
INTRODUCTION
Malignant gliomas are the most common type of primary tumors of the central nervous system in Europe and US, accounting for 80% of patients [1, 2]. Annual incidence of these neoplasms is approximately 4–7 per 100,000 in men and 3–5 in women. The number of patients is expected to increase in industrialized countries as the population ages. Tumor diagnoses are based on histopathological features and are graded according to the World Health Organization classification [3]. Among them, glioblastomas multiform (GBM) are the most frequent, and harbor the poorest prognosis [1, 2] as only 2 to 5% of patients survive after 2 years [4]. These gliomas are indeed highly refractory to any treatment.
Since the publication of the phase III study initiated by the European Organisation for Research and Treatment of Cancer (EORTC) and the National Cancer Institute of Canada Clinical Trials Group (NCIC CTG) groups in 2005, standard treatment is surgery followed by radiotherapy and temozolomide (TMZ)-based chemotherapy [5]. Concomitant radiotherapy and TMZ administration has significantly improved median survival of glioma patients, from 12.1 to 14.6 months, with 27% of patients alive at 2 years, instead of 10% without TMZ [6]. This improvement was confirmed during the five-year follow-up period with a median survival of 9.8% versus 1.9% [7]. The benefit offered by TMZ has been established in a systematic review [8]. Nevertheless, malignant gliomas prognosis remains poor and most of the patients relapse, thus there is a clear need for new treatments.
Some glioma hallmarks were demonstrated to be of clinical relevance and may offer opportunities for new therapies. Among them, some are responsible for a poorer prognosis (mutation/amplification of the epidermal growth factor receptor (EGFR) [9, 10]). and, on the other hand, some others are associated with a better one (O-6-methylguanine-DNA methyltransferase (MGMT) promoter methylation [11], mutations in isocitrate dehydrogenase (IDH) genes [12] and 1p/19q deletion [13]). The loss of heterozygosity (LOH) of these chromosomal arms was indeed correlated with the oligodendroglioma noticeable chemosensitivity [13, 14], suggesting that some key genes involved in chemosensitivity may be located on this area.
This fact consequently provides a list of more than 1,700 putative target genes to increase glioma sensitivity to chemotherapy, and among them some of the 150 DNA-repair genes inventoried by Wood et al. [15].
Prior to 2005, GBM patients had been treated for decades with chemotherapy agents. The failure of these drugs showed that intrinsic or acquired resistance mechanisms are of paramount importance in these high grade tumors. Since most drugs were alkylating agents, inducing a panel of DNA damages, DNA repair is a highly relevant resistance mechanism. Moreover, the DNA repair gene Mgmt was shown to be associated to patients’ outcome when treated with TMZ [11]. Mgmt was considered as a prognostic marker as well as a therapeutic opportunity [16]. Unfortunately, targeting Mgmt activity using chemical inhibitor O6BG was first associated with a crippling hematopoietic toxicity [17, 18]. Yet innovative strategies have emerged, allowing for reduction of side effects and transforming the hypothesis into medical practice [19].
We proposed the hypothesis that other DNA-repair genes would be involved in glioma resistance to TMZ. Using small interfering RNA (siRNA) we found that inhibition of 4 of them, namely ercc1, ercc2, mutyh and pnkp, showed a significant impact on cisplatin (CDDP) and/or TMZ cytotoxic effect in several human glioma cell lines. Furthermore, an ercc-1 siRNA-based adjuvant treatment was able to improve the efficacy of TMZ in glioma tumor bearing mice. This study offers a promising adjuvant therapy to improve the clinical management of malignant gliomas.
RESULTS
In silico identification of candidate genes
To select genes potentially involved in glioblastoma chemoresistance, the chromosomal areas commonly considered as correlated with oligodendroglioma chemo-sensitivity (1p36–1p32 and 19q13.2–19q13.4; [20]) were screened, using databases on the web (http://www.ncbi.nlm.nih.gov/PubMed/; http://www.ensembl.org; http://www.cgal.icnet.uk/DNA_Repair_Genes.html; Supplementary to the paper by Wood, 2005) and published data [15, 21, 22]. Nine genes located in these regions belong to different DNA-repair systems among the 1,700 genes (Table 1).
Table 1: List of DNA repair-associated genes located on 1p/19q LOH regions
DNA repair genes |
DNA repair system |
Reference |
Location |
ercc1 |
NER |
NM_001983 |
19q13.2–3 |
lig1 |
NER |
NM_000234 |
19q13.2–3 |
ercc2 |
NER |
NM_000400 |
19q13.3 |
pold1 |
NER and MMR |
NM_002691 |
19q13.3 |
ruvbl2 |
HR |
NM_006666 |
19q13.3 |
pnkp |
BER |
NM_007254 |
19q13.3–4 |
rad54L |
HR |
NM_003579 |
1p32 |
mutyh |
BER |
NM_012222 |
1p34.3 |
mad2L2 |
ADN polymerase |
NM_006341 |
1p36 |
According to Wood et al, 2005
NER, nucleotide excision repair; MMR, mismatch repair; HR, homologous recombination; BER, base excision repair.
Screening of DNA-repair genes involved in chemoresistance of astrocytoma cells
The in vitro part of the project was developed as a 3-stage strategy. SiRNAs targeting candidate genes were screened on one cell line (U373). We chose to achieve this step using CDDP because Ercc1 is associated with a chemotherapy resistance (especially to platinum drugs) in several experimental models (see discussion). Then, the study was extended to six astrocytoma-derived cell lines and validated at the molecular level. Finally, the study was expanded to TMZ. Nine DNA-repair genes were screened on the U373 cell line with up to 5 different siRNAs per gene (Table 1). Cell viability was measured in the absence and presence of CDDP. SiRNAs were selected on the basis of two criteria: the absence of basal toxicity and their ability to improve chemotherapy treatment. SiRNAs inducing more than 35% cell death in absence of drug were considered as toxic and discarded. On the other hand, those increasing sensitivity to the chemotherapy drug were retained. An siRNA was considered as chemosensitizing when its chemoresistance index (CI) was lower than the GFP siRNA’s CI (0.5). In Figure 1, the white square corresponds to siRNAs with low toxicity and chemosensitization properties. Nine siRNAs out of 46 matched with these criteria, corresponding to 6 genes: ercc1, ercc2, mutyh, pnkp, ruvbl2 and pold1.
