
INTRODUCTION
Aberrations in the hepatocyte growth factor (HGF)-mesenchymal-epithelial transition (MET) receptor tyrosine kinase axis are frequent in solid malignancies [1]. One such aberration is the overexpression of the MET protein as determined by immunohistochemistry (IHC) which may be associated with MET amplification. MET amplification is present in about 2.6% among 1,115 patient tumors assayed [2]. For example, the frequency of MET amplification is rare in gastroesophageal/gastric cancer [3] while MET protein overexpression has been reported in higher incidence [4]. The discordance between low MET amplification and high MET protein expression indicates there are other potential mechanisms that can lead to MET overexpression. One such mechanism is MET exon14 deletion (METex14del) where part of the transmembrane portion and region for the Casitas B-lineage lymphoma (Cbl) E3 ligase-mediated degradation is deleted leading to delay degradation of MET and hence its overexpression (Supplementary Figure S1) [5, 6].
METex14del was initially described in 2006 in non-small cell lung cancer (NSCLC) and was caused by mutation in the splice donor site in intron 14 and intronic sequence deletions around MET exon 14 [5]. The presence of METex14del in NSCLC has subsequently been confirmed by RNA sequencing and whole genome sequencing [7, 8]. Additionally, METex14del has been reported in gastric cancer (GC) cell line Hs746T [9, 10] and neuroblastoma [11] indicating this is a potential common mechanism for a variety of tumors to delay the ubiquitination and down-regulation of MET protein leading to its overexpression [5].
We investigated patients with metastatic solid malignancies primarily gastrointestinal (GI) and lung malignancies for the presence of METex14del using multiplexed fusion transcript detection assay and then confirmed with reverse transcription PCR (RT-PCR) correlated the MET protein expression and MET amplification in METex14del+ cases. We further generated patient derived tumor cell lines and screened them for the presence of METex14del and investigated the consequence of MET inhibition in these METex14del+ cells lines.
RESULTS
The patient cohort from the NEXT-1 trial (NCT02141152), which is an actively enrolling clinical trial for genomic profiling in cancer patients, was used (Figure 1). Of 428 patients enrolled and screened, sufficient RNAs for multiplexed fusion transcript detection analysis by nCounter assay were available in 230 patients (Table 1). The detailed probe design for multiplexed fusion transcript assay surveying for ALK, ROS1, RET, NTRK1, and NTRK3 is provided in Supplementary Table S1. Of the multiplexed fusion assay, a nanostring probe to detect any 141bp METex14del transcript (p.982_1028del47, c.2942 (Supplementary Table S1) was included. Of the 230 tumor specimens screened, 86 specimens were freshly frozen tissues and 144 specimens were from formalin-fixed paraffin-embedded (FFPE) tissues. In parallel, we screened fifty patient derived tumor cell (PDC) lines generated from the SMC Biomarker study (NCT01831609) for METex14del. The SMC Oncology Biomarker study is an ongoing study which enrolls metastatic solid cancer patients with malignant ascites, malignant pleural effusion, endoscopic biopsies or surgical specimens for PDC model establishments (Figure 1 for Study Flow Chart).
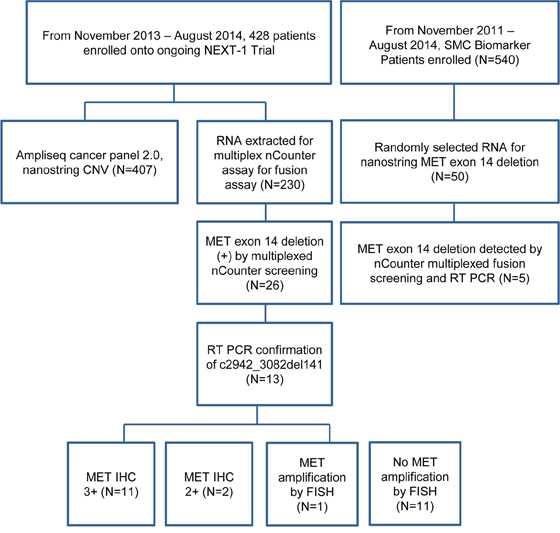
Figure 1: Study flow chart.
