
INTRODUCTION
The concept of ‘oncogene addiction’ [1] has given rise to targeted cancer therapy, which is effective in treating cancers dependent on the corresponding oncogenic driver [2]. Examples include the epidermal growth factor receptor (EGFR) and the androgen receptor (AR). However, the dramatic responses to targeted therapy are limited by pre-existing or acquired therapy resistance [3]. The best understood mechanisms are secondary missense mutations in driver genes. The T790M ‘gateway’ mutation in the EGFR produces erlotinib and gefitinib resistance [4–6], while the F876L mutation in the AR converts enzalutamide from an antagonist to an agonist [7–9]. Epigenetic mechanisms can alter the expression of splice variants, such as those encoding ligand-independent versions of the AR, which are expressed at increased levels in treatment resistant prostate cancers [10]. Non-genetic mechanisms are not as well understood, but may play a role in treatment failure. In this report, we show that androgen deprivation therapy can paradoxically activate a pro-tumorigenic signaling mechanism that is dependent on paracrine interactions within the tumor microenvironment.
We (SY and CC) recently discovered that androgen deprivation therapy (ADT) unexpectedly enhances the metastatic phenotype of castration resistant prostate cancer (CRPC), even as these therapies inhibit tumor growth [11–13]. Specifically, treatment with the anti-androgens bicalutamide and enzalutamide inhibited proliferation but induced macrophage migration and tumor cell invasion in vitro as well as distant metastases in orthotopic tumor models of CRPC [11]. This may reflect a divergence between the effects of AR signaling on proliferation compared to the effects on the metastatic phenotype. These pro-metastatic effects are mediated by CCL2 [11, 12], a chemokine also known as monocyte chemoattractant protein-1 that binds the cognate receptor CCR2 to induce chemotaxis [14, 15]. Monocytes are a major source of CCL2 [15], but CCL2 is also produced by a variety of cells in tumors, including epithelial tumor cells and the cellular components of the tumor microenvironment, such as endothelium, stroma and tumor-associated macrophages (TAMs) [14]. In addition, studies of prostate cancer (PCa) patient serum and/or tumor tissue samples support a role for CCL2 in ADT-induced metastasis [11, 12, 16]. We (KLN and JJK) have also previously demonstrated that TNF is negatively regulated by androgens [17]. Specifically, castration induces TNF mRNA in rodent prostatic stroma. Promoter analysis has shown that CCL2 is regulated by TNF via NFκB [18]. Indeed, it has been reported that TNF induces CCL2 expression in ovarian cancer cells [19] as well as sensory neurons [20–22], and vascular smooth muscle cells [23]. Given these two sets of previous findings from our laboratories, we tested the hypothesis that TNF signaling is required for enzalutamide induced metastasis of CRPC via CCL2.
RESULTS
Androgen deprivation induces TNF expression
To address the role of TNF in metastasis following androgen deprivation, we initially employed three cell lines, representing CRPC (C4-2), prostate stromal myofibroblasts (WPMY-1) and tumor associated macrophages (THP-1), either alone, in co-culture or via conditioned media, to simulate the in vivo context of PCa. C4-2, a sub-line of the human androgen-dependent LNCaP prostate cancer cell line, derived by selecting for growth as a xenograft in a castrated athymic nude mouse [24–26], is a well-established cell line model for CRPC. WPMY-1 is an SV40 large-T antigen-immortalized myofibroblast cell line (expressing smooth muscle α-actin and vimentin), derived from a cancerous human prostate [26]. THP-1 [27] is derived from a human acute monocytic leukemia, displays monocytic markers, has phagocytic activity and expresses CCR2 [28], indicating that it is a model for TAMs [29].
Following treatment with dihydrotestosterone (DHT), TNF secretion was reduced in C4-2 (Figure 1a). Conversely, treatment of C4-2 with the anti-androgen enzalutamide induced TNF secretion and an increase in mRNA expression (Figure 1b–1c). The coordinate increase in both protein and mRNA is consistent with transcriptional repression of the TNF gene by the AR. However, we cannot exclude effects on mRNA stability. Neither DHT nor enzalutamide affected TNF expression in the TAM-like THP-1 or stromal myofibroblast-like WPMY-1 cell lines. Similarly, in the rat stroma-derived PS-1 cells, there was no change in TNF expression in response to enzalutamide (data not shown). The anti-androgen bicalutamide and the synthetic androgen R1881 had analogous effects to enzalutamide and DHT, respectively (Supplementary Figure S1). Similar levels of TNF secretion, following DHT withdrawal or enzalutamide treatment, were observed when C4-2 cells were co-cultured with THP-1 and/or WPMY-1 cells (Figure 2).
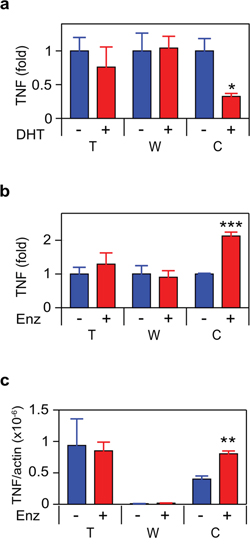
Figure 1: ADT induces TNF expression in CRPC. a. b. THP1 (T), WPMY-1 (W) or C4-2 (C) cells were treated for 72 h with vehicle (–) or 10 nM DHT (+) (a) or 10 μM enzalutamide (Enz; +) (b) TNF was assayed by ELISA (n ≥ 3). c. mRNA from cultures identical to (b) was quantitated by RT-PCR. TNF mRNA levels, normalized to ß-actin, are shown (n = 3). Treated (red) and untreated (blue) cultures compared by Student’s unpaired t-test. *p < 0.05, ***p < 0.001.
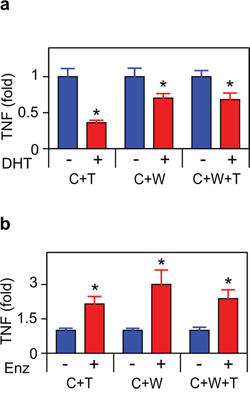
Figure 2: ADT induces TNF secretion in tumor/microenvironment co-cultures. Co-cultures, as indicated in Figure 1, were treated for 72 h with vehicle (–) or 10 nM DHT (+) a. or 10 μM enzalutamide (Enz) (+) b. TNF was assayed by ELISA (n = 3). Treated (red) and untreated (blue) cultures compared by Student’s unpaired t-test. *p < 0.05.
