
INTRODUCTION
Neurotensin (NTS), a 13-amino acid peptide, functions as a primary neurotransmitter as well as a neuromodulator in the central nervous system (CNS) and as a hormone in the periphery [1–3]. NTS contributes to numerous physiologic functions in the gastrointestinal (GI) tract including GI secretion, gut motility, and growth of various normal tissues [1, 2]. Moreover, NTS stimulates the growth of several cancer types including neuroendocrine tumors (NETs) that, compared to other cancers, are increasing in incidence [1, 3, 4].
The actions of NTS are mediated through three receptors (i.e., NTSR1, NTSR2 and NTSR3/sortilin), named according to the order in which they were cloned [2, 3]. In particular, high-affinity NTSR1, which is found in various regions of the CNS, in the small and large intestine, and in a variety of solid tumors, is considered a predominant mediator of the effects of NTS on cell proliferation, migration, and invasion [3, 5]. In contrast, the low-affinity NTSR2, which shares 60% homology with NTSR1, demonstrates a more localized distribution; the expression of NTSR2 has been recently reported in prostate cancers and B cell lymphomas [6, 7]. Different from NTSR1 and NTSR2, which are G protein-coupled receptors, NTSR3/sortilin is a single transmembrane receptor, which binds various neurotrophic factors and neuropeptides and is not specific for NTS [3, 8].
Epigenetic alterations involving DNA methylation or histone modifications can vary the expression patterns of genes that are important for cancer development and progression [9–11]. For example, it is well known that hypermethylation of CpG islands in the promoter region of tumor suppressor genes results in gene silencing, which can lead to the facilitation of tumor progression in certain tissues [9, 12]. In addition, DNA hypomethylation, which is also observed in many cancers, induces transcriptional activation of oncogenes and contributes to cancer progression [9, 13, 14].
Diverse expression levels of the NTSRs, especially increased expression of NTSR1, have been reported in various types of cancers (e.g., colon, pancreas, breast, lung and prostate) [15–21]; however, the molecular mechanisms for this altered expression pattern are not entirely known. Although NTS can stimulate the growth of NET cells [4], the expression profiles and cellular functions of the NTSRs have not been well-delineated in NETs. In our present study, we analyzed the expression of NTSR1 protein in normal and NET tissues for GI, lung and thymus, and endogenous expression of NTSRs and transcriptional repression of NTSR1 and NTSR2 genes in NET cell lines. We demonstrate the epigenetic alteration of NTSR1 and NTSR2 by methylation analyses of their promoters in NET cells and in clinical tissues. Furthermore, we show that NTSR1 knockdown suppresses cell proliferation, anchorage-independent growth, attachment, migration and invasion of NET cells.
RESULTS
Expression of NTSR1 in normal and NET tissues by immunohistochemistry
NTS and NTSR1 complexes have been frequently observed in progression of several types of tumors. Although the expression of NTS which is mainly released by endocrine cells in the small bowel is broadly known in NETs, NTSR1 expression has not been well-studied in NETs. To evaluate the expression of NTSR1, immunohistochemical analysis was performed in clinical NET patient samples (i.e., 12 GI, 2 thymus and 6 lung NET tissues) used in our previous study [22]. Compared to normal tissues (5 GI, 2 thymus and 5 lung tissues) in which NTSR1 was not or barely detected, increased expression of NTSR1 was observed in all tested GI (12 of 12 NETs, Fig. 1A), and thymus (2 out of 2 NETs, Fig. 1B), and a majority of lung (5 out of 6 NETs, Fig. 1C) NET samples. These data suggest that NTSR1 is highly expressed in NETs but not in normal tissues.
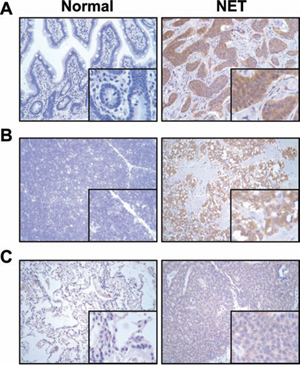
Figure 1: Expression of NTSR1 in normal and clinical NET tissues. Immunohistochemical analysis of NTSR1 was performed in human normal and NET tissues. Representative images for NTSR1 protein expression in GI A. thymus B. and lung C. were shown at 200 × magnification. While the staining of NTSR1 was absent or barely detected in normal tissues (left), strong NTSR1 labeling was observed in clinical NET tissues (right).
Endogenous expression of NTS and NTSRs, and induction of NTSR1 and NTSR2 by 5-aza-CdR treatment in NET cells
To elucidate expression profiles of NTS signaling components in NET cells, we first analyzed the expression of NTS and NTSRs in four human NET cell lines (BON, QGP-1, NCI-H727 and UMC-11) by RT-PCR. Expression of NTS and NTSR3 mRNA was noted in all four cell lines (Fig. 2A); in contrast, NTSR2 expression was not detected (data not shown). Variable expression of NTSR1 was noted with the greatest expression in NCI-H727 cells and moderate expression in BON; very little or no NTSR1 transcripts were demonstrated in QGP-1 and UMC-11 cells (Fig. 2A).