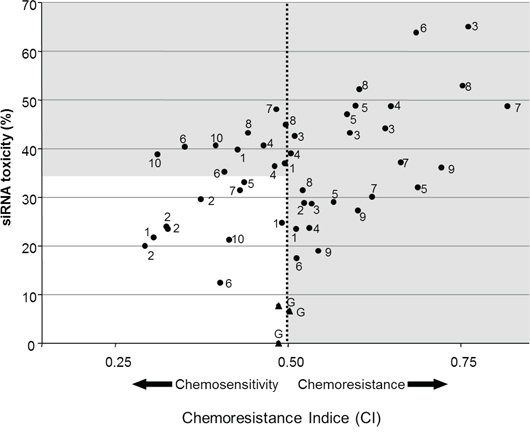
Figure 1: Efficiency and toxicity of siRNAs. Cell viability (%) was measured in the presence (x-axis) and absence (y-axis) of CDDP (5.10−6 M). SiRNA (150 nM) inducing less than 35% of cell death without drug, and a chemosensitization were selected for further analyses.
Highlighted genes were further analyzed to validate preliminary screening, using only the most efficient siRNA for each gene. The experiments were extended to 5 other cell lines and we confirmed that 4 siRNAs, ercc1#2, ercc2#1, mutyh#5 and pnkp#1, had significant chemosensitization effects (Table 2). Inhibition of ercc1 was the most effective in sensitizing cells to CDDP (up to 24.9%). Moreover, its effect was the most widespread since 4 out of the 6 cell lines were sensitized. The knock-down induced by the 3 other siRNAs was equally effective (up to 17%) but only on 2 (mutyh and pnkp) or 3 (ercc2) cell lines.
Table 2: SiRNA-induced drug sensitization in vitro
Drug |
Cell line |
Ercc1 |
Ercc2 |
Mutyh |
Pnkp |
CDDP |
U373 |
19.9** |
15.8** |
16.9** |
17.6** |
GHD |
24.9** |
10.1* |
10.8* |
12.1* |
|
LN229 |
24.7** |
16.9** |
n.s. |
n.s. |
|
U318 |
14.1** |
n.s |
n.s. |
n.s. |
|
TMZ |
U373 |
24.5** |
17.6** |
26.0** |
25.1** |
GHD |
9.5** |
n.s |
n.s. |
n.s. |
Drug sensitization index (DS) was measured for the 4 siRNA candidates on 6 cell lines (U373, U87, GHD, LN229, U138 and CCF). Data are the mean of 3 independent experiments. Only significant values are indicated. n.s., non-significant.
In order to validate these effects at molecular level, siRNA-induced down-regulation of the mRNAs was measured (Table 3). The target specificity was confirmed by the absence of an effect on either cyclophiline A or other gene expression (data not shown). There was no obvious link between the efficiency of siRNA in decreasing mRNA levels (nor with residual mRNA content; data not shown) and functional impact on cell viability. This is reinforced by the observation that the ercc1 mRNA level actually decreased (by 65%) in U87 cells which were not yet chemosensitized.
Table 3: siRNA-induced downregulation of mRNA measured by RT-qPCR
Cell line |
Quantified mRNA |
Targeted siRNA |
siRNA GFP |
Inhibition (%) |
p-value (ANOVA) |
||
mean (fMol) |
sem |
mean (fMol) |
sem |
||||
GHD |
ercc1 |
8.6E-06 |
2.0E-06 |
2.1E-05 |
5.0E-06 |
58.6 |
0.019 |
ercc2 |
1.7E-07 |
3.9E-08 |
4.3E-07 |
7.1E-08 |
61.6 |
0.011 |
|
mutyh |
5.0E-05 |
1.3E-05 |
1.7E-04 |
3.6E-05 |
71.0 |
0.004 |
|
pnkp |
8.0E-06 |
1.6E-06 |
5.0E-05 |
1.6E-05 |
83.8 |
0.005 |
|
U373 |
ercc1 |
1.9E-06 |
8.2E-07 |
8.4E-06 |
2.3E-06 |
77.4 |
0.018 |
ercc2 |
1.2E-07 |
3.0E-08 |
2.8E-06 |
7.9E-07 |
95.7 |
0.003 |
|
mutyh |
2.6E-05 |
7.6E-06 |
9.5E-05 |
1.8E-05 |
72.9 |
0.007 |
|
pnkp |
3.6E-05 |
9.6E-06 |
6.5E-05 |
1.6E-05 |
44.2 |
0.013 |
|
mgmt |
4.2E-06 |
5.2E-07 |
2.1E-05 |
1.0E-06 |
80.4 |
0.001 |
|
All used siRNAs significantly reduced expression of their respective targets, when compared to the control siRNA (GFP). Measures were performed twice from each cell sample and results were expressed as the mean of at least three independent samples (originating from independent in vitro experiments)
Sensitization to temozolomide
Since TMZ is the gold standard in chemotherapy for gliomas, the study was extended to this drug using GHD and U373 cell lines. All siRNAs sensitized U373 cells to both CDDP and TMZ with a similar efficacy (Figure 2). The siRNAs were less efficient on GHD cells treated with TMZ compared to CDDP and only the effects of the ercc1 siRNA remained statistically significant (data not shown).
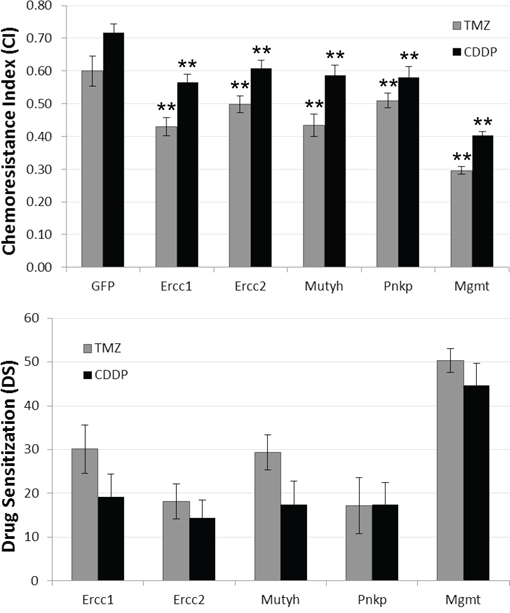
Figure 2: SiRNA-induced chemosensitization. U373 cells were transfected with siRNA targeting GFP, Ercc-1, Ercc-2, Mutyh or Pnkp, and treated with CDDP (grey) or TMZ (black). A. The Chemoresistance Index (CI) corresponds to the proportion of a cell population that survived chemotherapy. It was computed as follows: cell number with chemotherapy/cell number (OD) in control condition. B. DS corresponds to the chemosensitivity induced by siRNA. Data represented the mean of 3 independent experiments. Statistically significant difference between effects induced by siRNA GFP and other siRNA were indicated by 2 asterisks (p < 0.01). Error bars represent the bootstrapped standard errors.
