journal. 2014; 37:3–9.
18. Yazdi PT, Wang Y, Zhao S, Patel N, Lee EY, Qin J. SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev. 2002; 16:571–582.
19. Boohaker RJ, Cui X, Stackhouse M, Xu B. ATM-mediated Snail Serine 100 phosphorylation regulates cellular radiosensitivity. Radiother Oncol. 2013; 108:403–408.
20. Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005; 102:13944–13949.
21. Aldaz B, Sagardoy A, Nogueira L, Guruceaga E, Grande L, Huse JT, Aznar MA, Diez-Valle R, Tejada-Solis S, Alonso MM, Fernandez-Luna JL, Martinez-Climent JA, Malumbres R. Involvement of miRNAs in the differentiation of human glioblastoma multiforme stem-like cells. PLoS One. 2013; 8:e77098.