TNF is required for CCL2 secretion
Since we previously observed that androgen deprivation induced CCL2 [11], we measured CCL2 secretion and mRNA in the cultures examined in Figure 1 and observed an identical pattern, correlating TNF and CCL2 expression at the protein and mRNA levels (Figure 3). CCL2 and TNF secretion were also coincident in C4-2 treated with bicalutamide or R1881 (Supplementary Figure S2). To determine if CCL2 expression was regulated by TNF, we employed etanercept, a soluble receptor composed of the extracellular domain of the p75 TNF receptor (TNFR2) fused to the Fc portion of the immunoglobulin protein. Etanercept binds TNF avidly and with high specificity [30], making this an ideal reagent for testing the role of secreted or membrane bound TNF in mediating intracellular signaling events. Figure 4 demonstrates that etanercept blocked enzalutamide induced secretion of the CCL2 protein (Figure 4a) as well as the induction of CCL2 mRNA levels (Figure 4b). This suggests autocrine TNF signaling can regulate transcription of the CCL2 gene (or, alternatively, CCL2 mRNA stability) in C4-2 cells. Etanercept also blocked CCL2 secretion, but not the mRNA level, in WPMY-1 myofibroblasts (Figure 4). However, enzalutamide did not induce TNF production at either the protein or mRNA level in WPMY-1 (Figure 1b–1c).
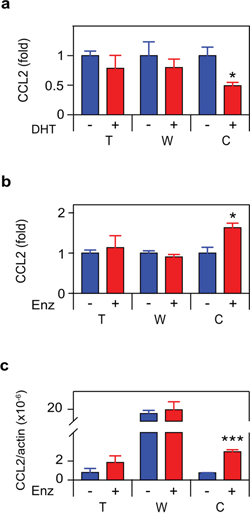
Figure 3: ADT induces CCL2 expression in CRPC cells. a. b. Cells as indicated in Figure 1 were treated for 72 h with vehicle (–) or 10 nM DHT (+) (a) or 10 μM enzalutamide (Enz) (+) (b) CCL2 was assayed by ELISA (n ≥ 3). c. mRNA from cultures identical to (b) were quantitated by RT-PCR. CCL2 mRNA levels, normalized to ß-actin, are shown (n = 3). Treated (red) and untreated (blue) cultures compared by Student’s unpaired t-test. *p < 0.05, **p < 0.01.
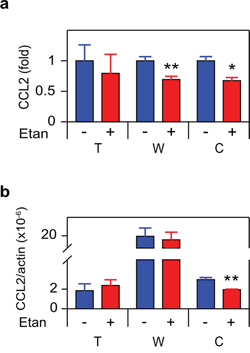
Figure 4: TNF is required for enzalutamide-induced CCL2 expression in human CRPC. a. Cells indicated as in Figure 1 were cultured in the presence of 10 μM enzalutamide and either vehicle (–) or 1 μg/mL etanercept (Etan) (+), and assayed for CCL2 by ELISA 72 h later (n ≥ 3). b. mRNAs from cultures identical to a. were quantitated by RT-PCR. CCL2 mRNA levels, normalized to ß-actin, are shown (n = 3). Treated (red) and untreated (blue) cultures compared by Student’s unpaired t-test. *p < 0.05, **p < 0.01.
Figures 1–4 indicate that TNF signaling mediates ADT induced transcription and secretion of CCL2 by C4-2 CRPC cells. To determine if our observations in C4-2 cells are representative of the response of CRPC to ADT, we examined PTEN-CaP8, a murine CRPC cell line derived from prostate-specific Pten null mice homozygously deleted for PTEN [31]. To do this, we replicated the experiments shown in Figures 3a–3b and Figure 4a, employing PTEN-CaP8 in place of C4-2 cells and found that PTEN-CaP8 cells responded the same as C4-2 cells (Figure 5a). Specifically, DHT reduced, and enzalutamide induced, CCL2 secretion in PTEN-CaP8 cells and etanercept blocked enzalutamide-induced CCL2 production (Figure 5a). Thus, TNF-mediated CCL2 induction may be a typical response of CRPC to ADT. To verify that this response is a feature of CRPC cell lines, we examined the isogenic parental lines that are the progenitors of PTEN-CaP8 and C4-2. These cell lines (PTEN-P8 and LNCaP, respectively) resemble localized prostate cancers; that is, these are less advanced versions of the corresponding CRPC cell line. For both PTEN-P8 and LNCaP, the effects of blocking androgen signaling were less pronounced. Importantly, even when regulated by ADT, etanercept did not block CCL2 production (Figure 5b, 5c), indicating that these less advanced cell lines are distinct from the CRPC cell lines. Taken together, this suggests that ADT-induced, TNF-dependent CCL2 production is a feature of CRPC.
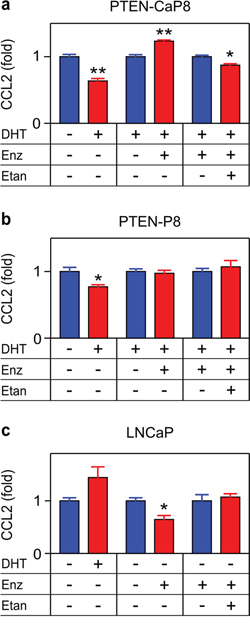
Figure 5: TNF is required for enzalutamide-induced CCL2 expression in a murine CRPC cell line. PTEN-CaP8 a. PTEN-P8 b. and LNCaP c. were treated with vehicle (–), 10 nM DHT (DHT, +), 10 μM enzalutamide (Enz, +) or 1 μg/mL etanercept (Etan, +) as indicated. Secreted CCL2 was measured by ELISA after 72 h (n ≥ 3). Treated (red) and untreated (blue) cultures compared by Student’s unpaired t-test. *p < 0.05, **p < 0.01.
TNF blockade reduces paracrine CCL2 secretion
To directly test the requirement for TNF, C4-2, THP-1 and WPMY-1 cells were treated with TNF, and CCL2 secretion and mRNA was measured (Figure 6). The two microenvironment-derived cell lines (THP-1 and WPMY-1, representing TAMs and myofibroblast stromal cells, respectively) demonstrated increased CCL2 expression at both the protein and mRNA levels, in response to TNF (Figure 6a–6d).