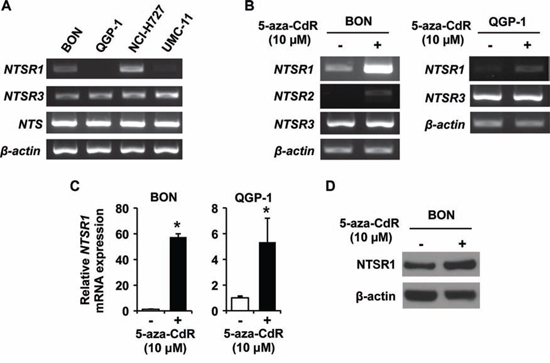
Figure 2: Expression analysis of NTSRs in endogenous and 5-aza-CdR treated NET cell lines. A. RT-PCR analysis of NTS, NTSR1, NTSR2, NTSR3 and β-actin expression in NET cells. B. RT-PCR analysis of NTSRs and β-actin expression in BON and QGP-1 cells treated with 0 (DMSO) or 10 μM 5-aza-CdR. The media containing 5-aza-CdR were replaced every 24 h for 4 d. C. Quantitative RT-PCR (qRT-PCR) analysis confirmed that treatment with 5-aza-CdR increased the expression of NTSR1 gene in BON and QGP-1 cells. The reaction was performed using a TaqMan Gene Expression Master Mix and TaqMan probes for human NTSR1 and GAPDH as internal control (Applied Biosystems). Expression levels were assessed by evaluating threshold cycle (Ct) values. The relative amount of mRNA expression was calculated by the comparative ΔΔCt method (*p < 0.05 vs. DMSO). D. Western blot analysis showing induction of NTSR1 by 5-aza-CdR treatment for 96 h in BON cells. The protein extracts for cell lysates were analyzed with the indicated antibodies. β-actin was used as a loading control.
Previously, we found that repression of Wnt inhibitory genes (SFRP-1, Axin-2, DKK-1, DKK-3 and WIF-1) results from promoter methylation or histone modification in NET cells [22]. To determine whether alterations in expression levels of NTSR1 and NTSR2 were due to epigenetic mechanisms (e.g., promoter methylation), we treated BON and QGP-1 cells with a demethylating agent, 5-aza-2’-deoxycytidine (5-aza-CdR), and examined the expression of NTSRs using RT-PCR (Fig. 2B). Treatment with 5-aza-CdR increased the expression of NTSR1 and NTSR2 in BON and the expression of NTSR1 in QGP-1 cells. To confirm these results, mRNA expression levels were also investigated by qRT-PCR (Fig. 2C). Treatment of the cells with 5-aza-CdR resulted in an approximate 57-fold induction of NTSR1 expression in BON and an approximate 5-fold induction in QGP-1 cells. Western blot analysis confirmed that the level of NTSR1 protein was augmented in BON cells treated with 5-aza-CdR (Fig. 2D). Collectively, our findings of mRNA and protein expression suggest that NTSR1 and NTSR2 are targets of epigenetic modulation through methylation in NET cells.
Methylation status of NTSR1 and NTSR2 promoters in NETs
To test whether the induction of NTSR1 and NTSR2 by 5-aza-CdR was due to promoter methylation, we examined the methylation status of the 5’ regions of these genes using methylation-specific PCR (MSP) and bisulfite sequencing in three NET cell lines. Using two primer pairs, MSP analysis showed partial methylation of the NTSR1 promoter in all tested cell lines (Fig. 3A). The methylation profile of the 5’ region of NTSR1 was further analyzed by the direct sequencing of the MSP products (Supplementary Fig. 1A) and bisulfite sequencing (Fig. 3B). The CpG sites of the NTSR1 promoter were partially methylated in tested NET cells. Moreover, promoter methylation of NTSR2 was also noted by MSP analysis (Fig. 3A). Similar sequencing analyses for the NTSR2 promoter confirmed hypermethylation of the CpG islands consistent with the MSP data (Fig. 3C and Supplementary Fig. 1B).
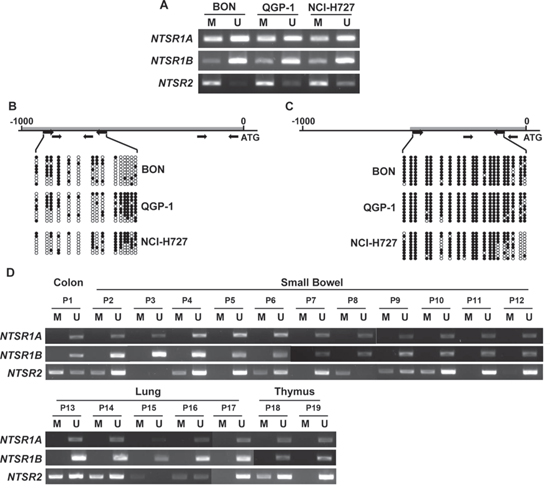
Figure 3: DNA methylation analysis of the NTSR1 and NTSR2 promoters in NETs. A. MSP analysis of NTSR1 and NTSR2 promoters with respective two primer pairs (NTSR1A and NTSR1B) and primers (NTSR2) specific for the methylated (M) and unmethylated (U) DNA in three NET cell lines. The PCR products were visualized by 2% agarose gel. B. Bisulfite genomic sequencing analysis of NTSR1 promoters in BON, QGP-1 and NCI-H727 cells. Each row of circles represents the DNA sequence of an individual clone; closed and open circles indicate methylated and unmethylated CpG sites, respectively. Bold grey lines are candidate CpG islands searched by the software of Applied Biosystems. The thicker and upper, and thinner and lower arrows below the CpG islands represent the primers for bisulfite sequencing and MSP, respectively. C. Bisulfite genomic sequencing analysis of NTSR2 CpG islands in the NET cells. D. MSP analysis of NTSR1 and NTSR2 promoters with the same primers described above in clinical NET samples.