Table 1: Patient characteristics
Variable |
N (%) |
MET exon 14 deletion (+) |
Patient tumor specimens (N = 230) |
||
Age-year |
||
Median |
57 |
|
Range |
20–87 |
|
Sex, no. (%) |
||
Male |
134 |
|
Female |
96 |
|
Stage |
230 (100) |
|
Tumor type, no. (%)*** |
||
Gastric cancer |
42 |
3(7.1) |
NSCLC |
51 |
5(9.8) |
Colon cancer |
43 |
4(9.3) |
Rectal cancer |
23 |
0(0.0) |
Hepatocellular carcinoma |
15 |
0(0.0) |
Sarcoma |
9 |
0(0.0) |
Pancreatic cancer |
5 |
0(0.0) |
Cholangiocarcinoma |
6 |
0(0.0) |
Melanoma |
5 |
0(0.0) |
ACUP* |
3 |
1(33.3) |
Esophageal squamous carcinoma |
1 |
0(0.0) |
Renal cell carcinoma |
1 |
0(0.0) |
Others |
15 |
0(0.0) |
Patient Derived Cells (N = 50) |
||
Patient derived cells (N = 50) |
||
Gastric cancer |
22 |
1 |
Colon cancer |
5 |
1 |
NSCLC |
4 |
1 |
Melanoma |
2 |
1 |
Cholangiocarcioma |
3 |
0 |
HCC** |
4 |
0 |
Duodenal carcinoma |
1 |
0 |
Esophageal squamous cell |
1 |
1 |
Sarcoma and other rare cancer |
8 |
0 |
*(Adenocarcinoma of unknown primary had met exon 14 skipping and MET amplification).
**HCC, hepatocellular carcinoma.
***219 included for final analysis from 230.
Of the 230 tumor cohort (86 fresh frozen tissue and 144 formalin-fixed paraffin-embedded tissues), 219 were finally included in the analysis as 11 samples failed to pass QC (quality control). With initial screening of multiplexed nCounter fusion transcript analysis, 26 were detected as potential positive cases for METex14del with high fusion transcript mRNA expression (Supplementary Figure S2) and 13 (5.7%) patients were eventually confirmed to be METex14del+ by quantitative reverse transcriptase-polymerase chain reaction (RT-PCR): 3 gastric carcinoma (GC), 4 colon, 5 non-small cell lung cancer (NSCLC) and one adenocarcinoma of unknown primary (ACUP). Among these 13 METex14del cases, 11 cases were MET IHC 3+ and 2 cases were MET IHC 2+. Only one of the 13 METex14del+ cases had concomitant MET amplifications (Table 2). All METex14del cases were negative for ALK, ROS1, RET, NTRK1, and NTRK3 fusion.
Table 2: Characteristics of MET exon 14 deletion (METex14) patients according to tumor types
Colon |
Gastric |
ACUP |
Lung |
|
N |
4 |
3 |
1 |
5 |
Age |
||||
Median |
63 |
53 |
49 |
|
(range) |
42–87 |
27–67 |
36–60 |
|
Gender |
||||
Male |
2 |
2 |
1 |
1 |
Female |
2 |
1 |
0 |
4 |
Smoking history |
||||
Never-smoker |
NC |
NC |
NC |
4 |
Ever-smoker |
1 |
|||
Histology |
||||
Adenocarcinoma |
4 |
3 |
1 |
4 |
Squamous cell |
0 |
0 |
0 |
1 |
Large cell neuroendocrine |
0 |
0 |
0 |
0 |
Undifferentiated |
0 |
0 |
0 |
0 |
Tumor differentiation |
||||
Well |
1 |
0 |
0 |
0 |
Moderate |
2 |
0 |
0 |
1 |
Poor |
1 |
3 |
1 |
4 |
MET IHC |
||||
0 |
0 |
0 |
0 |
0 |
1+ |
0 |
0 |
0 |
0 |
2+ |
1 |
0 |
0 |
0 |
3+ |
3 |
3 |
1 |
5 |
Concomitant MET amplification |
||||
Yes |
0 |
1 |
0 |
0 |
No |
4 |
2 |
0 |
5 |
Confirmed by RT-PCR |
||||
Yes |
4 |
3 |
1 |
5 |
No |
0 |
0 |
0 |
0 |
NC, not contributable; wt, wild type.
All 13 METex14del cases were further confirmed by qualitative RT-PCR using probes overlapping an exon 13–15 junction, a fusion transcript caused by exon 14 skipping. In all cases, although the absolute Ct (cycles to threshold) values of RT-PCR showed relatively high around 32, there was definite amplification of target sequences. Deep sequencing targeting whole MET gene including intron using DNAs from GI cancers, there were many mutations in the introns (Table 3). Interestingly, all our GI samples harbored c.3082+811A TTTTAACA > GGTTTGAT mutations on intron 14 region of MET.