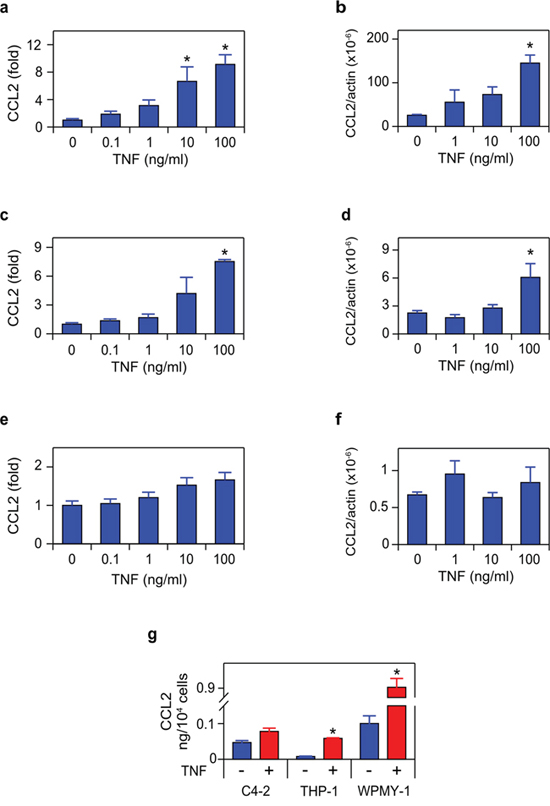
Figure 6: TNF induces CCL2 in microenvironment-derived cells. a. c. e. WPMY-1 (a) THP1 (c) and C4-2 (e) were treated with TNF for 72 h, as indicated. Secreted CCL2 was measured by ELISA (n = 3). b. d. f. RNA was extracted from WPMY-1 (b) THP-1 (d) and C4-2 (f) treated as in (a, c, e) CCL2 mRNA levels, normalized to ß-actin, are shown (n = 3). g. The amount of CCL2 secreted by the indicated cell lines, following 72 h treatment with vehicle (red) or 100 ng/mL TNF (blue). Data re-plotted from (a, c, e) Differences were assessed by one-way ANOVA, followed by Tukey-Kramer HSD test (a–f). *p < 0.05.
Surprisingly, C4-2 cells, which produce CCL2 in a TNF-dependent, autocrine manner (Figures 3–4), did not show significant modulation of CCL2 in response to TNF (Figure 6e–6f), even though exogenous TNF was employed at doses that exceed the concentration of soluble TNF detected in C4-2 media following androgen deprivation (5–10 pg/mL; data not shown). This suggests that TNF concentrations determined by ELISA in cell culture media might not reflect the concentration available to receptors on the C4-2 cell surface. Indeed, TNF is expressed in a membrane-bound form, which is cleaved by the extracellular protease TACE to generate the soluble (‘secreted’) form of the cytokine [32]. Since membrane-bound TNF is biologically active and cannot be detected in ELISA assays of cell culture media, we may have significantly underestimated the biologically effective TNF concentration that is required to trigger the autocrine effects in the single cell culture conditions we employed in Figures 3 and 4. This suggests the possibility that microenvironment cell lines, which are more responsive to TNF (see Figure 6a–6d versus Figure 6e–6f), might be the relevant source of CCL2 in the in vivo tumor context. Thus, the production of CCL2 may be mediated by paracrine interactions between the tumor cells and the microenvironment, rather than autocrine production of CCL2 by the C4-2 prostate cancer tumor cells. To test this, we mixed C4-2 tumor cells with either THP-1 or WPMY-1 cells, and blocked androgen binding and signaling in these co-cultures, using the androgen antagonist enzalutamide. In both mixed cultures (C4-2 tumor cells plus WPMY-1 myofibroblasts (Figure 7a), and C4-2 tumor cells plus THP-1 TAM-like cells (Figure 7b)), enzalutamide induced CCL2 and etanercept blocked CCL2 induction, consistent with our proposed paracrine mechanism.
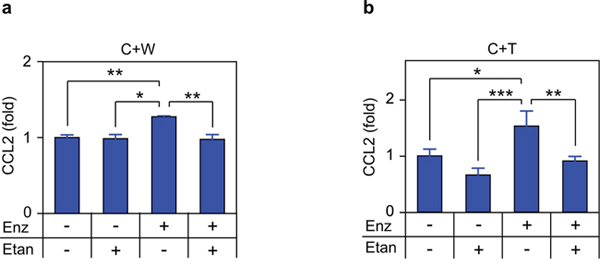
Figure 7: TNF is required for enzalutamide-induced CCL2 secretion in co-cultures. Mixed cultures of C4-2 plus WPMY-1 (C+W;) a. or C4-2 PCa plus THP1 (C+T;) b. were treated with vehicle (–), 10 μM enzalutamide (Enz, +) or 1 μg/mL etanercept (Etan, +) for 72 h, as indicated. Secreted CCL2 was measured by ELISA (n ≥ 3). Differences were assessed by one-way ANOVA followed by Tukey-Kramer HSD test. *p < 0.05, **p < 0.01, ***p < 0.001.
TNF is required for migration and invasion
As noted, we have previously demonstrated that enzalutamide induces a metastatic phenotype in both cell culture and mouse models of CRPC [11]. To determine if TNF regulation of CCL2 is required for the metastatic phenotype, we performed three experiments. First, we demonstrated that TNF induces migration of tumor associated macrophage-like THP-1 and CRPC C4-2 cells in a cell culture model of the tumor microenvironment (Figure 8). Specifically, conditioned media (CM) from TNF-treated WPMY-1 cells, expected to contain biologically active concentrations of CCL2 (Figure 6g), was placed in the bottom of a transwell chamber (Figure 8a). Both THP-1 and C4-2 cells migrated towards the CM, and migration was reduced by a CCR2 inhibitor (Figure 8b–8d). Notably, neither C4-2 nor THP-1 cells migrated towards media plus recombinant TNF (Supplementary Figure S3). Also, TNF did not enhance C4-2 proliferation (Supplementary Figure S4), arguing that increased cell numbers do not account for increased migration. Second, paracrine CCL2, produced by enzalutamide treatment, induced migration of C4-2 (Figure 9). In these experiments, CM was from enzalutamide-treated C4-2/WPMY-1 (Figure 9a, 9b) or C4-2/THP-1 (Figure 9c, 9d) mixed cultures. Again, the enzalutamide-treated CM is expected to contain higher levels of CCL2 (relative to the vehicle-treated control), in this case due to enzalutamide induction of TNF (Figure 7). C4-2 migration towards the CM was inhibited when etanercept was present during CM production, thereby blocking TNF-mediated paracrine CCL2 secretion. Finally, we replicated the experiment in Figure 9, but measured invasion, i.e., migration through a Matrigel-coated membrane. C4-2 cells were induced to invade, in a TNF-dependent manner, by C4-2/WPMY-1 CM (Figure 10a, 10b) but not by C4-2/THP-1 CM (Figure 10c, 10d), consistent with the relatively higher level of TNF-dependent CCL2 production by WPMY-1 cultures (Figure 6g).