CpG island methylation of NTSR1 and NTSR2 was further investigated in the above clinical specimens used in immunohistochemical analyses. By MSP analysis, methylation of the NTSR1 promoter was not noted in any of the NET specimens, and methylation of NTSR2 was observed in 12 out of 19 NET samples (Fig. 3D). Surprisingly, promoter methylation of NTSR1 was shown in 11 out of 12 normal tissues samples (Supplementary Fig. 2). These data demonstrate that reduction or silencing of NTSR gene expression was strongly associated with DNA methylation of the respective gene promoters in NET cell lines and patient samples. In particular, the absence of NTSR1 promoter methylation is in line with NTSR1 protein expression (Fig. 1) and leads to a strong expression of the protein in tested clinical NET samples. In addition, Dong et al. [25, 26] in our laboratory reported that DNA methylation contributes to NTS expression in human liver and colon cancer cells. Therefore, based on our current study and previous reports, it is clear that DNA methylation can control NTS signaling by regulation of expression levels for the agonist (i.e., NTS) and its receptors (NTSR1 and NTSR2).
NTSR1 knockdown inhibits NET cell growth and migration
Recently, we showed that expression and secretion of NTS are directly regulated by the Wnt/β-catenin pathway in NET cells [4]. We also found that inhibition of NTS signaling suppressed cell proliferation and anchorage-independent growth in these cells [4]. To further delineate the possible proliferative effect of NTSR1, we used small interfering RNA (siRNA) against NTSR1 in BON cells, which express NTSR1 mRNA and have been widely utilized as a novel carcinoid cell model [27]. Compared with cells transfected with non-targeting control, siRNA-mediated knockdown of NTSR1 suppressed cell proliferation (Fig. 4A). In addition, NTSR1 knockdown significantly inhibited the expression of c-Myc and Cyclin D1 which play integral roles in cell proliferation (Fig. 4B) [4, 28, 29]. Next, we performed soft agar assays to determine the effect of NTSR1 knockdown on anchorage-independent growth. The number of BON cell colonies transfected with NTSR1 siRNA was significantly lower than those of control cells (Fig. 4C). These findings suggest that a growth-stimulating function of NTS may be mediated mainly by NTSR1 in NET cells.
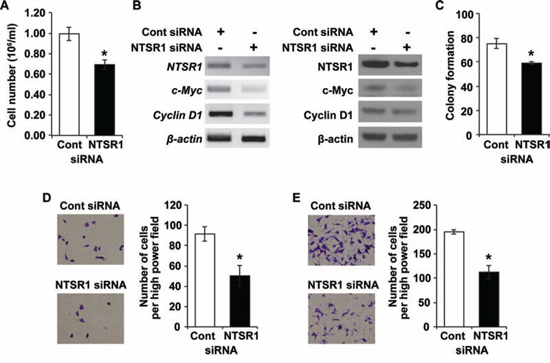
Figure 4: Knockdown of NTSR1 affects NET cell growth and migration. A. Equal numbers of BON cells transfected with siRNA against non-targeting control or NTSR1 were plated in 24 well plates. The cell numbers were counted after 48 h incubation using a cell counter (*p < 0.05 vs. control siRNA). B. RT-PCR (left) and western blot (right) analyses showing expression of NTSR1, c-Myc and Cyclin D1 in BON cells transfected with control or NTSR1 siRNA. β-actin was used as a loading control. C. The number of colonies compared with the control siRNA in soft agar assay. Colony formation of representative control or NTSR1 knockdown BON cells was assessed over a period of 4 wks (*p < 0.05 vs. control siRNA). D. Boyden chamber migration assay with type I collagen-coated Transwells was carried out with control or NTSR1 knockdown BON cells over 24 h. Phase-contrast microscopic images (left) and quantification of migrated cells (right), which were counted in four different fields with an inverted microscope (*p < 0.05 vs. control siRNA), are shown. E. Transwell migration assay performed with respective siRNA-transfected BON cells over 48 h as described above.