Table 3: Clinicopathologic characteristics of the gastrointestinal cancer and adenocarcinoma of unknown primary with METex14del
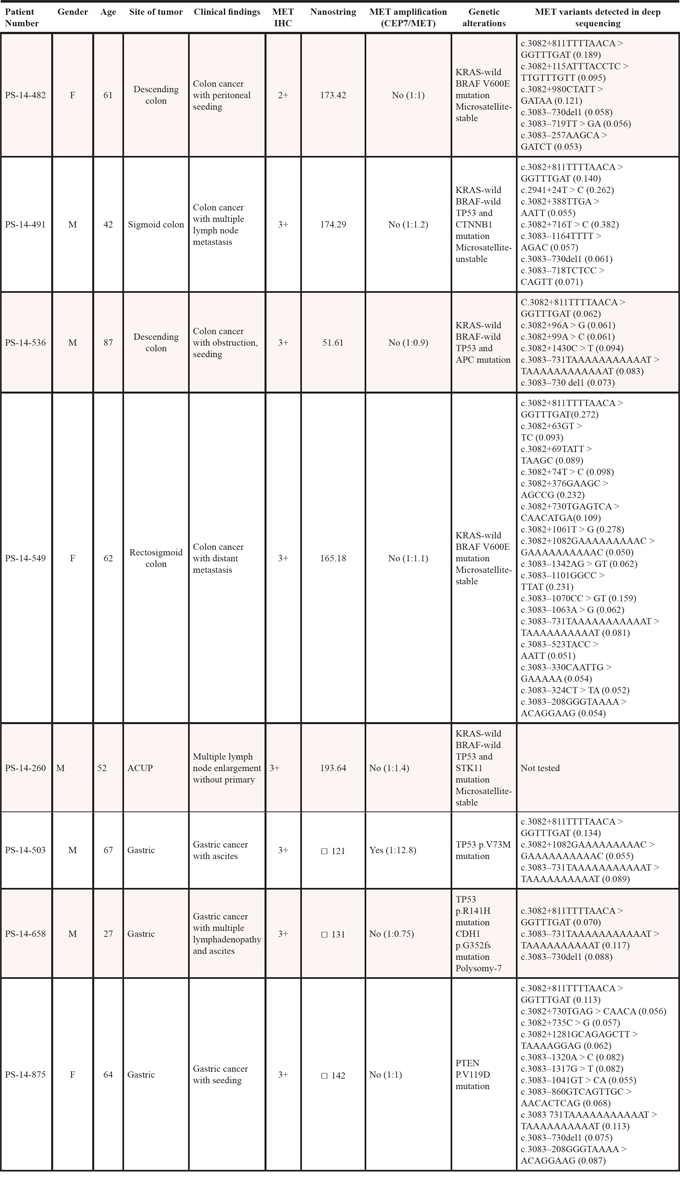
A total of 3 out of 42 GC patients were METex14del positive (Table 3). All GC cases were MET IHC 3+ and the only case in the series with MET amplification. For example, one case was a 27-year old male patient who presented with poorly differentiated adenocarcinoma and massive malignant ascites and died shortly after diagnosis. His tumor showed strong MET overexpression by IHC (3+) but no MET amplification by FISH (Figure 2a and 2b (with both amplification and METex14+), Table 3). PDC cell lines were generated from his malignant ascites and investigated for anti-tumor activity by MET inhibitors (below). The second METex14del case was a 67-year old male patient who also presented with poorly differentiated adenocarcinoma with concomitant MET amplification (MET/CEP7 ~12.8) and strong MET overexpression. For colon cancer, 4 patients were METex14del positive (Tables 2 and 3). All of the METex14del+ (or positive) colon cancer patients were not MET amplified and all but one were MET IHC 3+. KRAS was wild-type in all 4 colon cancer patients but BRAF V600E was detected in two of the 4 cases. Interestingly, all 4 colon cancer METex14+cases were left-sided colon cancer. For NSCLC, 5 of 51 (9.8%) patients were METex14del+ and none of the patients had concomitant MET amplifications. The median age for the five patients was 49 years and four patients (80%) were never-smokers (Table 2). Of the METex14del+ NSCLC patients, one patient had concomitant EGFR deletion mutation in exon 19 and T790M within exon 20. None of the METex14del NSCLC patients had concomitant KRAS mutations or MET amplification.
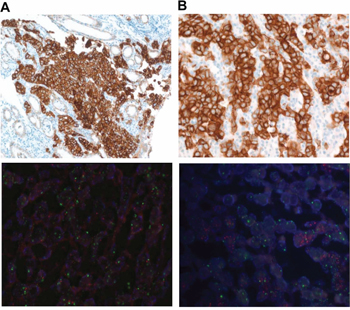
Figure 2: A. METex14+ GC with MET protein overexpression by IHC (upper panel) and no MET amplification by FISH (lower panel) B. METex14+ GC with MET protein overexpression by IHC (upper panel) and concomitant MET amplification by FISH (lower panel).