In vivo therapeutic effect of Ercc1-siRNA
The chemosensitive effect of siRNA targeting Ercc1 was assessed in Nude mice carrying human glioma xenografts in 3 independent experiments. In all of them, Ercc1-siRNA #3 was highly effective in inhibiting tumor growth. Its effect was significantly different from the control treatment (GFP-siRNA) and greater than a siRNA targeting Mgmt (Figure 3). None of the groups experienced side effects such as body weight loss.
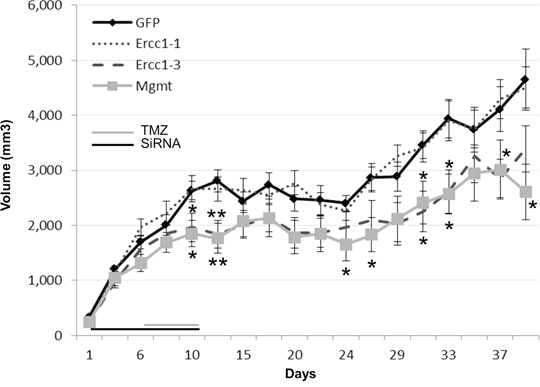
Figure 3: Therapeutic effects of the combined effect with TMZ and Ercc1-siRNA in vivo. In vivo combined effects of Ercc1#1-, Ercc1#3- or Mgmt-siRNA (4 μg) with TMZ (4.2 mg/kg) on U373 xenografts growth. The combined therapy with TMZ and Ercc1#3 (dash line) provided a significantly stronger reduction of tumor growth than TMZ with a control siRNA (GFP) (*, p < 0.05). The first day of administration was defined as ‘day 0′. The days on which the treatments were administered are indicated by grey (TMZ) and black (siRNA) lines at the bottom of the graphs. Mean tumor volumes +/− SE are shown. 2-tailed Student’s T-tests were used to evaluate results significance.
DISCUSSION
Despite the demonstration of the improvement in survival following radio- and chemotherapy regimens (TMZ) for glioblastoma, this malignancy remains an incurable disease.
A variety of cancer cell resistance mechanisms have been described, including decreased drug uptake, increased drug efflux, intracellular drug inactivation or drug-induced damage repair. The high activity of these mechanisms in gliomas have hampered the use of chemotherapy and thereby prevented widespread its acceptance as an effective treatment modality. Most chemotherapy drugs commonly employed in the past such as the PCV regimen (Procarbazine, Lomustine (CCNU), and Vincristine), CDDP, fotemustine or the presently used TMZ are DNA-alkylating molecules which damage DNA. These data have led many studies to focus on the link between DNA-repair and chemoresistance [23–26]. However it is only recently that the response to TMZ was found strongly correlated with mgmt promoter methylation status [11]. Down-regulation of Mgmt expression would explain sensitivity to therapy due to the absence of alkylating adducts repair. Mgmt is considered as a major DNA-repair enzyme involved in resistance to several chemotherapy drugs since the end of the twentieth century [27]. Hegi et al. have shown a correlation between the DNA-repair gene mgmt and response to TMZ [11]. Indeed, several clinical trials have addressed the possibility of increasing chemotherapy efficacy while blocking mgmt activity [28–31]. Using an innovative strategy, Adair et al. have recently demonstrated that Mgmt blockade is associated with an improvement of TMZ efficacy [19]. Although these results are preliminary with a small cohort of patients, they consist of the first dual clinical and functional demonstration of the link between Mgmt and TMZ resistance. These results indicate that DNA-repair inhibition is a highly attractive and plausible therapeutic avenue.
Thus Mgmt is to date the main molecular pathway related to TMZ response in glioblastoma.
As discussed above, the decrease in mgmt expression has often been reported, as a consequence of its promoter methylation. In this study, we observed both that siRNAs were responsible for a decrease of expression and a chemosensitizing impact. Similar observations were made using a second siRNA duplex (data not shown). Our results suggest that mgmt expression was sufficient for supporting a significant DNA repair activity.
As discussed below, we demonstrated that targeting other DNA-repair genes can provide similar efficacy compared to Mgmt invalidation.
Gliomas cytogenetic and clinical management
In contrast to glioblastomas, oligodendrogliomas constitute a chemoresponsive entity. The correlation between 1p/19q LOH and oligodendroglioma chemosensitivity [13] prompted us to perform an in silico analysis of these chromosomal areas in order to identify genes potentially involved in chemosensitivity. Jenkins et al. demonstrated that 1p/19q LOH was mediated by a translocation [32]. This early cytogenetic event was also associated with a longer overall survival. These data led to a better understanding of the cytogenetic alterations mechanisms frequently associated. But, above all, they reinforce the hypothesis that expression of genes located in both regions can be altered, being finally responsible for chemosensitivity of oligodendroglioma.
Among the 1,700 genes located on the 1p and 19q chromosomal areas commonly considered as correlated with oligodendroglioma chemosensitivity, we found 9 DNA-repair genes. We confirmed the in silico analysis with a functional genomic approach using siRNAs to knock down gene expression, mimicking LOH.
In vitro study
For validating candidates at functional level, we developed an adequate in vitro chemosensitivity assay in 96-well plates to test all the designed siRNA. The temporal sequence of treatments was crucial since the siRNA impact on DNA repair activity had to line up with the chemotherapy treatment. We succeeded in interfering with drug-induced DNA adduct repair using an siRNA transfection performed 24 hours prior to chemotherapy. This result is consistent with data previously published by Parker et al. showing that drug effect occurs in the first 6 hours of treatment [33].
SiRNAs found to induce cellular toxicity were discarded from the study to prevent erroneous conclusions. The observed toxicity could be the result of a critical gene inhibition and, though possibly interesting, it would have masked any intended chemosensitization. Similarly, siRNAs that did not have any impact on cell numeration were rejected. In the end, only 9 out of the 46 tested sequences in this study had a significant chemosensitivity impact, demonstrating the importance of a validated assay to bring out efficient siRNA targets.
The most prominent functional impact was observed with the two most studied genes of the NER system: ercc1 and ercc2, a.k.a. xeroderma pigmentosum group D (XPD). The NER system has a critical role in repairing DNA and has been extensively studied in cancer cells.
The most important chemosensitization occurred when ercc1 expression was inhibited: Sensitization was not larger but was found in a greater number of cell lines (4 vs 3 for ercc2, and 2 for mutyh and pnkp). These results emphasize previous reports on the major role of ercc1 in repairing DNA alterations related to alkylating chemotherapy. Thanks to its nuclease activity, the ercc1 protein plays a crucial role in the early excision step of damaged DNA [34, 35]. It is involved in CDDP-induced adduct repair in vitro [36, 37]. Reduction of its expression enhances CDDP cytotoxicity in ovarian cancer cell lines [38, 39], and was generally found involved in drug resistance of cultured cells [40, 41].