In addition to cell growth, activation of NTSR1 induces cell migration, invasion, and metastasis in head and neck squamous cell carcinomas, glioblastomas and breast cancer cells [19, 30, 31]. Based on these studies, we evaluated the migration of BON cells transfected with NTSR1 siRNA using a Boyden chamber migration assay with type I collagen-coated Transwells. Knockdown of NTSR1 decreased the migratory potential of BON cells at 24 (Fig. 4D) and 48 h (Fig. 4E), respectively. Additionally, the effect of pharmacologic blockade of NTSR1 using SR-48692 on NET cell migration was assessed by Transwell migration assays. Treatment with SR-48692, which represses NET cell growth [4], significantly decreased BON cell migration in a dose-dependent manner (Supplementary Fig. 3A). Taken together, these findings show that knockdown of NTSR1 through siRNA or treatment with a selective NTSR1 antagonist inhibits cell growth and migration of NET cells.
Stable silencing of NTSR1 suppresses proliferation, adhesion, migration and invasion in NET cells
To further investigate the contribution of NTSR1 on NET cell proliferation, adhesion, migration and invasion, we utilized NTSR1 small hairpin RNA (shRNA) to establish stable BON cell clones (N-2 and N-3) with low levels of NTSR1 expression following puromycin selection (Fig. 5A). The stable shRNA-mediated knockdown of NTSR1 decreased mRNA levels and promoter activity of interleukin-8 (IL-8), which can be induced by NTS signaling (Fig. 5A and Supplementary Fig. 4A) [32]. Treatment with SR-48692 also inhibited the promoter activity of IL-8 (Supplementary Fig. 4B). Consistent with results obtained from the above experiments using siRNA, the inhibition of cell growth and suppression of anchorage-independent growth was also noted in the cell lines expressing NTSR1 shRNA compared with control cells (Fig. 5B and 5C). In addition, an adhesion assay was performed to assess cell binding ability to the extracellular matrix which is crucial for maintaining cell viability and migration [33]. The stably-silenced NTSR1 clones demonstrated lower numbers of attached cells on type I collagen-coated plates compared with control cells (Fig. 5D and Supplementary Fig. 3B). Furthermore, BON cell clones expressing shRNA targeting NTSR1 showed significantly decreased cell migration compared to control cells (Fig. 5E). Finally, to examine the intravasation effect of NTSR1, the human Alu sequence PCR-based chick chorioallantoic membrane (CAM) assay was performed as described previously [34]. Compared to that of control, amplified bands of lower CAM inoculated with NTSR1-transfected BON cells were significantly decreased indicating that the invasive capacity of BON cells was diminished in NTSR1 knockdown clones (Fig. 5F). Collectively, these findings confirm that stable silencing of NTSR1 markedly inhibits cell proliferation, anchorage-independent growth, cell adhesion, migration and invasion suggesting an oncogenic function for NTSR1 in NET cells.
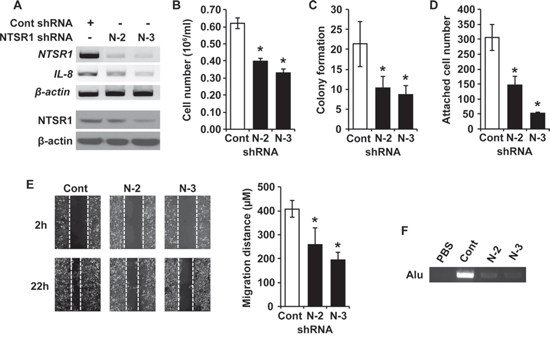
Figure 5: Stable silencing of NTSR1 suppresses cell growth, adhesion, migration and invasion of NET cells. A. RT-PCR (upper) and western blot (bottom) analyses showing NTSR1 expression in stable non-targeting control or two NTSR1 knockdown BON cell clones (N-2 and N-3). B. The stable BON cell clones were incubated for 48 h; cell numbers were counted using a cell counter (*p < 0.05 vs. control shRNA). C. The number of colonies compared with control shRNA in soft agar assay. Colony formation of representative BON stable cell clones was assessed over a period of 4 wks (*p < 0.05 vs. control shRNA). D. The same number of stable BON cells was added onto type I collagen-coated plate for 15 min. The attached cells were fixed, and then stained with crystal violet. The number of attached cells was counted and the mean values were determined (*p < 0.05 vs. control shRNA). E. Phase-contrast microscopic images showing stable BON cell clones 2 h (upper) and 22 h (bottom) after removing the Culture-Insert (Ibidi, Munich, Germany). Wounding migration assay performed with control and the two BON cell clones expressing NTSR1 shRNA over 22 h. Quantification was carried out by measuring the migrated distance (right, *p < 0.05 vs. control shRNA). F. Reduction of invasive activity by stable silencing of NTSR1 in BON cells. To determine the invasive effect of NTSR1, human Alu sequence PCR-based assay was performed. Genomic DNA isolated from lower CAMs was used as a template of PCR amplification. PBS was used as negative control. The PCR product was electrophoresed on a 2% agarose gel.