METex14del patient derived tumor cell lines
We identified 5 with high METex14 transcript expressions and further confirmed by RT-PCR (Table 1, Supplementary Figure S2). Of the 5 PDC cell lines, four METex14del+ cell lines were tested for potential anti-tumor efficacy of c-MET inhibitors, crizotinib (small molecule) and SAIT301 (monoclonal antibody) [6]. In Figure 3D, the expressions of MET protein in GC and CRC PDC cell lines were confirmed using Western blot analysis. Crizotinib, a small molecule targeting MET as well as ALK [12–14], led to dose-dependent growth inhibition both in GC and CRC PDCs (Figure 3A). We further tested otherMET inhibitors such as PHA-665752 and cabozantinib (XL184) (Supplementary Figure 3A and 3B); PHA-665752 is a small molecule inhibitor that specifically targets MET, and cabozantinib is a small molecule inhibitor that targets the MET, VEGFR2, and Ret kinases [15]. PHA-665752 and cabozantinib demonstrated potent growth inhibition in METex14+ GC and CRC PDCs whereas lapatinib, an EGFR and HER2 inhibitor, and cetuximab (Erbitux), an EGFR targeting monoclonal antibody exhibited no such effects (Supplementary Figure S3C). Other PDC line data including melanoma and esophageal squamous cell carcinoma are provided in Supplementary Figure S4. Taken together, these experimental results implicate anti-MET drugs specific inhibition of METex14del+ patient derived cell lines.
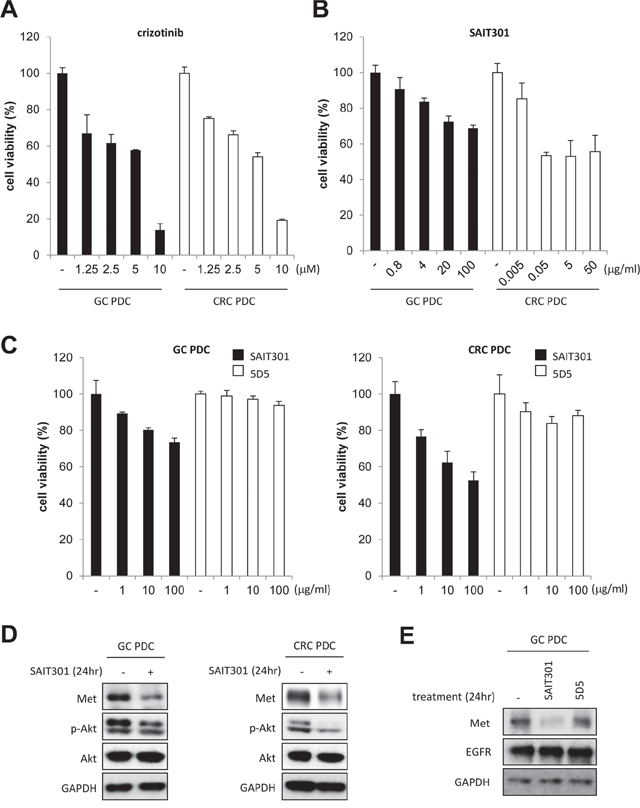
Figure 3: The anti-tumor efficacy of crizotinib and SAIT301 in METex14+ GC and CRC PDCs. A. and B. The viability of METex14+ GC (■) and CRC (□) PDCs by CTG assay after treating with indicated concentrations of crizotinib (A) and SAIT301 (B) for 5 days. C. The viability of PDCs was measured by CTG assay after treatment with various concentrations of SAIT301 (■) and 5D5 (□) for 5 days. The relative cell viability (%) represents the percent growth as compared to the control group (no treatment). D. The protein levels of MET and p-Akt were measured by Western blot in GC and CRC PDCs after 24 h treatment of SAIT301. E. The MET protein levels were measured by Western blot in GC PDC after 24 h treatment of SAIT301 and 5D5.
Previously reported anti-MET antibody, SAIT301 promotes a Cbl-independent MET degradation pathway and internalization of MET without ubiquitination [6]. Splice mutations of exon 14 have been associated with a deletion of the juxtamembrane domain of MET resulting in the loss of interaction with Cbl and Cbl-dependent MET degradation through ubiquitination. We tested SAIT301 in the METex14del+ GC and CRC PDCs to further confirm its Cbl-independent Met degradation mechanism. As shown in Figure 3B and 3C, SAIT301 demonstrated potent growth inhibition of both GC and CRC PDCs whereas 5D5, another bivalent Met targeting antibody, exhibited no proliferation inhibitory effects. To determine antibody mediated down-regulation of MET, we measured the total MET levels in GC and CRC METex14del+ PDCs following SAIT301 treatment (Figure 3D). SAIT301 antibody dramatically reduced MET protein levels. In addition, phosphorylation of Akt, one of major signaling mediators of MET RTK, was also significantly inhibited by treatment with SAIT301. In conclusion, these results confirm that SAIT301 induces degradation of MET in METex14+ PDCs by down-regulating MET in Cbl-independent manners and subsequent inhibition of tumor cell growth.