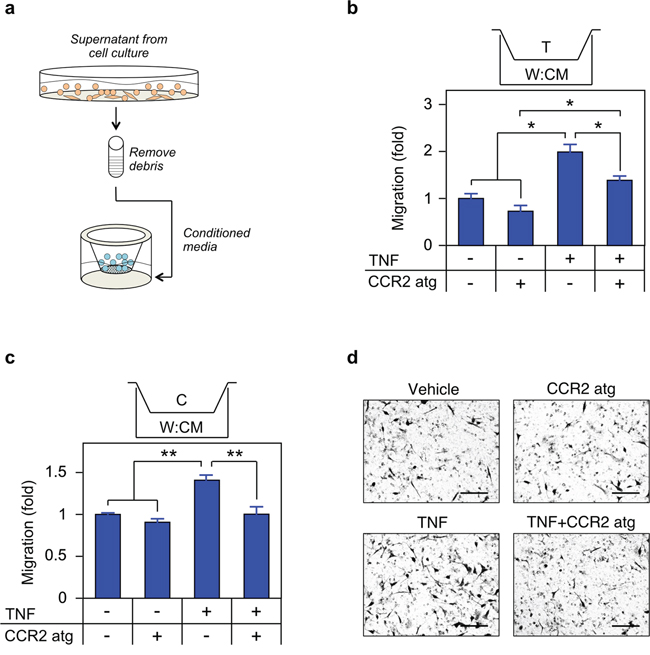
Figure 8: TNF induced CCL2 signaling is required for migration. a. Schematic of migration assay. b. c. THP1 (b) or C4-2 (c) were seeded in the upper chamber and migration determined as in Materials and Methods (n = 3). CM was from cells grown for 72 h in media plus vehicle (–) or 100 ng/mL TNF (TNF, +). The CCR2 antagonist (CCR2 atg, +) was added to the CM, as indicated. d. Representative photomicrographs of migrated C4-2 cells from (c) Magnification, 460 ×; scale bar, 100 μm. Differences were assessed by one-way ANOVA followed by Tukey-Kramer HSD test. *p < 0.05, **p < 0.01.
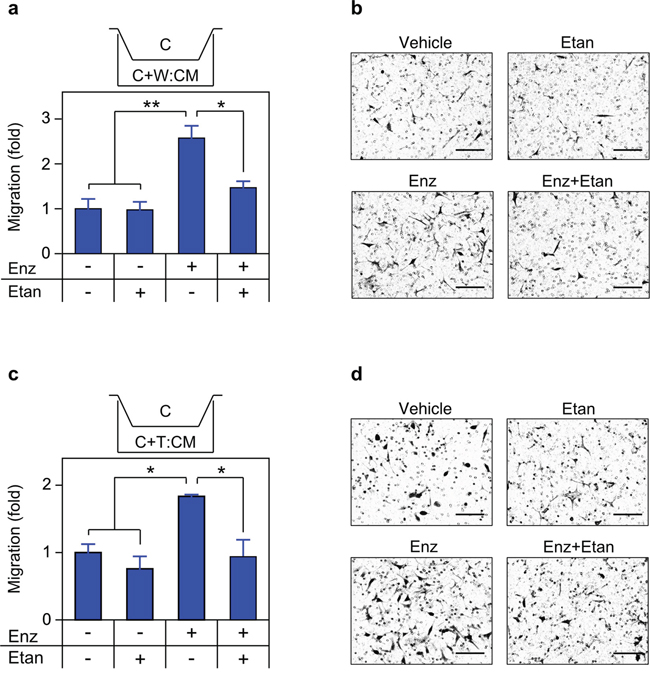
Figure 9: Secreted TNF is required for enzalutamide-induced migration. a. c. Transwell migration assays were performed as in Figure 8, with C4-2 cells seeded in the upper chamber and migration determined as in Materials and Methods (n = 3). CM derived from mixed cultures of C4-2/WPMY-1 (a) or C4-2/THP1 (c) grown for 72 h in the presence of vehicle (–), 10 μM enzalutamide (Enz, +) or 1 μg/mL etanercept (Etan; +), as indicated. b. d. Representative photomicrographs of migrated C4-2 cells from (a) and (c) respectively. Magnification, 460x; scale bar, 100 μm. Differences were assessed by one-way ANOVA followed by Tukey-Kramer HSD test. *p < 0.05, **p < 0.01
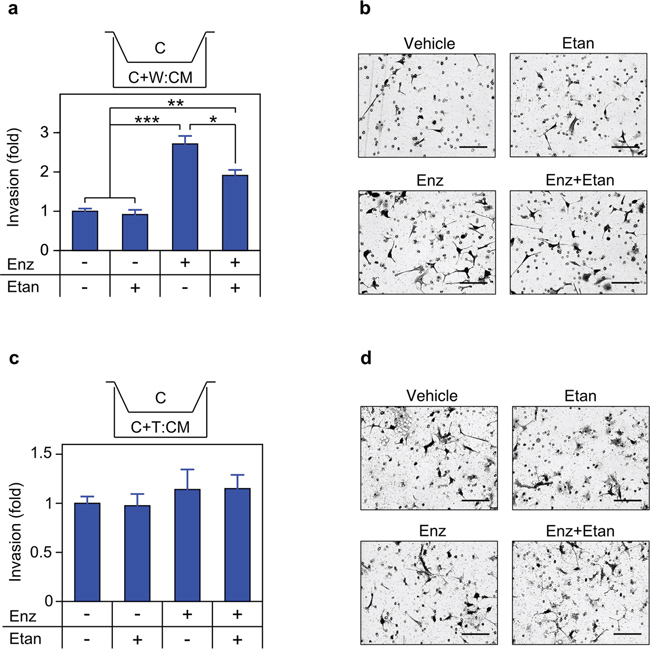
Figure 10: Secreted TNF is required for enzalutamide-induced invasion. a. c. Transwell invasion assays were performed, essentially identical to Figure 9, except that transwell membranes were coated with Matrigel, (n = 3). b. d. Representative photomicrographs of cells that have invaded the Matrigel coated membrane, from (a) and (c) respectively. Magnification, 460 ×; scale bar, 100 μm. Statistical comparison by one-way ANOVA followed by Tukey-Kramer HSD test. *p < 0.05, **p < 0.01, ***p < 0.001
TNF and CCL2 expression in prostate cancers
It is important to consider our findings in the context of human cancers. A key study [16] sampled PCa patients within three months of initiating ADT, when AR signaling is still likely suppressed (i.e., prior to the onset of CRPC), and found elevated serum TNF and CCL2 correlated with reduced survival. To gain additional insight into the correlation between PCa metastasis, CCL2 and TNF, we interrogated two sets of metastatic CRPC patient samples [33, 34] using Oncomine [35]. In these patients AR signaling is typically reactivated, and indeed AR mRNA expression levels are increased in both data sets (Figure 11a). TNF mRNA was significantly elevated in CRPC patient samples (Figure 11b; see also Supplementary Figure S5). In contrast, CCL2 mRNA was significantly reduced in both data sets (Figure 11c), which may reflect induction of CCL2 at the primary tumor site only [48], rather than also at the metastatic tumor site (see Discussion). Moreover, a similar pattern (AR increased, CCL2 significantly decreased) is observed in metastases from patients who did not receive ADT (Supplementary Figure S6, Supplementary Table S1), supporting the idea that CCL2 is only required at the primary tumor site.