Moreover, its expression or polymorphisms were correlated with stage or clinical outcome in several cancers e.g. glioma [42], stomach [43], ovary [44] and lung [45, 46], (for review, see: [47, 48]). Thus, it appears as a very consistent if not universal component of tumor drug resistance and thus as a spread diagnostic/prognostic marker and/or therapeutic target.
Ercc2 expression was correlated with resistance to alkylating compounds in numerous cell lines [49, 50], including glioma cell lines [51], but no link has been established between this expression and NER activity [50]. This suggests that ercc2 is not an NER rate-limiting enzyme as in primary lymphocytes [52]. It could also act via the homologous recombination repair system as in SKMG-4 glioma cells [53]. Another level of complexity is the number of ercc2 polymorphisms without clear associated phenotypes [54]. These polymorphisms obviously contribute to governing ercc2 cellular function. So, while a previous report showed that its overexpression increased DNA repair in a glioma cell line [55], the present results are the first demonstration of its functional involvement in drug resistance.
We also found two genes belonging to the BER system: mutyh and pnkp. Only the systematic bio-informatics analysis prompted us to analyze them, as no previous publication involved them in chemosensitivity.
Mutyh is known to repair 8-oxo-7,8-dihydro2′deoxyguanosine (8-oxodG) caused by oxidation. Gene mutations and variants were associated with development of multiple colorectal adenomas and cancers [56]. This is the first time that mutyh has been implicated in chemosensitivity, warranting further studies to investigate its mechanisms of action.
Pnkp was shown to be involved in repairing DNA strand breaks caused by reactive oxygen species, ionizing radiation or alkylating agents [57, 58]. It has been related to susceptibility to genotoxic agents but not to chemosensitivity [59]. Nevertheless, interactions of pnkp with another DNA-repair protein such as Xrcc1, which is related to tumoral processes, could account for our results [60].
Heterogeneity
We observed heterogeneity in responses to siRNAs among the 6 cell lines. While no siRNA improved chemotherapeutic effects on CCF and U87, all siRNAs sensitized U373 to both CDDP and TMZ. Such heterogeneous results were already observed for glioma cell lines in an unrelated study [61]. These differences were not related to known differences in cytogenetic or genetic status e.g. p53 mutations. Heterogeneity in cell line response to siRNAs may result from the strong variability in gene expression. However, we did not find any correlation between the mRNA levels (in basal or CDDP conditions) and the siRNAs’ ability to chemosensitize. Such a correlation was not either found for Mgmt. Indeed, while the methylation status of the promoter is a routinely used marker of response to TMZ, Mgmt expression failed to be as useful [62]. It is important to note that among cell lines with opposite behaviors, U373 and U87 are the most frequently used for studying gliomas, as shown by the number of publications (search «glioma»» and «U87» or «U373» or «U138» or «LN229» or «CCF»: 1,534; 513; 63; 127; 94 respectively on Pubmed in March 2015). Thus, no cell line can be sufficiently representative for constituting an in vitro model, and results obtained with only one cell line should be considered very cautiously for clinical applications.
SiRNA
The delivery of drugs to the central nervous system (CNS) is a crucial issue in medical management. We and others have demonstrated the poor capacity of siRNAs packaged with polyethylenimine (PEI) to pass through the blood brain barrier [63–65] and among the numerous other vectors or strategies investigated for targeting brain and intracranial tumors, none has been validated to date. This is the reason why we chose to use ectopically implanted tumor instead of intracranial ones. As a second consequence of using siRNAs in this study, our results cannot be easily transferred to bedside in absence of a validated procedure at the clinical level. Novel chemical agents may be developed that will enable translation of this technique to clinical practice. Indeed, the Ercc1-XPF interaction can be successfully prevented in vitro [66, 67], providing hope for future clinical use. But the development of chemical inhibitors directed to Ercc2, Mutyh and Pnkp proteins is needed.
The aim of our study was to elucidate a part of the mechanisms of astrocytoma drug resistance. We have identified and functionally validated that Ercc1, Ercc2, Mutyh and Pnkp participate to TMZ resistance in gliomas. Our results suggest that survival of glioma patients may be improved when targeting these genes.
MATERIALS AND METHODS
Cell lines and medium
U373, U138, U87, CCF and LN229 cell lines derived from primary human astrocytomas, purchased from American Type Culture Collection (ATCC, Rockville, MD), maintained in DMEM (Cambrex Biosciences, New Jersey, USA) supplemented with 10% fetal calf serum (v/v; AbCys, Paris, France). Cells were maintained in 5% CO2 at 37°C in a humidified incubator.
Since the experiments were performed the ATCC found that the U251 and U373 cell lines had a common origin [68]. We then cannot be sure of the cell-line we actually worked on. Nevertheless, both cell-lines derived from glioblastomas.
The GHD cell line was obtained in our laboratory from a human glioblastoma (genotype was checked with fluorescence in situ hybridization and contained chromosome 7 polysomy and chromosome 10 monosomy).