MATERIALS AND METHODS
Patients
Patients with metastasis of solid cancers were enrolled onto the NEXT-1 trial [NCT#02141152] at Samsung Medical Center. The study was approved by the institutional review board of the Samsung Medical Center. All study participants provided written informed consent before study entry. Briefly, patients with metastatic solid cancer were eligible to enter the study. From November 2013 to August 2014, 428 patients were enrolled. of 428, 230 patients cohort with available tissue specimens for RNA extractions were included in this screening project.
NanoString-Based multiplexed MET exon 14 deletion and ALK, ROS1, NTRK1, NTRK3 or RET fusion transcripts assay
We performed nCounter assays (NanoString, Seattle, WA) according to the manufacturer's instruction in duplicate. Total RNA was extracted from fresh tumor tissue (tumor content > 70%) or from one to four FFPE tissue sections (4 μm thick) using the High Pure RNA Paraffin kit (Roche Diagnostic, Mannheim, Germany). After RNA extraction according to the manufacturer's protocol, we added additional DNase treatment. RNA concentration was measured with the Nanodrop 8000 (Thermo-Scientific, Wilmington, DE) and stored -80°C until use. Briefly, 300 ng of total RNA was hybridized to nCounter probe sets for 16 hours at 65°C. The target sequence for METex14del transcript was 5′-ATTACTACTTGGGTTTTTCCTGTGG CTGAAAAAGAGAAAGCAAATTAAAGATCAGTTTC CTAATTCATCTCAGAACGGTTCATGCCGACAAGTG CAGTAT (Supplementary Figure S1A). For control, we used GAPDH exon 1–2 (accession number NM_002046.3), GUSB exon 4–5 (accession number NM_000181.1), OAZ1 (accession number NM_004152.2) and POLR2A (accession number NM_000937.2). Samples were processed using an automated nCounter Sample Prep Station (NanoString Technologies, Inc., Seattle, WA) as previously described [9]. Full probe sets are provided in Supplementary Table S1. Probe designs are described in previous work [16, 17].
Validation of nanostring results by quantitative RT-PCR
RNAs were reverse transcribed using a superscript III first-strand synthesis system (Invitrogen, Carlsbad, CA). For validation of nanostring results, we designed forward primer for exon 13 of MET, GFPT1 F (5′-TGGGTTTTTCCTGTGGCTGAA-3′), reverse primer for exon 15 of MET (5′-GCATGAACCGTTCTGAG ATGAATT-3′) and probes overlapping an exon 13-exon 15 junction (5′-AAGCAAATTAAAGATCAGTTTCC-3′). GAPDH gene (ID; Hs99999905_m1) was used as an endogenous control. TaqMan probes were labelled with the reporter dye molecule FAM (6-carboxyfluorescein) at the 5′ end and with TaqMan minor groove binder non-fluorescent quencher (MGB-NFQ) probe at the 3 the 3in) at the 5′ end and with GATCAGTT TaqMan Universal PCR master mix with AmpErase UNG (Applied Biosystems), 900 nm primers (forward and reverse), 250 nm TaqMan probe, and 5 μl of cDNA sample in a total reaction volume of 20 μl. PCR conditions were 95°C for 10 min followed by 40 cycles of amplification at 95°C for 15 s and 60°C for 1 min on the ABI PRISM 7500HT Fast Real-time PCR. Ct values < 33 were considered as METex14del+ and ≥ 34 were negative for METex14del.
Additional validation of nanostring results by CustomDx-Met001
To validate our results additionally, we used a qRT-PCR based kit for detection of alternatively spliced variant of MET to detect METex14del. The kit is intended to detect the presence of alternatively spliced (METex14del) MET transcript in RNA from FFPE tissue sections in accordance with the provided protocol (Custom Diagnostics, Irvine, CA). For the MET WT control, Ct from MET WT P/P mix should be in range between 22 and 28, and Ct from METex14del P/P mix should be “undetermined”. For the METex14del Control, Ct from MET WT P/P mix should be between 16 and 22; the Ct from METex14del P/P mix should be between 26 and 32. If the Ct values for controls fall outside the expected range then that run should not be used for evaluation of test samples.
MET immunohistochemistry
For MET immunohistochemistry, we used CONFIRM anti-Total MET (SP44) rabbit monoclonal primary antibody (Ventana Medical Systems, Tucson, AZ, USA) with a Ventana BenchMark XT automated slide processing system according to the manufacturer's protocol as previously described [4, 18]. Both membranous and cytoplasmic staining was scored as follows: 0, no reactivity or faint staining; 1+, faint or weak staining; 2+, moderate staining; 3+, strong staining in > 10% of tumor cells. Membranous alone staining was scored by consensus recommendation on HER2 scoring for gastric carcinoma [19]: 0, no reactivity; 1+, faint/barely perceptible membranous reactivity; 2+, weak to moderate complete or basolateral membranous reactivity; 3+, moderate to strong complete or basolateral membranous reactivity in > 10% of tumor cells. MET overexpression was defined as 2+ or 3+ by membranous and cytoplasmic interpretation and only 3+ by membranous interpretation as previously described [4, 18].