DISCUSSION
There is emerging evidence that either NTS or NTSR1 can be utilized as a prognostic marker for various cancers due to aberrant expression noted in tumors and not detected in normal tissues, and that silencing of the genes can inhibit the tumorigenic activities in some cancer cells [15–21]. In particular, NTSR1 expression is strongly associated with a worse survival and a higher incidence of distant metastases in lung and breast cancers [17, 20]. However, the mechanisms for the expression of NTSR1 in cancer tissues including NETs, have not been well-defined. Here, we showed that NTSR1 protein is not or barely detected in 12 normal tissues but is strongly expressed in 95% of clinical NET samples (19 of 20 NETs). Similar to the above described cancers, high expression of NTSR1, noted in this study, and NTS are associated with NET progression indicating that NTSR1 may be a useful prognostic marker for NETs.
We also found that differential expression of NTSR1 and silencing of NTSR2 in NET cells is a result of promoter methylation. Previously, it was reported that DNA methylation plays an important role in the regulation of NTS expression in human cancer cells [25, 26]. Similar to the Wnt/β-catenin pathway which regulates the expression of both NTS and NTSR1 [4, 35], promoter methylation demonstrates a complicated regulatory process for NTS signaling at the level of the agonist as well as the simultaneous regulation of its receptors. In addition to hypermethylation, which is commonly found with tumor initiation and progression, hypomethylation of certain genes (e.g., urokinase plasminogen activator) is a common mechanism for the aberrant gene expression patterns in tumors, and plays a central role in human tumor progression [14, 36–38]. Similarly, the lack of NTSR1 promoter methylation is observed in all NET tissues and the promoter methylation was shown in most normal tissues in this study. These data represent that the absence of promoter methylation closely correlates with overexpression of NTSR1 in NET clinical samples. Histone modification could also be related to its transcriptional regulation similar to epigenetic repression of DKK-1, DKK-3 and WIF-1 noted in our previous study showing that silencing of these genes occurs through histone modification in NET cells, although the Wnt inhibitors are silenced by methylation of their promoters in most tumors [22]. Further work is required to elucidate the detailed mechanisms for epigenetic silencing of NTSR1 and NTSR2 in diverse tumor types.
NTS and NTSR1 are neuropeptide-receptor complexes which are frequently deregulated during tumor progression [3, 5]. In particular, NTSR1 activation induces cell proliferation, survival, migration and invasion through multiple oncogenic pathways in various cancers [3, 5]. Recently, we proposed that NTS is a mediator for NET cell growth through inhibition of NTS signaling [4]. In our current study, we confirmed the high expression of NTSR1 protein in clinical NET tissues as described above and analyzed the effect of NTSR1 on NET cell growth, adhesion, migration and invasion to better delineate the significance of NTSR1 in NET progression. In addition to cell proliferation and anchorage-independent growth, which were suppressed by NTS knockdown, NTSR1 silencing resulted in reduction of NET cell migration and invasion indicating that NTSR1 is mainly engaged in NET progression.
In summary, we have identified promoter methylation as an important molecular mechanism for the regulation of NTSR1 and NTSR2 expression. In addition, we also demonstrate high expression of NTSR1 protein in clinical NETs, and the oncogenic function of NTSR1 in NET cells. Our findings identify a potential role for NTSR1 in the growth and progression of NETs and provide a rationale for further exploration of NTSR1 as a therapeutic target for NET treatment.
MATERIALS AND METHODS
Immunohistochemistry
Immunostaining with NTSR1 antibody, obtained from Abcam (ab117592, Cambridge, MA), was performed as described previously [22]. The slides for paraffin-embedded tissue blocks of NETs from GI (n = 12), lung (n = 6) and thymus (n = 2) and normal tissues from GI (n = 5), lung (n = 5) and thymus (n = 2) were provided from Department of Pathology and Markey Biospecimen and Tissue Procurement Shared Resource Facility, University of Kentucky. Assessment of the stained slides was performed blindly by an experienced pathologist (EYL).
Cell culture and treatment, siRNA transfections and lentiviral transductions
Four human NET cell lines, BON (pancreatic carcinoid), QGP-1, (pancreatic somatostatinoma), NCI-H727 (bronchial carcinoid) and UMC-11 (bronchial carcinoid) were used in this study. The cell lines were authenticated with 17 autosomal short tandem repeat loci and the sex identity locus in May 2012 at Genetica DNA Laboratories (Cincinnati, OH). BON cells were maintained in a 1:1 ratio of DMEM and F12K, supplemented with 5% FBS. QGP-1, NCI-H727 and UMC-11 cells were cultured in RPMI1640 medium with 10% FBS. Cells were grown at 37°C in a humidified 5% CO2 incubator. Reagents for cell treatment, a demethylating agent, 5-aza-2’-deoxycytidine and an NTSR1 antagonist, SR-48692 were purchased from Sigma (St Louis, MO) and dissolved in dimethyl sulfoxide (DMSO). Transfection with nontargeting control and SMARTPool NTSR1 siRNA (Dharmacon, Lafayette, CO) was performed using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA) as previously described [4]. For generation of NTSR1-silenced BON cells, the shRNA lentiviruses were produced using shRNA vectors (SHGLY-NM_002531, Sigma). Cells were transduced with each virus and then selected with puromycin (2 μg/mL) as previously reported [4, 39].