Inhibition of gene expression by siRNA
SiRNAs (three to five per gene) were designed (Table 4) and delivered in duplex form (Eurogentec, Belgium). SiRNA targeting Green Fluorescent Protein (GFP) were used as a control. In vivo-jetPEI® was from Polyplus-transfection (Illkirch, France)
Table 4: Sequences of siRNAs
Gene |
SiRNA #1 |
SiRNA #2 |
SiRNA #3 |
SiRNA #4 |
SiRNA #5 |
ERCC1 |
5′-AUC-CCG-UAC-UGA-AGU-UCG-U-3′ |
5′-GGA-GCU-GGC-UAA-GAU-GUG-U-3′ |
5′-CAA-GGC-CUA-UGA-GCA-GAA-A-3′ |
5′-ACA-GCU-CAU-CGC-CGC-AUC-A-3′ |
5′-AGA-GAA-GAU-CUG-GCC-UUA-U-3′ |
ERCC2 |
5′-GGA-CGU-CGA-UGG-GAA-AUG-C-3′ |
5′-AGA-CGG-UGC-UCA-GGA-UCA-A-3′ |
5′-UCA-UCA-UCG-AGC-CCU-UUG-A-3′ |
5′-GGA-ACA-AGC-UGC-UCU-UUA-U-3′ |
5′-UGA-CUU-UCU-UAC-CUU-CGA-U-3′ |
GFP |
5′-GAC-GUA-AAC-GGC-CAC-AAG-UUC-3′ |
/ |
/ |
/ |
|
LIG1 |
5′-AGA-CGC-UCA-GCA-GCU-UCU-U-3′ |
5′-GAA-GAU-AGA-CAU-CAU-CAA-A-3′ |
5′-AGA-CAG-CAG-AGG-CCA-GAA-A-3′ |
5′-GCA-GAC-GUU-CUG-CGA-GGU-U-3′ |
5′-GCA-GAU-CCA-GCC-AUU-CCA-A-3′ |
MAD2L2 |
5′-GAA-GAA-UGA-UGU-GGA-GAA-A-3′ |
5′-GAC-UCG-CUG-UUG-UCU-CAU-G-3′ |
5′-CUC-GCA-ACA-UGG-AGA-AGA-U-3′ |
5′-GAA-GAU-CCA-GGU-CAU-CAA-G-3′ |
5′-UGA-GCA-GGA-UGU-CCA-CAU-G-3′ |
MGC13170 |
5′-CAA-GGA-CUU-GGC-UGC-UGA-G-3′ |
5′-GGA-GAA-GGU-GGA-UAA-GUG-G-3′ |
5′-GAA-GGU-GGA-UAA-GUG-GGC-U-3′ |
/ |
/ |
MUTYH |
5′-GAA-GCA-UGC-UAA-GAA-CAA-C-3′ |
5′-UGG-GAU-GAU-UGC-UGA-GUG-U-3′ |
5′-GCA-CCC-UUG-UUU-CCC-AGC-A-3′ |
5′-GGU-UGU-CCA-CAC-CUU-CUC-U-3′ |
5′-GCU-GAC-AUA-UCA-AGU-AUA-U-3′ |
PNKP |
5′-CAC-ACU-GUA-UUU-GGU-CAA-U-3′ |
5′-AGA-GAC-CCG-CAC-ACC-AGA-A-3′ |
5′-GAA-UCU-UGU-ACC-CAG-AGA-U-3′ |
5′-AGU-CCA-CCU-UUC-UCA-AGA-A-3′ |
5′-CAA-CCG-GUU-UCG-AGA-GAU-G-3′ |
POLD1 |
5′-GGA-GAU-GGA-GGC-AGA-ACA-C-3′ |
5′-GUU-GGA-GAU-UGA-CCA-UUA-U-3′ |
5′-UCA-CCG-GUU-ACA-ACA-UCC-A-3′ |
5′-CUU-AGA-CUC-CAC-CAG-CUG-C-3′ |
5′-AUU-CAG-AUG-GGA-UAC-CUC-C-3′ |
RAD54L |
5′-CCA-GCA-UUG-UGA-AUA-GAU-G-3′ |
5′-UCA-CCU-CGC-UAA-AGA-AGC-U-3′ |
5′-GGA-GCU-GUU-UAU-CCU-GGA-U-3′ |
5′-UGA-UCU-GCU-UGA-GUA-UUU-C-3′ |
5′-GCA-GUG-AGA-CCC-AGA-UCC-A-3′ |
RUVBL2 |
5′-AUC-UUC-UCC-CUG-GAG-AUG-A-3′ |
5′-ACU-GAC-CCU-CAA-GAC-CAC-A-3′ |
5′-ACG-CAA-GGG-UAC-AGA-AGU-G-3′ |
/ |
/ |
For in vitro experiments, cells were transfected with siRNA duplexes at a concentration of 150 nM in the culture medium by using Oligofectamine (Invitrogen, Cergy-Pontoise, France) 24 h after cell seeding according to the manufacturer’s instructions. Each condition (siRNA) was tested in 3 independent experiments, with six replicates each time.
For in vivo delivery, siRNA were diluted in 5% glucose (final) using 4 μl of glucose stock solution per μg of siRNA. 0.1 μl of in vivo-jetPEI® were used per μg of siRNA, and were diluted in the same volume of glucose stock solution. The transfection reagent solution was added to the siRNA solution and incubated for 15 min at room temperature. The mixture was diluted in 200 μl (final) of water.
Evaluation of siRNA effect on drug sensitivity
We developed an in vitro assay designed to screen siRNAs. Cells were seeded in 96-well plates. SiRNAs (150 nM) were transfected the second day. The next day, cells were treated with drugs: CDDP (5.10−6 M) (Merck, NJ, USA) or TMZ (10 mg/ml) (Schering-Plough, Levallois-Perret, France) and post-incubated with a drug-free medium for 96 hours. Cell survival was then evaluated by measuring mitochondrial succinate dehydrogenase activity with 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma; 0.5 mg/ml) added to the culture medium. Culture medium was discarded after 4 h and formazan crystals were dissolved in DMSO/ethanol (50/50). Optical density (OD) was read at 540 nm.
Chemoresistance was related to an index (CI) corresponding to the proportion of a cell population that survived chemotherapy. It was computed as follows: cell number (OD) with chemotherapy/cell number (OD) in control condition. The benefit of transfection was represented by the siRNA-induced drug sensitization index (DS), which corresponds to the cell population (%) that survived a simple chemotherapy treatment but died in response to the same treatment following siRNA transfection. It was computed as follows: (CI siRNA GFP – CI siRNA X)/CI siRNA GFP × 100. Standard errors of this index were computed using a bootstrap, technique implemented in the boot library [69] in R [70]. Significant differences between series were tested by ANOVA using Statview (SAS institute). Differences were considered significant when p < 0.05 (*) and highly significant when p < 0.01 (**).
Real-time quantitative reverse transcription-PCR
The NucleoSpin kit (Macherey-Nagel, Germany) was used to extract RNA and cDNA was purified after reverse transcription (Mini Elute, PCR Purification Kit, Qiagen, France).
Real-time quantitative PCR was performed on a Light Cycler (Roche Diagnostic, France) using SYBR Green.
Measures were performed twice from each cell sample and results were expressed as the mean of at least three independent samples (originating from independent in vitro experiments). Specific primers (Eurogentec, Belgium) were as follows: Cyclophiline A: forward primer, 5′-TTC ATC TGC ACT GCC AAG AC-3′; reverse primer, 5′-TCG AGT TGT CCA CAG TCA GC-3′; Ercc1: forward primer 5′-GGC GAC GTA ATT CCC GAC TA-3′; reverse primer, 5′-AGT TCT TCC CCA GGC TCT GC-3′; Ercc2: forward primer, 5′-CGG AAC TAT GGG AAC CTC CT-3′; reverse primer, 5′-TAC TTC TCC AGG GCG ACA CT-3′; Mutyh: forward primer, 5′-GTC CTG ACG TGG AGG AGT GT-3′; reverse primer, 5′-CCT CTG CAC CAG CAG AAT TT-3′; Pnkp: forward primer, 5′-TCG AGA GAT GAC GGA CTC CT-3′; reverse primer, 5′-TTT ATT GTG GAG GGG AGC TG-3′; Mgmt: forward primer, 5′-AGCTGATGCCGTGGAGGT-3′; reverse primer, 5′-ACGACTCTTGCTGGAAAACG-3′.
