
INTRODUCTION
The diagnosis of “lower-grade” diffusely infiltrative glioma (WHO grade II and III) is a histologic diagnosis largely based upon neoplastic cell morphology and the presence or absence of features of anaplasia (e.g. mitoses, microvascular proliferation, and necrosis), according to the 2007 WHO Classification of Tumours of the Central Nervous System [1]. Tumors with oligodendroglial and/or astrocytic morphologies are classified as oligodendroglioma (O), oligoastrocytoma (OA), and astrocytoma (A), respectively, and graded as either II or III. There is considerable interobserver variability in assessing both morphology and grade, which has significant clinical implications [2]. In particular, the diagnosis of grade II is extremely important for current management approaches. Unfortunately, molecular diagnostic information for this strictly defined group has not been clearly delineated in recent studies, which have combined the entire spectrum of grade II and grade III gliomas [3]. In recent years, there have been major advances in our understanding of the epigenetic, genetic, and molecular alterations in glioma [4–6]. Isocitrate dehydrogenase (IDH) gene mutations, estimated to occur in 70% to 90% of diffuse lower-grade gliomas, affect the epigenetic regulation of the genome and are strongly implicated in both tumorigenesis and prognosis [3, 7–11]. Secondary analysis of clinical trial RTOG 9402 retrospectively validated IDH mutation as both a prognostic and a predictive marker of response to PCV (procarbazine, lomustine, and vincristine) chemotherapy and radiation independent of 1p19q codeletion (1p19q codel) [12, 13]. Loss of heterozygosity of 1p19q, occurring in 60% to 80% of O and up to 45% of OA [14], is an established genetic marker of glioma with predominant oligodendroglial morphology [15]. Codeletion of 1p19q has also been demonstrated to be a prognostic marker [13] as well as predictive of responsiveness to PCV chemotherapy and radiation [16, 17]. Inactivating mutations of alpha-thalassemia/mental retardation syndrome X-linked (ATRX) gene, correlated with the alternative lengthening of telomeres (ALT) phenotype, are highly associated with IDH and TP53 mutations but mutually exclusive with 1p19q codel [12, 18]. Prevalence of ATRX loss, which in some studies has been associated with favorable prognosis in anaplastic gliomas, ranges from 33% to 72% of IDH mutated noncodel gliomas [18, 19]. Mutation of the tumor suppressor gene TP53, frequently resulting in p53 protein overexpression, is also mutually exclusive with 1p19q codel [20, 21]. Approximately 60% of diffuse A and 40% of OA carry TP53 mutation [20].
Here, we report the frequency of IDH mutations, 1p19q codeletion, ATRX expression loss, and p53 overexpression in strictly defined WHO grade II O, OA, and A, treated at the Mayo Clinic over a 10-year period (2003–2012) and define their diagnostic and prognostic value.
RESULTS
Clinical data
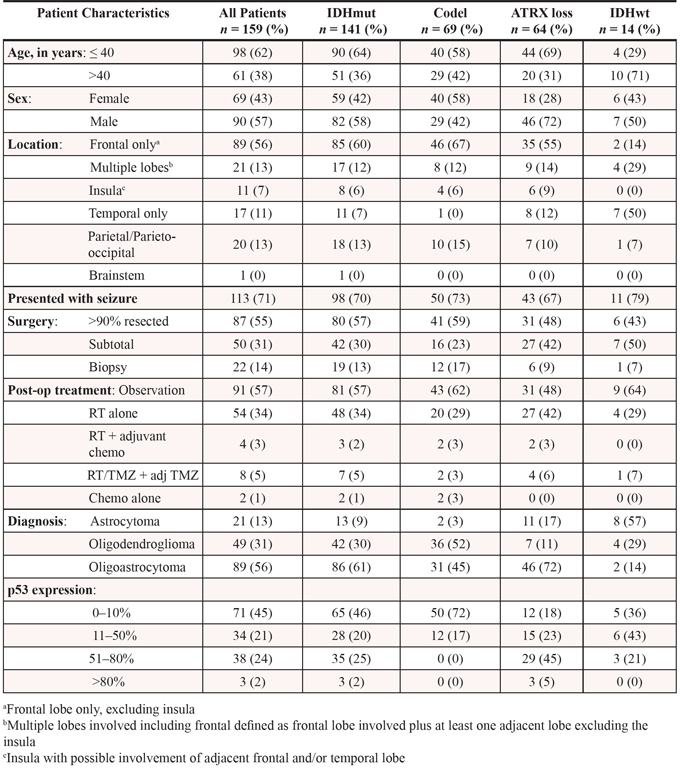
Patient characteristics, treatment history, and pathologic data are summarized in Table 1. The median age of all patients was 37 years (interquartile range 29–46), ranging from 16 to 84 years of age. There were 70 females and 89 males. The median follow-up in subjects not known to be deceased is 4 years (range: 4 days to 23.6 years). A total of 31 patients were known to be deceased due to tumor. The majority (87 of 159) (55%) had >90% resection of tumor as defined by T2/FLAIR hyperintensity, while 50 (31%) had subtotal resection, and 22 (14%) had stereotactic biopsy only. Most patients (81 of 159) (57%) were observed after initial surgery, and 54 (34%) had radiation without chemotherapy after initial surgery. Only 14 (9%) patients received upfront chemotherapy, 12 of whom also received radiation.
Table 1: Patient clinical data, treatment history and pathology
Molecular marker results
The results of the IDH, 1p19q, ATRX testing are summarized in Figures 1 and 2 as well as Table 1.