RNA isolation, reverse transcription-PCR (RT-PCR) and quantitative reverse transcription-PCR (qRT-PCR) analysis
Total RNA was isolated from the cultured NET cells using RNeasy kits according to the manufacturer’s instructions (Qiagen, Valencia, CA). Each cDNA was synthesized using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) and total RNA for NET cells. RT-PCR analysis was performed using synthesized cDNA, HotStarTaq DNA Polymerase (Qiagen) and the primers described in Supplementary Table S1. The PCR products were analyzed on a 2% agarose gel. β-actin was used as an internal control. qRT-PCR reaction was performed using a TaqMan Gene Expression Master Mix and TaqMan probes for human NTSR1 and GAPDH according to the manufacturer’s protocol (Applied Biosystems). Expression levels were assessed by evaluating threshold cycle (Ct) values. The relative amount of mRNA expression was calculated by the comparative ΔΔCt method.
Western blot analysis
The protein extracts for cell lysates were prepared in a Cell Lysis Buffer (Cell Signaling, Danvers, MA) containing 1 mM PMSF. Total cell lysates containing equivalent amounts of protein were separated on NuPAGE 4–12% Bis-Tris gels (Invitrogen) and transferred to PVDF membranes. The membranes were incubated with specific primary antibodies and subsequently horseradish peroxidase-conjugated secondary antibody. Following incubation with the antibody, proteins were visualized using ECL detection system (Buckinghamshire, UK). The anti-NTSR1 antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The antibodies for c-Myc and Cyclin D1 were obtained from Epitomics (Burlingame, CA). The antibody for β-actin used as a loading control was from Cell Signaling.
Methylation analysis
Methylation of 5’ regions of NTSR1 and NTSR2 was analyzed using MSP (methylation-specific PCR) and bisulfite sequencing analyses. Briefly, PCR was performed using bisulfite-modified genomic DNA by MethylCode Bisulfite Conversion Kit (Invitrogen) and the primers which were designed using Methyl Primer Express Software v1.0 (Applied Biosystems) and shown in Supplementary Table S1. The PCR products for MSP were visualized by 2% agarose gel. For bisulfite-sequencing, PCR products were cloned into the TOPO TA cloning vector (Invitrogen) and the plasmids from individual bacterial colonies were sequenced.
Cell proliferation
Equal numbers of BON cells transfected with siRNA or shRNA were plated in 24-well plates. Cell proliferation was assessed at 48 h after seeding directly by cell counting using a Beckman Coulter Cell Viability Analyzer (Beckman-Coulter, Fullerton, CA).
Soft agar assay
To measure anchorage-independent growth, BON cells were plated in growth medium containing 0.4% agarose in six-well plates onto a bottom layer of solidified 0.8% agarose. After incubation for 4 weeks, colonies were stained with crystal violet solution, washed repeatedly with distilled water and quantified using AlphaEaseFC software (Alpha Innotech Corporation, San Leandro, CA).
Transwell migration assay
A Boyden chamber migration assay with type I collagen-coated Transwells was carried out with control and NTSR1 knockdown BON cells or BON cells treated with different concentration of SR-48692. The chambers were incubated at 37°C for 24 or 48 h, respectively, and the cells were fixed with methanol and stained with 0.5% crystal violet in 20% methanol. Activity of cell migration was quantified by counting cell numbers in four different fields under an inverted microscope.
Luciferase reporter assays
BON cells were plated in 24-well plates and transiently transfected with the IL-8 reporter (0.4 μg) and the Renilla luciferase reporter (0.05 μg) using Lipofectamine 2000 according to the manufacturer’s instructions (Invitrogen). For SR-48692 treatment, 0 (DMSO) or 5 μM SR-48692 were treated into BON cells one day after transfection. The cells were harvested and luciferase activity was measured using a Dual-Luciferase Reporter Assay System (Promega, Madison, WI).
Adhesion assay
A cell adhesion assay was performed to assess cell binding ability to the extracellular matrix. Briefly, 48-well plates were coated with type I collagen and washed with PBS. Equal numbers of detached BON cell clones were plated to each coated well and incubated for 15 min. After 37°C incubation, unattached cells were removed by washing with PBS, and the adherent cells were fixed, and then stained with crystal violet. The plate was washed and dried completely. Quantification was performed through counting the number of attached cells or measuring absorbance for crystal violet-stained cells at 550 nm by adding and solubilizing with 2% SDS.
Wounding migration assay
To compare the migratory activity of stable knockdown of NTSR1, a wounding migration assay was performed with control and the two BON cell clones expressing NTSR1 shRNA. The wounded monolayers using the Culture-Insert (Ibidi, Munich, Germany) were incubated for 22 h. The cells were fixed with ice-cold methanol and stained with crystal violet. Quantification was carried out by measuring the migrated distance.
Human Alu sequence PCR-based assay
To determine the invasive effect of NTSR1, human Alu sequence PCR-based assay was performed as described previously [34]. Stable BON cells in PBS were inoculated on artificially-generated air sacs at 1 × 107 cells per chick chorioallantoic membrane (CAM) of 9 day chick embryos. After 3 d, genomic DNA was isolated from lower CAMs and then used as a template of PCR amplification. The primer sequences are shown in Supplementary Table S1. The PCR product was electrophoresed on a 2% agarose gel.