Ethics and animal care
All procedures related to animal care are conform to the guidelines of the French government and were approved by our institutional ethics committee (authorization n° 201503261330924). All animals were Nude mice (Harlan, France) of 5 weeks-old upon arrival.
Human xenografts assays
The U373 glioma cells (ATCC, MD, USA) were subcutaneously implanted one week after the mice arrival. A 100 μL solution containing 0.75 × 106 U373 cells in DMEM were injected subcutaneously (s.c.) into the upper leg of nude mice (Charles River). When tumors are visible, mice were randomized into groups of 6 mice each. Ercc1, Mgmt or GFP siRNA were injected daily into the peritoneal cavity of each mouse on Days 20–29 (4 μg siRNA in 5% final Glucose 200 μL of H20). TMZ treatment began on the 5th siRNA administration day. It was orally administrated daily for 5 days at a dose of 4.2 mg/kg in 200 μL of orange juice. Tumors were measured every other day with a dial caliper. Results are presented as the mean ± SE with significance calculated by 2-tailed Student’s t test. Significance was assigned for a p-value < 0.05.
ACKNOWLEDGMENTS
We thank Mrs Virginie Pautre for her help in qRT-PCR, Mrs Danièle Adamski, Mrs Loubna Larag and Mr Matthieu Ségura for their assistance in cell culture, Mr Brivael Trelhu for his work on chemotherapy test development, and Mrs Mariko Matsui, Mr Manuel François and Mr Sébastien Maréchal for their work on functional in vitro experiments.
We thank Prof Carlo Chiorri of University of Genoa for his advice and help in the statistical analyses.
This work is part of the national program “Carte d’Identité des Tumeurs® (CIT), funded and developed by the Ligue Nationale Contre le Cancer. SB was supported by La Ligue Contre le Cancer – comité de l’Isère. SM was supported by La Ligue Nationale Contre le Cancer.
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
REFERENCES
1. Ostrom QT, Gittleman H, Farah P, Ondracek A, Chen Y, Wolinsky Y, Stroup NE, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro Oncol. 2013; 15:ii1–56.
2. Ohgaki H. Epidemiology of brain tumors. Methods Mol Biol. 2009; 472:323–342.
3. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007; 114:97–109.
4. Grossman SA, Ye X, Piantadosi S, Desideri S, Nabors LB, Rosenfeld M, Fisher J. Survival of patients with newly diagnosed glioblastoma treated with radiation and temozolomide in research studies in the United States. Clin Cancer Res. 2010; 16:2443–2449.
5. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005; 352:987–996.
6. Stupp R, Hegi ME, Gilbert MR, Chakravarti A. Chemoradiotherapy in malignant glioma: standard of care and future directions. J Clin Oncol. 2007; 25:4127–4136.
7. Stupp R, van den Bent MJ, Hegi ME. Optimal role of temozolomide in the treatment of malignant gliomas. Curr Neurol Neurosci Rep. 2005; 5:198–206.
8. Yang LJ, Zhou CF, Lin ZX. Temozolomide and radiotherapy for newly diagnosed glioblastoma multiforme: a systematic review. Cancer Invest. 2014; 32:31–36.
9. Smith JS, Tachibana I, Passe SM, Huntley BK, Borell TJ, Iturria N, O’Fallon JR, Schaefer PL, Scheithauer BW, James CD, Buckner JC, Jenkins RB. PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J Natl Cancer Inst. 2001; 93:1246–1256.
10. Jansen M, Yip S, Louis DN. Molecular pathology in adult gliomas: diagnostic, prognostic, and predictive markers. Lancet Neurol. 2010; 9:717–726.
11. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005; 352:997–1003.
12. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009; 360:765–773.
13. Cairncross JG, Ueki K, Zlatescu MC, Lisle DK, Finkelstein DM, Hammond RR, Silver JS, Stark PC, Macdonald DR, Ino Y, Ramsay DA, Louis DN. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998; 90:1473–1479.
14. Perry JR, Louis DN, Cairncross JG. Current treatment of oligodendrogliomas. Arch Neurol. 1999; 56:434–436.
15. Wood RD, Mitchell M, Lindahl T. Human DNA repair genes. Mutat Res. 2005; 577:275–283.
16. Hegi ME, Liu L, Herman JG, Stupp R, Wick W, Weller M, Mehta MP, Gilbert MR. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J Clin Oncol. 2008; 26:4189–4199.
17. Quinn JA, Jiang SX, Reardon DA, Desjardins A, Vredenburgh JJ, Gururangan S, Sampson JH, McLendon RE, Herndon JE 2nd, Friedman HS. Phase 1 trial of temozolomide plus irinotecan plus O6-benzylguanine in adults with recurrent malignant glioma. Cancer. 2009; 115:2964–2970.
18. Quinn JA, Jiang SX, Reardon DA, Desjardins A, Vredenburgh JJ, Rich JN, Gururangan S, Friedman AH, Bigner DD, Sampson JH, McLendon RE, Herndon JE 2nd, Walker A, Friedman HS. Phase II trial of temozolomide plus o6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma. J Clin Oncol. 2009; 27:1262–1267.
19. Adair JE, Johnston SK, Mrugala MM, Beard BC, Guyman LA, Baldock AL, Bridge CA, Hawkins-Daarud A, Gori JL, Born DE, Gonzalez-Cuyar LF, Silbergeld DL, Rockne RC, Storer BE, Rockhill JK, Swanson KR, et al. Gene therapy enhances chemotherapy tolerance and efficacy in glioblastoma patients. J Clin Invest. 2014; 124:4082–4092.
20. Smith JS, Alderete B, Minn Y, Borell TJ, Perry A, Mohapatra G, Hosek SM, Kimmel D, O’Fallon J, Yates A, Feuerstein BG, Burger PC, Scheithauer BW, Jenkins RB. Localization of common deletion regions on 1p and 19q in human gliomas and their association with histological subtype. Oncogene. 1999; 18:4144–4152.
21. Harris AL. DNA repair: relationship to drug and radiation resistance, metastasis and growth factors. Int J Radiat Biol Relat Stud Phys Chem Med. 1985; 48:675–690.
22. Christmann M, Tomicic MT, Roos WP, Kaina B. Mechanisms of human DNA repair: an update. Toxicology. 2003; 193:3–34.
23. Bosken CH, Wei Q, Amos CI, Spitz MR. An analysis of DNA repair as a determinant of survival in patients with non-small-cell lung cancer. J Natl Cancer Inst. 2002; 94:1091–1099.