Fluorescent and bright-field double in situ hybridization
FISH was performed using dual-color DNA-specific MET/CEP7 probes (Abnova, Walnut, CA, USA) as described previously [11]. Two pathologists (S.A and M.H) counted the numbers of MET and chromosome 7 centromere probe (CEP7) signals (1 for individual signals, 6 for small clusters and 12 for big clusters) in 20 inter-phase tumor cell nuclei, and the mean number of MET and CEP7 copies per nucleus were determined, along with the ratio. Normal MET/CEP7 signals (one to two copies per cell) in the various non-neoplastic cells served as the internal positive control. We defined MET gene amplification as a MET/CEP7 ratio > 2.0 in 20 tumor nuclei and polysomy-7 were regarded as negative for gene amplification.
Immunoblot analysis
Total proteins from PDCs were isolated using RIPA buffer (Sigma-Aldrich, St. Louis, MO, USA) containing a protease inhibitor cocktail (Roche, Mannheim, Germany) and phosphatase inhibitor cocktail (Roche), and protein concentrations were determined according to Bradford procedure using a Quick Start Bradford Protein Assay (Bio-Rad, Hercules, CA, USA). Thirty μg of proteins were subjected to 10% SDS-polyacrylamide gel electrophoresis, and electro-transferred onto nitrocellulose membranes. The membranes were blocked with 5% nonfat dry milk in Tris-buffered saline containing 0.1% v/v Tween 20, and probed overnight at 4 °C with a Specific antibodies: pMET (Tyr 1234/1235), pAkt (Ser473), Akt(C67E7), pERK1/2 (Thr202/Tyr204), ERK1/2 (Thr202/Tyr204), GAPDH from Cell Signaling Technology (Beverly, MA, USA), and MET from Abcam (Abcam, Cambridge, UK) and MET (C-28) from Santa Cruz biotechnology (Santa Cruz, CA, USA), and beta actin from Sigma Aldrich. Horseradish peroxidase-conjugated anti-rabbit or mouse IgG (Vector, Burlingame, CA, USA) were used as a secondary antibody, and signals were detected by chemiluminescence using ECL Western Blotting Substrate (Thermo Scientific, Rockford, IL, USA), and visualized by using LAS-4000 (Fujifilm, Tokyo, Japan).
Reagents
SAIT301 was produced using a recombinant CHO stable cell line [20]. Crizotinib, PHA-665752, XL-184 and lapatinib were purchased from Selleck Chemicals (Houston, TX, USA).
Patient derived tumor cell culture and cell proliferation inhibition assay
Patient derived tumor cells (PDCs) were isolated from malignant effusions, surgical tissues or biopsies after obtaining informed consent form (the SMC Oncology Biomarker study (NCT#01831609, clinicaltrials.gov). The protocol was approved by the Institutional Review Board at Samsung Medical Center. The cells were cultured in RPMI media supplemented with 10% fetal bovine serum, 0.5 μg/ml of hydrocortisone (Sigma Aldrich), 5 μg/ml of insulin(PeproTech, Rocky Hill, NJ, USA), 5 ng of EGF and FGF (PeproTech). Cell proliferation in response to antibody treatment in vitro was assessed by a CTG (Promega, Madison, WI, USA) assay according to manufacturer instructions. Cells were plated at a density of 5 × 105 cells in FBS 10% (v/v) RPMI 1640 medium onto a 96-well plate (BD Biosciences, Palo Alto, CA, USA). After 24 h incubation, treated antibodies or small molecules diluted in 10% FBS (v/v) RPMI medium were added. After 5 days incubation, 100 μl of the CTG reagent was added to each well followed by incubation at RT for 30 min. The luminescence signal was recorded using Envision 2104 Multi-label Reader (Perkin Elmer, Foster City, CA, USA).
DISCUSSION
The Met proto-oncogene is encoded by 21 exons spanned by 20 introns [7]. The transmembrane domain of MET is encoded by the whole of exon 13 and part of exon 14. Met exon 14 deletion thus results in an in frame deletion of 47 amino acids in the juxtamembrane region which contains the domain. The MET deletion mutant, while displaying decreased Cbl binding, leads to prolonged protein stability, extended cell signaling on ligand stimulation, and increased tumorigenicity [9]. The incidence of METex14del was estimated to be 3.5% in NSCLC [5] and 1.9% in neuroblastoma [11]. Recently, analysis of tumor genomic profiles from 38,028 patients identified 221 cases with METex14 mutations (0.6%), including 126 distinct sequence variants in lung adenocarcinoma (3.0%), other lung neoplasms (2.3%), brain glioma (0.4%), and tumors of unknown primary origin (0.4%) [21]. To date, METex14del has not been reported in either gastric or colon cancer patients. This is the first report that identified METex14del at a frequency of approximately 5% in both gastric and colon cancer in addition to NSCLC. All METex14del+ cases also over-expressed MET protein with only one case showed MET amplification consistent with the hypothesis that METex14del leads to MET over-expression without the need for concurrent MET amplification. In addition, METex14del occurs exclusively to ALK, ROS1, NTRK1, NTRK3 or RET fusions indicating METex14del is likely a driver mutation and defines a unique molecular subset of gastric and colon cancers.