Statistical analysis
Descriptive statistics including mean and standard deviation (SD) were calculated to summarize mRNA levels, number of cells, colony formation, migrated and attached cell number, luciferase activity and migration distance for each experiment. Bar graphs were generated to represent mean (± SD) fold changes of increase or decrease in experimental groups relative to control. Within respective experiments, comparisons between two groups were performed using two-sample t-tests, whereas comparisons across groups were accomplished using one-way analysis of variance models and test for linear trend of increasing dose levels or pairwise comparisons with control group were subsequently performed using contrast statements. Normality assumptions of the analysis of variance and two-sample t-tests for each outcome were assessed. p-values < 0.05 were considered statistically significant.
ACKNOWLEDGMENTS
We thank Dana L. Napier and Cynthia Long for technical assistance, and the members of the Markey Cancer Center’s Research Communications Office for manuscript preparation. We further acknowledge support from the the Biospecimen and Tissue Procurement and Biostatistics and Bioinformatics Shared Resource Facilities of the University of Kentucky Markey Cancer Center (supported by NCI grant P30CA177558)
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
GRANT SUPPORT
This research was supported by a grant from the NIH (R37 AG010885) and the Markey Cancer Foundation.
REFERENCES
1. Evers BM. Neurotensin and growth of normal and neoplastic tissues. Peptides. 2006; 27:2424–2433.
2. Kalafatakis K, Triantafyllou K. Contribution of neurotensin in the immune and neuroendocrine modulation of normal and abnormal enteric function. Regul Pept. 2011; 170:7–17.
3. Wu Z, Martinez-Fong D, Tredaniel J, Forgez P. Neurotensin and its high affinity receptor 1 as a potential pharmacological target in cancer therapy. Front Endocrinol (Lausanne). 2012; 3:184.
4. Kim JT, Liu C, Zaytseva YY, Weiss HL, Townsend CM, Jr., Evers BM. Neurotensin, a novel target of Wnt/beta-catenin pathway, promotes growth of neuroendocrine tumor cells. Int J Cancer. 2015; 136:1475–1481.
5. Dupouy S, Mourra N, Doan VK, Gompel A, Alifano M, Forgez P. The potential use of the neurotensin high affinity receptor 1 as a biomarker for cancer progression and as a component of personalized medicine in selective cancers. Biochimie. 2011; 93:1369–1378.
6. Swift SL, Burns JE, Maitland NJ. Altered expression of neurotensin receptors is associated with the differentiation state of prostate cancer. Cancer Res. 2010; 70:347–356.
7. Saada S, Marget P, Fauchais AL, Lise MC, Chemin G, Sindou P, Martel C, Delpy L, Vidal E, Jaccard A, Troutaud D, Lalloue F, Jauberteau MO. Differential expression of neurotensin and specific receptors, NTSR1 and NTSR2, in normal and malignant human B lymphocytes. J Immunol. 2012; 189:5293–5303.
8. Nykjaer A, Willnow TE. Sortilin: a receptor to regulate neuronal viability and function. Trends Neurosci. 2012; 35:261–270.
9. Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004; 4:143–153.
10. Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem. 2006; 75:243–269.
11. Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006; 6:107–116.
12. Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol. 2005; 2:S4–11.
13. Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene. 2002; 21:5400–5413.
14. Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009; 1:239–259.
15. Maoret JJ, Pospai D, Rouyer-Fessard C, Couvineau A, Laboisse C, Voisin T, Laburthe M. Neurotensin receptor and its mRNA are expressed in many human colon cancer cell lines but not in normal colonic epithelium: binding studies and RT-PCR experiments. Biochem Biophys Res Commun. 1994; 203:465–471.
16. Wang L, Friess H, Zhu Z, Graber H, Zimmermann A, Korc M, Reubi JC, Buchler MW. Neurotensin receptor-1 mRNA analysis in normal pancreas and pancreatic disease. Clin Cancer Res. 2000; 6:566–571.
17. Souaze F, Dupouy S, Viardot-Foucault V, Bruyneel E, Attoub S, Gespach C, Gompel A, Forgez P. Expression of neurotensin and NT1 receptor in human breast cancer: a potential role in tumor progression. Cancer Res. 2006; 66:6243–6249.
18. Gui X, Guzman G, Dobner PR, Kadkol SS. Increased neurotensin receptor-1 expression during progression of colonic adenocarcinoma. Peptides. 2008; 29:1609–1615.
19. Dupouy S, Viardot-Foucault V, Alifano M, Souaze F, Plu-Bureau G, Chaouat M, Lavaur A, Hugol D, Gespach C, Gompel A, Forgez P. The neurotensin receptor-1 pathway contributes to human ductal breast cancer progression. PLOS ONE. 2009; 4:e4223.
20. Alifano M, Souaze F, Dupouy S, Camilleri-Broet S, Younes M, Ahmed-Zaid SM, Takahashi T, Cancellieri A, Damiani S, Boaron M, Broet P, Miller LD, Gespach C, et al. Neurotensin receptor 1 determines the outcome of non-small cell lung cancer. Clin Cancer Res. 2010; 16:4401–4410.