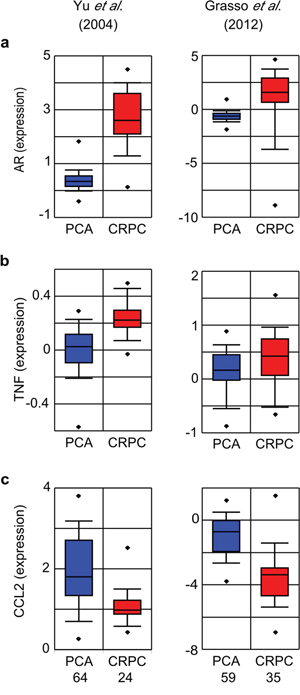
Figure 11: TNF, CCL2 and AR mRNA expression in metastatic CRPC. Oncomine was used to generate box plots of AR a. TNF b. and CCL2 c. mRNA expression in human primary PCa (blue) and CRPC (red) from the indicated data sets. Sample size as indicated (bottom), with the exception that the Grass et al. data set contains only 34 (rather than 35) samples for the TNF probe. PCa and CRPC sample sets were compared by Student’s unpaired t-test. p < 10−5, except b. Grasso et al., p = 0.02.
DISCUSSION
Observations that TNF can transcriptionally regulate the CCL2 gene [18], that NFκB is up-regulated in PCa [36–38], and that ADT induced metastasis is dependent on CCL2 [11–13], led us to test the hypothesis that TNF mediates the paradoxical induction of metastasis that occurs following ADT. The role of TNF in PCa has been uncertain [39]. Castration-induced regression (due to epithelial cell apoptosis) in normal rodent prostate requires TNF up-regulation [17] and down-regulation of the caspase-8 inhibitor c-FLIP [40], but in CRPC c-FLIP expression is increased [41], blocking the pro-apoptotic effects of TNF and inducing NFκB, which promotes cell survival [42]. The induction of metastases by ADT, even as the primary tumor regresses [11–13], suggests a stage-specific role for TNF in CRPC, similar to TGFß, which inhibits early cancers and promotes late stage cancer metastasis [43, 44]. First we demonstrated that in C4-2 CRPC tumor cells, various modes of androgen deprivation de-repress TNF mRNA expression, leading to secretion into the media (Figures 1–2). Enzalutamide-induced CCL2 mRNA and protein is dependent on secreted TNF in these cells (Figure 3–4), since it is blocked by a soluble receptor. Figure 5 demonstrates that ADT-induced TNF signaling can enhance CCL2 secretion in a second CRPC-derived cell line (PTEN-CaP8), but not in the less advanced parental cell lines that gave rise to both C4-2 and PTEN-CaP8. This suggests the possibility that a common mechanism driving the evolution of CRPCs also selects for CCL2 expression in response to TNF signaling (likely among other pro-tumorigenic features). In support of this concept, Qin et al. [45] found that ADT of LNCaP cells enriches for a minority LNCaP cell population (PSA-/lo cells) that displays a castration-resistant cancer phenotype, including the ability to grow in castrate hosts, and enhanced expression of cancer stem cell genes. A similar selection process may have been at work in the development of C4-2 and PTEN-CaP8 from the respective parental cell lines LNCaP and PTEN-P8 [24, 25, 31]. Thus, we speculate that the ADT-induced production of CCL2 via TNF signaling is a feature of a population resembling PSA-/lo (progenitor) cells, and contributes to the aggressive tumor phenotype of this cancer population, which expands following ADT [45].
Exogenously added TNF induced CCL2 mRNA and CCL2 protein in macrophage- and myofibroblast-like cells, suggesting paracrine regulation of CCL2 production (Figure 6). We confirmed the paracrine mechanism by employing mixed cultures of tumor and microenvironment cells (Figure 7) and, analogously, by measuring in vitro metastatic phenotypes (migration and invasion) that depend on CCL2 signaling. Again, exogenous cytokine addition or conditioned media – both mimicking possible paracrine effects within the microenvironment - demonstrated a functional role for TNF in the paracrine loop controlling metastasis in response to enzalutamide (Figures 8–10). We therefore propose that tumor cells secrete TNF, which induces CCL2 production in two microenvironment cells (myofibroblasts and macrophages) promoting tumor migration, invasion and extravasation [46] and monocyte recruitment and differentiation into tumor associated macrophages [12, 14]. Figure 12 illustrates the paracrine interactions mediating ADT induced metastasis via the chemotactic activities of CCL2.
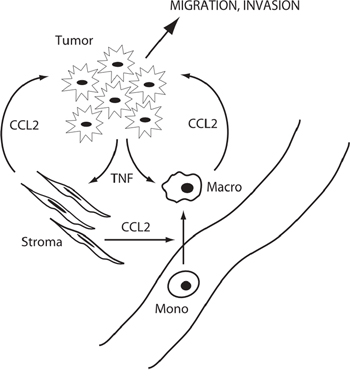
Figure 12: Schematic of microenvironment interactions. Tumor cells produce TNF in response to ADT, which induces CCL2 in stromal cells and macrophages, and promotes tumor cell migration and invasion and monocyte migration and differentiation to macrophages.