In GC, only Hs746T cell line exhibited both splice-site mutations and MET amplification with MET protein overexpression [9]. We are the first group to report on three GC cases (4.8%) with METex14del and strong MET protein overexpression by IHC. We performed MET exon 14 Sanger sequencing with gDNAs and cDNAs, but failed to detect mutations in our GI METex14del+cancer samples (data not shown). Given the low sensitivity of Sanger sequencing (<12%) and low Ct values in our RT-PCR results, we postulate that METex14del+ tumor population is present in small subpopulation of tumors. Furthermore, we also report c.3082+811A TTTTAACA > GGTTTGAT mutations on intron 14 region of MET with variant allele frequencies around 10%. This intronic mutation is a novel mutation, which has not been reported in COSMIC and TCGA lung adenocarcinomas (Supplementary Table S2). We found that this mutation site is important where proteins including well known splicing factors such as Jun, and Fox, etc. bind. ChIP-Seq dataset of ENCODE project provides strong evidences and the specific intronic mutation site reported here exists in the middle of the protein binding sites (Supplementary Figure S5). We assume that this 8bp mutation on this binding site would affect decreased protein binding affinity of these proteins and may cause exon 14 skipping in small subpopulations of tumor, especially given the tumor heterogeneity in GI cancer.
In two PDC cell lines with METex14del+ without concurrent MET amplification, tumor growths were profoundly inhibited by both MET tyrosine kinase inhibitors and a MET targeting monoclonal antibody-. Our study is the first proving efficacy of MET inhibitors or monoclonal antibody in human GI PDC lines with METex14del that over-expressed MET but did not have MET amplification. Furthermore the in vitro cell line inhibition data indicated that METex14del is potentially an actionable driver mutation in GI malignancies. This finding provides new opportunities for clinical trials on MET inhibitors in metastatic GC to include not only MET amplified GC, but also MET over-expressed, MET non-amplified and METex14del+ GC. So far all METex14del+ cases had concurrent MET over-expression, it remained to be determined if METex14del. Now, we developed screening algorithm to detect METex14del for screening oncology patients. First, we screen MET overexpression by IHC and select MET-positive (≥ 2+) cases.4 In IHC-positive cases, we perform FISH to exclude MET amplification as a cause of MET overexpression. In cases without MET amplification, we perform custom-designed and RT-PCR using mRNAs from tumor to detect METex14del transcripts. For RT-PCR positive cases, we sequence them to find underlying cause of METex14 alterations at the DNA level.
In summary this report supports that the aberration in Cbl-mediated negative regulation of MET can indeed result in MET protein overexpression and subsequent addiction of tumor cells to MET signaling and may serve as an actionable driver mutation in a subset of GI malignancies.
ACKNOWLEDGMENTS
We would like to thank Dr. James Christensen at Mirati Therapeutics, San Diego, California for his helpful scientific advises.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This study was supported by a grant from the 20 by 20 project of Samsung Medical Center (GF01140111). This research was supported by a grant of the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI14C2188 to Jeeyun Lee; grant number: HI14C3418 to J.M.L, B.K, J.C, K.K, Y.S.P and J.L).
REFERENCES
1. Ferracini R, Longati P, Naldini L, Vigna E, Comoglio PM. Identification of the major autophosphorylation site of the Met/hepatocyte growth factor receptor tyrosine kinase. J Biol Chem. 1991; 266:19558–19564.
2. Jardim DL, Tang C, Gagliato Dde M, Falchook GS, Hess K, Janku F, Fu S, Wheler JJ, Zinner RG, Naing A, Tsimberidou AM, Holla V, Li MM, et al. Analysis of 1, 115 patients tested for MET amplification and therapy response in the MD Anderson Phase I Clinic. Clin Cancer Res. 2014; 20:6336–6345.
3. Lennerz JK, Kwak EL, Ackerman A, Michael M, Fox SB, Bergethon K, Lauwers GY, Christensen JG, Wilner KD, Haber DA, Salgia R, Bang YJ, Clark JW, et al. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J Clin Oncol. 2011; 29:4803–4810.