21. Valerie NC, Casarez EV, Dasilva JO, Dunlap-Brown ME, Parsons SJ, Amorino GP, Dziegielewski J. Inhibition of neurotensin receptor 1 selectively sensitizes prostate cancer to ionizing radiation. Cancer Res. 2011; 71:6817–6826.
22. Kim JT, Li J, Jang ER, Gulhati P, Rychahou PG, Napier DL, Wang C, Weiss HL, Lee EY, Anthony L, Townsend CM, Jr., Liu C, Evers BM. Deregulation of Wnt/beta-catenin signaling through genetic or epigenetic alterations in human neuroendocrine tumors. Carcinogenesis. 2013; 34:953–961.
23. Demirci I, Herold S, Kopp A, Flasshove M, Klosterhalfen B, Janssen H. Overdiagnosis of a typical carcinoid tumor as an adenocarcinoma of the lung: a case report and review of the literature. World J Surg Oncol. 2012; 10:19.
24. Gridelli C, Rossi A, Airoma G, Bianco R, Costanzo R, Daniele B, Chiara GD, Grimaldi G, Irtelli L, Maione P, Morabito A, Piantedosi FV, Riccardi F. Treatment of pulmonary neuroendocrine tumours: state of the art and future developments. Cancer Treat Rev. 2013; 39:466–472.
25. Dong Z, Wang X, Zhao Q, Townsend CM, Jr., Evers BM. DNA methylation contributes to expression of the human neurotensin/neuromedin N gene. Am J Physiol. 1998; 274:G535–543.
26. Dong Z, Wang X, Evers BM. Site-specific DNA methylation contributes to neurotensin/neuromedin N expression in colon cancers. Am J Physiol Gastrointest Liver Physiol. 2000; 279:G1139–1147.
27. Evers BM, Townsend CM, Jr., Upp JR, Allen E, Hurlbut SC, Kim SW, Rajaraman S, Singh P, Reubi JC, Thompson JC. Establishment and characterization of a human carcinoid in nude mice and effect of various agents on tumor growth. Gastroenterology. 1991; 101:303–311.
28. Kerkhoff E, Rapp UR. Cell cycle targets of Ras/Raf signalling. Oncogene. 1998; 17:1457–1462.
29. Smalley MJ, Dale TC. Wnt signalling in mammalian development and cancer. Cancer Metastasis Rev. 1999; 18:215–230.
30. Servotte S, Camby I, Debeir O, Deroanne C, Lambert CA, Lapiere CM, Kiss R, Nusgens B, Decaestecker C. The in vitro influences of neurotensin on the motility characteristics of human U373 glioblastoma cells. Neuropathol Appl Neurobiol. 2006; 32:575–584.
31. Shimizu S, Tsukada J, Sugimoto T, Kikkawa N, Sasaki K, Chazono H, Hanazawa T, Okamoto Y, Seki N. Identification of a novel therapeutic target for head and neck squamous cell carcinomas: a role for the neurotensin-neurotensin receptor 1 oncogenic signaling pathway. Int J Cancer. 2008; 123:1816–1823.
32. Wang X, Jackson LN, Johnson SM, Wang Q, Evers BM. Suppression of neurotensin receptor type 1 expression and function by histone deacetylase inhibitors in human colorectal cancers. Mol Cancer Ther. 2010; 9:2389–2398.
33. Ruoslahti E, Pierschbacher MD. New perspectives in cell adhesion: RGD and integrins. Science. 1987; 238:491–497.
34. Kim JT, Kim MS, Bae MK, Song HS, Ahn MY, Kim YJ, Lee SJ, Kim KW. Cloning and characterization of tissue inhibitor of metalloproteinase-3 (TIMP-3) from shark, Scyliorhinus torazame. Biochim Biophys Acta. 2001; 1517:311–315.
35. Souaze F, Viardot-Foucault V, Roullet N, Toy-Miou-Leong M, Gompel A, Bruyneel E, Comperat E, Faux MC, Mareel M, Rostene W, Flejou JF, Gespach C, Forgez P. Neurotensin receptor 1 gene activation by the Tcf/beta-catenin pathway is an early event in human colonic adenomas. Carcinogenesis. 2006; 27:708–716.
36. Pakneshan P, Tetu B, Rabbani SA. Demethylation of urokinase promoter as a prognostic marker in patients with breast carcinoma. Clin Cancer Res. 2004; 10:3035–3041.
37. Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases. Biochim Biophys Acta. 2007; 1775:138–162.
38. Pulukuri SM, Estes N, Patel J, Rao JS. Demethylation-linked activation of urokinase plasminogen activator is involved in progression of prostate cancer. Cancer Res. 2007; 67:930–939.
39. Li J, Liu J, Song J, Wang X, Weiss HL, Townsend CM, Jr., Gao T, Evers BM. mTORC1 inhibition increases neurotensin secretion and gene expression through activation of the MEK/ERK/c-Jun pathway in the human endocrine cell line BON. Am J Physiol Cell Physiol. 2011; 301:C213–226.