24. Bellmunt J, Paz-Ares L, Cuello M, Cecere F, Albiol S, Guillem V, Gallardo E, Carles J, Mendez P, de la Cruz J, Taron M, Rosell R, Baselga J. Gene expression of ERCC1 as a novel prognostic marker in advanced bladder cancer patients receiving cisplatin-based chemotherapy. Ann Oncol. 2007; 18:522–528.
25. McNeill DR, Friedman DM 3rd. A dominant-negative form of the major human abasic endonuclease enhances cellular sensitivity to laboratory and clinical DNA-damaging agents. Mol Cancer Res. 2007; 5:61–70.
26. Garcia-Campelo R, Alonso-Curbera G, Anton Aparicio LM, Rosell R. Pharmacogenomics in lung cancer: an analysis of DNA repair gene expression in patients treated with platinum-based chemotherapy. Expert Opin Pharmacother. 2005; 6:2015–2026.
27. Gerson SL. MGMT: its role in cancer aetiology and cancer therapeutics. Nat Rev Cancer. 2004; 4:296–307.
28. Friedman HS, McLendon RE, Kerby T, Dugan M, Bigner SH, Henry AJ, Ashley DM, Krischer J, Lovell S, Rasheed K, Marchev F, Seman AJ, Cokgor I, Rich J, Stewart E, Colvin OM, et al. DNA mismatch repair and O6-alkylguanine-DNA alkyltransferase analysis and response to Temodal in newly diagnosed malignant glioma. J Clin Oncol. 1998; 16:3851–3857.
29. Quinn JA, Pluda J, Dolan ME, Delaney S, Kaplan R, Rich JN, Friedman AH, Reardon DA, Sampson JH, Colvin OM, Haglund MM, Pegg AE, Moschel RC, McLendon RE, Provenzale JM, Gururangan S, et al. Phase II trial of carmustine plus O(6)-benzylguanine for patients with nitrosourea-resistant recurrent or progressive malignant glioma. J Clin Oncol. 2002; 20:2277–2283.
30. Quinn JA, Desjardins A, Weingart J, Brem H, Dolan ME, Delaney SM, Vredenburgh J, Rich J, Friedman AH, Reardon DA, Sampson JH, Pegg AE, Moschel RC, Birch R, McLendon RE, Provenzale JM, et al. Phase I trial of temozolomide plus O6-benzylguanine for patients with recurrent or progressive malignant glioma. J Clin Oncol. 2005; 23:7178–7187.
31. Weingart J, Grossman SA, Carson KA, Fisher JD, Delaney SM, Rosenblum ML, Olivi A, Judy K, Tatter SB, Dolan ME. Phase I trial of polifeprosan 20 with carmustine implant plus continuous infusion of intravenous O6-benzylguanine in adults with recurrent malignant glioma: new approaches to brain tumor therapy CNS consortium trial. J Clin Oncol. 2007; 25:399–404.
32. Jenkins RB, Blair H, Ballman KV, Giannini C, Arusell RM, Law M, Flynn H, Passe S, Felten S, Brown PD, Shaw EG, Buckner JC. A t(1,19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. 2006; 66:9852–9861.
33. Parker RJ, Poirier MC, Bostick-Bruton F, Vionnet J, Bohr VA, Reed E. Peripheral blood leukocytes as a surrogate marker for cisplatin drug resistance: studies of adduct levels and the repair gene ERCC1. Basic Life Sci. 1990; 53:251–261.
34. Mu D, Park CH, Matsunaga T, Hsu DS, Reardon JT, Sancar A. Reconstitution of human DNA repair excision nuclease in a highly defined system. J Biol Chem. 1995; 270:2415–2418.
35. Aboussekhra A, Biggerstaff M, Shivji MK, Vilpo JA, Moncollin V, Podust VN, Protic M, Hubscher U, Egly JM, Wood RD. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell. 1995; 80:859–868.
36. Chaney SG, Sancar A. DNA repair: enzymatic mechanisms and relevance to drug response. J Natl Cancer Inst. 1996; 88:1346–1360.
37. Li Q, Yu JJ, Mu C, Yunmbam MK, Slavsky D, Cross CL, Bostick-Bruton F, Reed E. Association between the level of ERCC-1 expression and the repair of cisplatin-induced DNA damage in human ovarian cancer cells. Anticancer Res. 2000; 20:645–652.
38. Selvakumaran M, Pisarcik DA, Bao R, Yeung AT, Hamilton TC. Enhanced cisplatin cytotoxicity by disturbing the nucleotide excision repair pathway in ovarian cancer cell lines. Cancer Res. 2003; 63:1311–1316.
39. Chen H, Shao C, Shi H, Mu Y, Sai K, Chen Z. Single nucleotide polymorphisms and expression of ERCC1 and ERCC2 vis-a-vis chemotherapy drug cytotoxicity in human glioma. J Neurooncol. 2006;.
40. Chang IY, Kim MH, Kim HB, Lee do Y, Kim SH, Kim HY, You HJ. Small interfering RNA-induced suppression of ERCC1 enhances sensitivity of human cancer cells to cisplatin. Biochem Biophys Res Commun. 2005; 327:225–233.
41. Cummings M, Higginbottom K, McGurk CJ, Wong OG, Koberle B, Oliver RT, Masters JR. XPA versus ERCC1 as chemosensitising agents to cisplatin and mitomycin C in prostate cancer cells: role of ERCC1 in homologous recombination repair. Biochem Pharmacol. 2006; 72:166–175.
42. Chen P, Wiencke J, Aldape K, Kesler-Diaz A, Miike R, Kelsey K, Lee M, Liu J, Wrensch M. Association of an ERCC1 polymorphism with adult-onset glioma. Cancer Epidemiol Biomarkers Prev. 2000; 9:843–847.
43. Metzger R, Leichman CG, Danenberg KD, Danenberg PV, Lenz HJ, Hayashi K, Groshen S, Salonga D, Cohen H, Laine L, Crookes P, Silberman H, Baranda J, Konda B, Leichman L. ERCC1 mRNA levels complement thymidylate synthase mRNA levels in predicting response and survival for gastric cancer patients receiving combination cisplatin and fluorouracil chemotherapy. J Clin Oncol. 1998; 16:309–316.
44. Dabholkar M, Bradshaw L, Parker RJ, Gill I, Bostick-Bruton F, Muggia FM, Reed E. Cisplatin-DNA damage and repair in peripheral blood leukocytes in vivo and in vitro. Environ Health Perspect. 1992; 98:53–59.
45. Lord RV, Brabender J, Gandara D, Alberola V, Camps C, Domine M, Cardenal F, Sanchez JM, Gumerlock PH, Taron M, Sanchez JJ, Danenberg KD, Danenberg PV, Rosell R. Low ERCC1 expression correlates with prolonged survival after cisplatin plus gemcitabine chemotherapy in non-small cell lung cancer. Clin Cancer Res. 2002; 8:2286–2291.