4. Ha SY, Lee J, Kang SY, Do IG, Ahn S, Park JO, Kang WK, Choi MG, Sohn TS, Bae JM, Kim S, Kim M, Kim S, et al. MET overexpression assessed by new interpretation method predicts gene amplification and poor survival in advanced gastric carcinomas. Mod Pathol. 2013; 26:1632–1641.
5. Kong-Beltran M, Seshagiri S, Zha J, Zhu W, Bhawe K, Mendoza N, Holcomb T, Pujara K, Stinson J, Fu L, Severin C, Rangell L, Schwall R, et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006; 66:283–289.
6. Lee JM, Kim B, Lee SB, Jeong Y, Oh YM, Song YJ, Jung S, Choi J, Lee S, Cheong KH, Kim DU, Park HW, Han YK, et al. Cbl-independent degradation of Met: ways to avoid agonism of bivalent Met-targeting antibody. Oncogene. 2014; 33:34–43.
7. Seo JS, Ju YS, Lee WC, Shin JY, Lee JK, Bleazard T, Lee J, Jung YJ, Kim JO, Shin JY, Yu SB, Kim J, Lee ER, et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012; 22:2109–2119.
8. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014; 511:543–550.
9. Zang ZJ, Ong CK, Cutcutache I, Yu W, Zhang SL, Huang D, Ler LD, Dykema K, Gan A, Tao J, Lim S, Liu Y, Futreal PA, et al. Genetic and structural variation in the gastric cancer kinome revealed through targeted deep sequencing. Cancer Res. 2011; 71:29–39.
10. Asaoka Y, Tada M, Ikenoue T, Seto M, Imai M, Miyabayashi K, Yamamoto K, Yamamoto S, Kudo Y, Mohri D, Isomura Y, Ijichi H, Tateishi K, et al. Gastric cancer cell line Hs746T harbors a splice site mutation of c-Met causing juxtamembrane domain deletion. Biochem Biophys Res Commun. 2010; 394:1042–1046.
11. Yan B, Lim M, Zhou L, Kuick CH, Leong MY, Yong KJ, Aung L, Salto-Tellez M, Chang KT. Identification of MET genomic amplification, protein expression and alternative splice isoforms in neuroblastomas. J Clin Pathol. 2013; 66:985–991.
12. Christensen JG, Zou HY, Arango ME, Li Q, Lee JH, McDonnell SR, Yamazaki S, Alton GR, Mroczkowski B, Los G. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther. 2007; 6:3314–3322.
13. McDermott U, Iafrate AJ, Gray NS, Shioda T, Classon M, Maheswaran S, Zhou W, Choi HG, Smith SL, Dowell L, Ulkus LE, Kuhlmann G, Greninger P, et al. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res. 2008; 68:3389–3395.
14. Settleman J. Cell culture modeling of genotype-directed sensitivity to selective kinase inhibitors: targeting the anaplastic lymphoma kinase (ALK). Semin Oncol. 2009; 36:S36–41.
15. Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, Qian F, Chu F, Bentzien F, Cancilla B, Orf J, You A, Laird AD, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. 2011; 10:2298–2308.
16. Lira ME, Choi YL, Lim SM, Deng S, Huang D, Ozeck M, Han J, Jeong JY, Shim HS, Cho BC, Kim J, Ahn MJ, Mao M. A single-tube multiplexed assay for detecting ALK, ROS1, and RET fusions in lung cancer. J Mol Diagn. 2014; 16:229–243.
17. Lee J, Sohn I, Do IG, Kim KM, Park SH, Park JO, Park YS, Lim HY, Sohn TS, Bae JM, Choi MG, Lim do H, Min BH, et al. Nanostring-based multigene assay to predict recurrence for gastric cancer patients after surgery. PLoS One. 2014; 9:e90133.
18. Ha SY, Lee J, Jang J, Hong JY, Do IG, Park SH, Park JO, Choi MG, Sohn TS, Bae JM, Kim S, Kim M, Kim S, et al. HER2-positive gastric cancer with concomitant MET and/or EGFR overexpression: a distinct subset of patients for dual inhibition therapy. Int J Cancer. 2015; 136:1629–1635.
19. Hofmann M, Stoss O, Shi D, Buttner R, van de Vijver M, Kim W, Ochiai A, Ruschoff J, Henkel T. Assessment of a HER2 scoring system for gastric cancer: results from a validation study. Histopathology. 2008; 52:797–805.
20. Oh YM, Song YJ, Lee SB, Jeong Y, Kim B, Kim GW, Kim KE, Lee JM, Cho MY, Choi J, Nam DH, Song PH, Cheong KH, et al. A new anti-c-Met antibody selected by a mechanism-based dual-screening method: therapeutic potential in cancer. Mol Cells. 2012; 34:523–529.
21. Framton GM, Ali SJ, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, Akimov M, Bufill J, Lee C, Jentz D, Hoover R, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discovery. 2015; Published OnlineFirst May 13, 2015.