In our model system, enzalutamide induces TNF modestly and only in CRPC cells, but is sufficient to up-regulate CCL2 mRNA expression and CCL2 protein secretion, particularly in the WPMY-1 myofibroblast cells. Previously, we had shown that castration of rodents induced TNF mRNA expression in stromal, but not the epithelial cells of the normal prostate [17]. The observations reported here, that androgen deprivation induces TNF mRNA expression in an epithelial tumor cell line, but not the myofibroblast stromal cell line, may reflect differences inherent in tumor and tumor-associated cells as a result of the process of tumorigenesis. As expected, there is a correlation between the level of TNF secretion and the extent of invasion, which is a more stringent criteria of the metastatic phenotype (Figure 10). Although cell culture models facilitated our identification of key paracrine interactions in the cancer microenvironment, there are limitations in extrapolation to in vivo situations. In particular, our culture conditions may not reproduce the proportions of cellular constituents or the effective concentration of the secreted ligands seen in vivo. However, while our data derives from the co-culture of a set of relevant cell lines, many aspects of this signaling paradigm have been confirmed in murine models. Specifically, our prior data demonstrate that conditional deletion or silencing of AR in macrophages and PCa xenografts induces CCL2, macrophage infiltration of xenografts and metastasis [12]. Previously we showed that enzalutamide induces CCL2 in TRAMP-C1 xenografts, and that castration similarly induces CCL2 in normal mouse prostate [12]. Moreover, enzalutamide-enhanced metastasis has been observed in other CRPC models [47]. Future studies in mouse models are needed to confirm the microenvironment source(s) of TNF and verify a functional role for TNF in ADT induced metastasis in vivo.
Finally, a variety of data from PCa patients support our hypothesis. For example, we found CCL2 expression in human PCa samples correlated with PSA levels, the number of infiltrating macrophages and poor prognosis [12]. In a small patient study, anti-androgens enhance the growth of metastases, indicating that ADT-induced metastasis occurs in human PCa [13]. The strongest human data in support of a TNF-CCL2 paracrine loop comes from PCa patients assayed within three months of initiation of ADT [16]. At this stage of treatment, androgen signaling will be fully repressed (patient serum PSA levels low). In this cohort, patients with increased TNF and CCL2 fared worse, consistent with therapy-induced TNF/CCL2 enhancing metastasis even though ADT inhibited the growth of primary tumors. As mentioned above, this implies a divergence between AR signaling effects on proliferation and the metastatic phenotype.
While immunohistochemical staining of primary (localized) PCa tumors showed CCL2 levels correlated with Gleason score and tumor stage [48], analysis of metastasized CRPC sample data sets in Oncomine reveals reduced CCL2 (Figure 11). Specifically, in CRPC patients (where AR signaling is likely reactivated, and patient serum PSA levels are rising) CCL2 mRNA in the metastases was clearly reduced. Similarly, metastases in patients who were not given ADT also have decreased CCL2 mRNA (Supplementary Figure S6). Since the tumor cells have already metastasized and escaped the primary tumor microenvironment, CCL2 may no longer be necessary. That is, once metastatic seeding has occurred, the role of CCL2 in monocyte differentiation and tumor cell migration is diminished. Moreover, the stromal tumor microenvironment that was present in the primary tumor is unlikely to be replicated at the site of the metastatic lesion(s), which lack the cellular interactions that we believe generate CCL2. Furthermore, TNF is significantly elevated in the CRPC metastatic samples (Figure 11), but not consistently elevated in non-CRPC samples. The comparisons of non-CRPC patient samples include primary tumors, which have low versus high Gleason scores (Supplementary Figure S7), as well as primary versus metastasis from patients who did not undergo ADT (Supplementary Figure S6). Taken together, these latter datasets suggest that TNF induction is only consistently seen in response to ADT therapy, rather than as a consequence of tumor progression.
We therefore propose that CCL2 primarily acts via a therapy-induced paracrine loop, such as the one we document in cell culture models, at the site of the primary tumor but not at the site(s) of metastases. Another way to view this is that the process of metastasis interrupts the paracrine loop (TNF®CCL2) by relocating tumor cells away from the original tumor microenvironment. This further suggests that therapies directed against CCL2 or TNF might be effective only during a narrow window of time; that is, only if administered in conjunction with the initial course of androgen deprivation therapy given to patients still possessing a primary tumor, prior to the onset of CRPC and metastases. Thus, our mechanistic observations may explain the recent failure of CCL2 directed monoclonal therapy in PCa [49] since these trials were performed using anti-CCL2 as a single agent and in post-ADT patients who had already developed metastatic CRPC.
MATERIALS AND METHODS
Reagents
The following reagents were used: DHT (Sigma, St. Louis, MO, USA), enzalutamide (Selleckchem, Houston, TX, USA), etanercept (Amgen, Thousand Oaks, CA, USA), R1881 (PerkinElmer, Waltham, MA, USA), CCR2 antagonist BMS-741672 (Torcis Bioscience, Bristol, UK), and TNF (BioLegend, San Diego, CA, USA).
Cell culture
The C4-2 cell line was from Dr. Jer-Tsong Hsieh, UT Southwestern Medical Center, Dallas, TX. LNCaP, THP1, WPMY-1, PTEN-P8 and PTEN-CaP8 cell lines were from the American Type Culture Collection (Rockwell, MD, USA). C4-2, WPMY-1, THP1 and LNCaP, were cultured in RPMI-1640 media supplemented with 10% FCS and 1% penicillin/streptomycin. For THP1, the media also contained 10 mM HEPES and 2 mM glutamine; LNCaP culture media also contained 1 mM sodium pyruvate. PTEN-P8 and PTEN-CaP8 were cultured in DMEM supplemented with 10% FCS and 1% penicillin/streptomycin.
Enzyme-linked immunosorbent assays (ELISA)
Cells were cultured at a density of 1 × 105/ml and supplemented with enzalutamide, DHT or etanercept as indicated. For TNF treatment, cells were cultured at 4 × 104/ml. Cells were seeded at 5 × 104/ml for C4-2/WPMY-1 co-cultures and 1 × 105/ml for C4-2/THP1 co-cultures. TNF and CCL2 were quantitated with ELISA kits (eBioscience, San Diego, CA, USA), per the manufacturer’s instructions.
Metastasis assays
Two in vitro transwell assays (migration, invasion) were performed to assess metastatic phenotypes. For migration assays, cells were serum starved for 24 h, trypsinized if required, washed and resuspended in serum-free media. Approximately 1 × 105 cells were placed in the upper chamber of a 24-transwell plate (Corning Inc., Tewksbury, MA, USA). Matrigel coated chambers (Biocoat, Corning, NY, USA) was added to wells used for invasion assays. Conditioned medium was prepared by diluting culture supernatants with an equal amount of fresh media plus 5% FCS. For C4-2 cells, after 22 h incubation, chambers were fixed and stained with 1% toluidine blue for manual counting of photomicrographs under the microscope (Life Technologies, Carlsbad, CA, USA). For THP-1 cells, after 16 h incubation, media was collected and cells counted by Coulter counter. Counts were normalized to vehicle control.