46. Olaussen KA, Dunant A, Fouret P, Brambilla E, Andre F, Haddad V, Taranchon E, Filipits M, Pirker R, Popper HH, Stahel R, Sabatier L, Pignon JP, Tursz T, Le Chevalier T, Soria JC. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N Engl J Med. 2006; 355:983–991.
47. Altaha R, Liang X, Yu JJ, Reed E. Excision repair cross complementing-group 1: gene expression and platinum resistance. Int J Mol Med. 2004; 14:959–970.
48. Goode EL, Ulrich CM, Potter JD. Polymorphisms in DNA repair genes and associations with cancer risk. Cancer Epidemiol Biomarkers Prev. 2002; 11:1513–1530.
49. Chen ZP, Malapetsa A, Monks A, Myers TG, Mohr G, Sausville EA, Scudiero DA, Panasci LC. Nucleotide excision repair protein levels vis-a-vis anticancer drug resistance in 60 human tumor cell lines. Ai Zheng. 2002; 21:233–239.
50. Xu Z, Chen ZP, Malapetsa A, Alaoui-Jamali M, Bergeron J, Monks A, Myers TG, Mohr G, Sausville EA, Scudiero DA, Aloyz R, Panasci LC. DNA repair protein levels vis-a-vis anticancer drug resistance in the human tumor cell lines of the National Cancer Institute drug screening program. Anticancer Drugs. 2002; 13:511–519.
51. Chen ZP, McQuillan A, Mohr G, Panasci LC. Excision repair cross-complementing rodent repair deficiency gene 2 expression and chloroethylnitrosourea resistance in human glioma cell lines. Neurosurgery. 1998; 42:1112–1119.
52. Vogel U, Dybdahl M, Frentz G, Nexo BA. DNA repair capacity: inconsistency between effect of over-expression of five NER genes and the correlation to mRNA levels in primary lymphocytes. Mutat Res. 2000; 461:197–210.
53. Aloyz R, Xu ZY, Bello V, Bergeron J, Han FY, Yan Y, Malapetsa A, Alaoui-Jamali MA, Duncan AM, Panasci L. Regulation of cisplatin resistance and homologous recombinational repair by the TFIIH subunit XPD. Cancer Res. 2002; 62:5457–5462.
54. Benhamou S, Sarasin A. ERCC2/XPD gene polymorphisms and lung cancer: a HuGE review. Am J Epidemiol. 2005; 161:1–14.
55. Chen Z, Zhang J, Mohr G. Enhancing alkylating agent resistance through ERCC2 gene transfection in human glioma cell line. Chin Med J (Engl). 2003; 116:1171–1174.
56. Chow E, Thirlwell C, Macrae F, Lipton L. Colorectal cancer and inherited mutations in base-excision repair. Lancet Oncol. 2004; 5:600–606.
57. Whitehouse CJ, Taylor RM, Thistlethwaite A, Zhang H, Karimi-Busheri F, Lasko DD, Weinfeld M, Caldecott KW. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell. 2001; 104:107–117.
58. Chappell C, Hanakahi LA, Karimi-Busheri F, Weinfeld M, West SC. Involvement of human polynucleotide kinase in double-strand break repair by non-homologous end joining. Embo J. 2002; 21:2827–2832.
59. Rasouli-Nia A, Karimi-Busheri F, Weinfeld M. Stable down-regulation of human polynucleotide kinase enhances spontaneous mutation frequency and sensitizes cells to genotoxic agents. Proc Natl Acad Sci U S A. 2004; 101:6905–6910.
60. Plo I, Liao ZY, Barcelo JM, Kohlhagen G, Caldecott KW, Weinfeld M, Pommier Y. Association of XRCC1 and tyrosyl DNA phosphodiesterase (Tdp1) for the repair of topoisomerase I-mediated DNA lesions. DNA Repair (Amst). 2003; 2:1087–1100.
61. Saino M, Maruyama T, Sekiya T, Kayama T, Murakami Y. Inhibition of angiogenesis in human glioma cell lines by antisense RNA from the soluble guanylate cyclase genes, GUCY1A3 and GUCY1B3. Oncol Rep. 2004; 12:47–52.
62. Melguizo C, Prados J, Gonzalez B, Ortiz R, Concha A, Alvarez PJ, Madeddu R, Perazzoli G, Oliver JA, Lopez R, Rodriguez-Serrano F, Aranega A. MGMT promoter methylation status and MGMT and CD133 immunohistochemical expression as prognostic markers in glioblastoma patients treated with temozolomide plus radiotherapy. J Transl Med. 2012; 10:250.
63. Garin D AM, Gauchez A-S, Bohic S, Boccard S, Cloetens P, Fagret D, Berger F, Briat A, Pelletier L, Ghezzi C. In vivo siRNA distribution and pharmacokinetics assessed by nuclear imaging are modulated according to radiolabelling site. Nucmedbio. 2015;. 2015-04-007.
64. Braasch DA, Paroo Z, Constantinescu A, Ren G, Oz OK, Mason RP, Corey DR. Biodistribution of phosphodiester and phosphorothioate siRNA. Bioorg Med Chem Lett. 2004; 14:1139–1143.
65. van de Water FM, Boerman OC, Wouterse AC, Peters JG, Russel FG, Masereeuw R. Intravenously administered short interfering RNA accumulates in the kidney and selectively suppresses gene function in renal proximal tubules. Drug Metab Dispos. 2006; 34:1393–1397.
66. Jordheim LP, Barakat KH, Heinrich-Balard L, Matera EL, Cros-Perrial E, Bouledrak K, El Sabeh R, Perez-Pineiro R, Wishart DS, Cohen R, Tuszynski J, Dumontet C. Small molecule inhibitors of ERCC1-XPF protein-protein interaction synergize alkylating agents in cancer cells. Mol Pharmacol. 2013; 84:12–24.
67. Barakat KH, Jordheim LP, Perez-Pineiro R, Wishart D, Dumontet C, Tuszynski JA. Virtual Screening and Biological Evaluation of Inhibitors Targeting the XPA-ERCC1 Interaction. PLoS One. 2012; 7.
68. Healey JH. Editorial: The imperative to authenticate cell lines. Clin Orthop Relat Res. 2010; 468:3413–3414.
69. Canty AJ, Ripley B. boot: Bootstrap R (S-Plus) Functions. R package version 1. 2014;:3–11.
70. RCoreTeam: R: A language and environment for statistical computing. R Foundation for Statistical ComputingVienna, Austria: 2014.