RNA isolation and quantitative RT-PCR
Cells were cultured at the same densities used for the ELISA assays. Total RNA was extracted and reverse transcribed using pdN15 with Superscript III reverse transcription kit (Invitrogen, Waltham, MA, USA) to generate single-strand cDNA. PCR was performed using SYBR® Green qPCR Master Mix (Molecular Probes, Eugene, OR, USA) in a StepOnePlus instrument (Applied Biosystems, Waltham, MA, USA). PCR primers, spanning at least one intron, were: TNF-forward: 5′-CCTCTCTCTAATCAGCCCTCTG-3′; TNF-reverse: 5′-GAGGACCTGGGAGTA-GATGAG-3′; CCL2-forward: 5′-CAGCCAGATGCAATCAATG-CC-3′; CCL2-reverse: 5′-TGGAATCCTGAACCCACTTCT-3′; β-actin-forward: 5′-CATGTAC-GTTGCTATCCAGGC-3′ and β-actin reverse: 5′-CTCCTTAATGTCACGCACGAT-3′. Standard curves were generated using linearized plasmids encoding the corresponding cDNAs. β-Actin was used to normalize target gene expression. At least three biological replicates were analyzed (unless otherwise indicated), and at least three trials were conducted for each sample.
Cell proliferation assay
C4-2 cells at 50%-75% confluence were trypsinized and seeded into 96-well plates at 4.5 × 103 cells/well. Cells were cultured in complete medium supplemented with 100 ng/ml TNF for 72 h. Viability was measured by WST-1 assay, as per the manufacturer (Roche Applied Science, Indianapolis, IN, USA).
Microarray database analysis
Plots of gene expression levels were obtained from Oncomine (Life Technologies, http://www.oncomine.org). For statistical analysis individual data values were extracted from the Oncomine database. Homology regions of microarray probes were determined using UCSC genome browser (http://genome.ucsc.edu).
Statistics
Data are presented as the mean ± SEM. Statistical analyses were conducted using JMP-Pro10 (SAS, Cary, NC, USA). Differences between two means was assessed by Student’s unpaired t-test and differences among multiple means by one-way ANOVA followed by Tukey-Kramer HSD test.
ACKNOWLEDGMENTS
We thank Dr. Hsieh for providing C4-2 cells.
FUNDING
This research was supported by grants from the NIH (CA151753 to JJK; and CA156700 to C.C. and S.Y.).
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Weinstein IB. Cancer. Addiction to oncogenes—the Achilles heal of cancer. Science. 2002; 297:63–64.
2. Sawyers C. Targeted cancer therapy. Nature. 2004; 432:294–297.
3. Lackner MR, Wilson TR, Settleman J. Mechanisms of acquired resistance to targeted cancer therapies. Future oncology. 2012; 8:999–1014.
4. Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR, Fidias P, Lynch TJ, Rabindran SK, McGinnis JP, Wissner A, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A. 2005; 102:7665–7670.
5. Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS medicine. 2005; 2:e73.
6. Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005; 352:786–792.
7. Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, Yuan J, Kovats SG, Kim S, Cooke VG, Monahan JE, Stegmeier F, Roberts TM, et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer discovery. 2013; 3:1030–1043.
8. Balbas MD, Evans MJ, Hosfield DJ, Wongvipat J, Arora VK, Watson PA, Chen Y, Greene GL, Shen Y, Sawyers CL. Overcoming mutation-based resistance to antiandrogens with rational drug design. eLife. 2013; 2:e00499.
9. Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, Moon M, Maneval EC, Chen I, Darimont B, Hager JH. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer discovery. 2013; 3:1020–1029.
10. Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, Lotan TL, Zheng Q, De Marzo AM, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014; 371:1028–1038.
11. Lin TH, Izumi K, Lee SO, Lin WJ, Yeh S, Chang C. Anti-androgen receptor ASC-J9 versus anti-androgens MDV3100 (Enzalutamide) or Casodex (Bicalutamide) leads to opposite effects on prostate cancer metastasis via differential modulation of macrophage infiltration and STAT3-CCL2 signaling. Cell death & disease. 2013; 4:e764.
12. Izumi K, Fang LY, Mizokami A, Namiki M, Li L, Lin WJ, Chang C. Targeting the androgen receptor with siRNA promotes prostate cancer metastasis through enhanced macrophage recruitment via CCL2/CCR2-induced STAT3 activation. EMBO molecular medicine. 2013; 5:1383–1401.
13. Lin TH, Lee SO, Niu Y, Xu D, Liang L, Li L, Yeh SD, Fujimoto N, Yeh S, Chang C. Differential androgen deprivation therapies with anti-androgens casodex/bicalutamide or MDV3100/Enzalutamide versus anti-androgen receptor ASC-J9(R) Lead to promotion versus suppression of prostate cancer metastasis. J Biol Chem. 2013; 288:19359–19369.
14. Zhang J, Patel L, Pienta KJ. CC chemokine ligand 2 (CCL2) promotes prostate cancer tumorigenesis and metastasis. Cytokine Growth Factor Rev. 2010; 21:41–48.
15. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 2009; 29:313–326.
16. Sharma J, Gray KP, Harshman LC, Evan C, Nakabayashi M, Fichorova R, Rider J, Mucci L, Kantoff PW, Sweeney CJ. Elevated IL-8, TNF-alpha, and MCP-1 in men with metastatic prostate cancer starting androgen-deprivation therapy (ADT) are associated with shorter time to castration-resistance and overall survival. Prostate. 2014; 74:820–828.
17. Davis JS, Nastiuk KL, Krolewski JJ. TNF is necessary for castration-induced prostate regression, whereas TRAIL and FasL are dispensable. Mol Endocrinol. 2011; 25:611–620.
18. Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, Yen CA, Schmitt AD, Espinoza CA, Ren B. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013; 503:290–294.
19. Szlosarek PW, Grimshaw MJ, Kulbe H, Wilson JL, Wilbanks GD, Burke F, Balkwill FR. Expression and regulation of tumor necrosis factor alpha in normal and malignant ovarian epithelium. Mol Cancer Ther. 2006; 5:382–390.
20. Jung H, Miller RJ. Activation of the nuclear factor of activated T-cells (NFAT) mediates upregulation of CCR2 chemokine receptors in dorsal root ganglion (DRG) neurons: a possible mechanism for activity-dependent transcription in DRG neurons in association with neuropathic pain. Molecular and cellular neurosciences. 2008; 37:170–177.
21. Twining CM, Sloane EM, Milligan ED, Chacur M, Martin D, Poole S, Marsh H, Maier SF, Watkins LR. Peri-sciatic proinflammatory cytokines, reactive oxygen species, and complement induce mirror-image neuropathic pain in rats. Pain. 2004; 110:299–309.
22. Milligan ED, Twining C, Chacur M, Biedenkapp J, O’Connor K, Poole S, Tracey K, Martin D, Maier SF, Watkins LR. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J Neurosci. 2003; 23:1026–1040.
23. Chen YM, Chiang WC, Lin SL, Wu KD, Tsai TJ, Hsieh BS. Dual regulation of tumor necrosis factor-alpha-induced CCL2/monocyte chemoattractant protein-1 expression in vascular smooth muscle cells by nuclear factor-kappaB and activator protein-1: modulation by type III phosphodiesterase inhibition. J Pharmacol Exp Ther. 2004; 309:978–986.
24. Thalmann GN, Anezinis PE, Chang SM, Zhau HE, Kim EE, Hopwood VL, Pathak S, von Eschenbach AC, Chung LW. Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res. 1994; 54:2577–2581.
25. Wu HC, Hsieh JT, Gleave ME, Brown NM, Pathak S, Chung LW. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: role of bone stromal cells. Int J Cancer. 1994; 57:406–412.
26. Webber MM, Trakul N, Thraves PS, Bello-DeOcampo D, Chu WW, Storto PD, Huard TK, Rhim JS, Williams DE. A human prostatic stromal myofibroblast cell line WPMY-1: a model for stromal-epithelial interactions in prostatic neoplasia. Carcinogenesis. 1999; 20:1185–1192.
27. Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, Tada K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int J Cancer. 1980; 26:171–176.
28. Arefieva TI, Kukhtina NB, Antonova OA, Krasnikova TL. MCP-1-stimulated chemotaxis of monocytic and endothelial cells is dependent on activation of different signaling cascades. Cytokine. 2005; 31:439–446.
29. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, Li MO. The cellular and molecular origin of tumor-associated macrophages. Science. 2014; 344:921–925.
30. Kaymakcalan Z, Sakorafas P, Bose S, Scesney S, Xiong L, Hanzatian DK, Salfeld J, Sasso EH. Comparisons, of, affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin Immunol. 2009; 131:308–316.
31. Jiao J, Wang S, Qiao R, Vivanco I, Watson PA, Sawyers CL, Wu H. Murine cell lines derived from Pten null prostate cancer show the critical role of PTEN in hormone refractory prostate cancer development. Cancer Res. 2007; 67:6083–6091.
32. Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997; 385:729–733.
33. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012; 487:239–243.
34. Yu YP, Landsittel D, Jing L, Nelson J, Ren B, Liu L, McDonald C, Thomas R, Dhir R, Finkelstein S, Michalopoulos G, Becich M, Luo JH. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004; 22:2790–2799.
35. Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ, Kincead-Beal C, Kulkarni P, Varambally S, Ghosh D, Chinnaiyan AM. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007; 9:166–180.
36. Fradet V, Lessard L, Begin LR, Karakiewicz P, Mes-Masson A-M, Saad F. NFkB nuclear localization is predictive of recurrence in patients with positive margin prostate cancer. Clin Cancer Res. 2004; 10:8460–8464.
37. Jin RJ, Lho Y, Connelly L, Wang Y, Yu X, Saint Jean L, Case TC, Ellwood-Yen K, Sawyers CL, Bhowmick NA, Blackwell TS, Yull FE, Matusik RJ. The nuclear factor-kappaB pathway controls the progression of prostate cancer to androgen-independent growth. Cancer Res. 2008; 68:6762–6769.
38. Jin R, Sterling JA, Edwards JR, DeGraff DJ, Lee C, Park SI, Matusik RJ. Activation of NF-kappa B signaling promotes growth of prostate cancer cells in bone. PLoS One. 2013; 8:e60983.
39. Tse BW, Scott KF, Russell PJ. Paradoxical roles of tumour necrosis factor-alpha in prostate cancer biology. Prostate Cancer. 2012; 2012:128965.
40. Nastiuk KL, Kim JW, Mann M, Krolewski JJ. Androgen regulation of FLICE-like inhibitory protein gene expression in the rat prostate. J Cell Physiol. 2003; 196:386–393.
41. Gao S, Wang H, Lee P, Melamed J, Li CX, Zhang F, Wu H, Zhou L, Wang Z. Androgen receptor and prostate apoptosis response factor-4 target the c-FLIP gene to determine survival and apoptosis in the prostate gland. J Mol Endocrinol. 2006; 36:463–483.
42. Nastiuk KL, Krolewski JJ. FLIP-ping out – death receptor signaling in the prostate. Cancer Biol Ther. 2008; 7:1171–1179.
43. Pickup M, Novitskiy S, Moses HL. The roles of TGFbeta in the tumour microenvironment. Nat Rev Cancer. 2013; 13:788–799.
44. Wendt MK, Tian M, Schiemann WP. Deconstructing the mechanisms and consequences of TGF-beta-induced EMT during cancer progression. Cell Tissue Res. 2012; 347:85–101.
45. Qin J, Liu X, Laffin B, Chen X, Choy G, Jeter CR, Calhoun-Davis T, Li H, Palapattu GS, Pang S, Lin K, Huang J, Ivanov I, et al. The PSA(-/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell stem cell. 2012; 10:556–569.
46. Wolf MJ, Hoos A, Bauer J, Boettcher S, Knust M, Weber A, Simonavicius N, Schneider C, Lang M, Sturzl M, Croner RS, Konrad A, Manz MG, et al. Endothelial CCR2 signaling induced by colon carcinoma cells enables extravasation via the JAK2-Stat5 and p38MAPK pathway. Cancer Cell. 2012; 22:91–105.
47. Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, Escara-Wilke J, Wilder-Romans K, Dhanireddy S, Engelke C, Iyer MK, Jing X, Wu YM, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014; 510:278–282.
48. Lu Y, Cai Z, Galson DL, Xiao G, Liu Y, George DE, Melhem MF, Yao Z, Zhang J. Monocyte chemotactic protein-1 (MCP-1) acts as a paracrine and autocrine factor for prostate cancer growth and invasion. Prostate. 2006; 66:1311–1318.
49. Pienta KJ, Machiels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, Li S, Seetharam S, Puchalski TA, Takimoto C, Elsayed Y, Dawkins F, de Bono JS. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2, in metastatic castration-resistant prostate cancer. Investigational new drugs. 2013; 31:760–768.