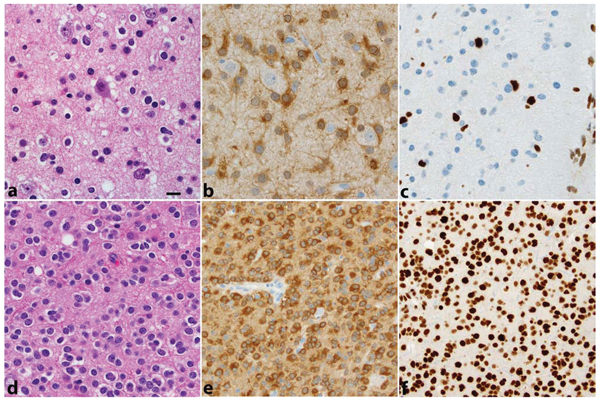
Figure 1: Two WHO grade II gliomas are illustrated. The first a. diagnosed as oligoastrocytoma displays infiltration of cortex by a low-density population of cells, in part, with round regular nuclei and clear halos (“oligodendroglial”), in part, with irregular elongated nuclei (“astrocytic”). The cells are IDH1R132H positive b. and lack ATRX expression c. In the second d. diagnosed as oligodendroglioma, tumor cells are IDH1R132H positive e. and maintain ATRX expression f. This second tumor showed 1p19q codeletion by FISH. All photos were taken at 400 × (magnification bar shown in panel a 10 microns).
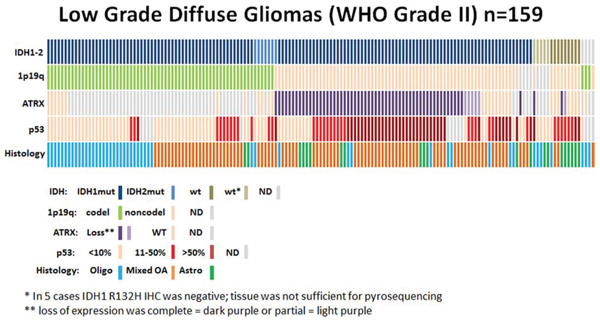
Figure 2: Low grade diffuse gliomas. Data regarding IDH status, 1p19q codeletion, ATRX status, p53 expression, and histological diagnosis are summarized for all patients (n = 159). Each column represents a patient.
IDH mutations: An IDH mutation was present in 141 of 155 (91%): 127 (90%) R132H mutations, 14 (10%) rare mutations including 2 R132C, 2 R132G, 1 R132S, 2 R132 unknown, 3 R172K, 1 R172H, and 3 R172 unknown mutations. Of 14 negative cases, 5 were tested only by IDH1R132H IHC due to insufficient tissue, and 9 were negative by both IHC and IDH1-IDH2 pyrosequencing. In the last 4 (of 159) tumors, sufficient tissue for testing was unavailable.
1p19q codeletion: Codeletion of 1p19q was present in 69 of 151 (46%) tumors tested: 2 of 18 (11%) A; 31 of 86 (36%) OA; and 36 of 47 (77%) O. Of the remaining 8 (of 159) tumors, 2 failed testing and 6 had insufficient tissue. All 1p19q codel tumors (n = 69) were IDH mutant (other than 3 tumors in which IDH status could not be determined).
ATRX loss: ATRX expression was lost in 61 of 70 (87%) IDHmut-noncodel tumors and 3 of 14 (21%) IDHwt tumors. Among the histologic diagnoses, ATRX expression was lost in 11 of 16 (69%) of A, 46 of 60 (77%) of OA, and 7 of 17 (41%) of O. None of the 12 IDHmut-codel tumors tested had ATRX loss. Nine cases had either insufficient tissue (n = 6) or failed testing (n = 3).
Expression of p53: Among the 66 IDHmut-codel tumors tested, p53 expression was limited to <10% of the cells in 48 (79%); and in the remaining 13 (21%), p53 expression was <50%. Of the 55 IDHmut-noncodel-ATRXloss tumors tested, 31 (56%) had p53 expression >50%. Indeed, the only 3 tumors with p53 expression >80% were also IDH mut-noncodel-ATRX loss. Among the 9 IDHmut-noncodel-ATRXwt tumors, p53 expression was <10% in 2 (22%), < 50% in 2 (22%), and >50% in 5 (56%). Of the 14 IDHwt tumors, p53 expression was <10% in 5 (36%), <50% in 6 (43%), and >50% in 3 (21%).
Four subgroups were identified: IDHmut-codel in 66 of 155 (43%), IDHmut-noncodel-ATRX loss in 60 (39%), IDHmut-noncodel-ATRXwt in 9 (6%), and 14 IDHwt (9%) (Figure 2). All histological diagnoses were found in each of the four subgroups, although O was most frequent among IDHmut-codel [33 of 66 (50%)], OA among IDHmut-ATRX loss [46 of 61 (75%)], and A among IDHwt [8 of 14 (57%)]. Among the 37 previously published [4], 17 were IDHmut-codel, 16 were IDHmut-noncodel-ATRX loss, and 4 were IDHmut-noncodel-ATRXwt. The median age of patients with IDH mutation was 36 years of age compared to 44 years of age for those with IDHwt tumors (p = 0.0554). There were no significant age differences among the remaining subgroups.
Clinical outcome
These patients presented with seizures 71% of the time, regardless of molecular group or tumor location. Frontal lobe presentation in isolation at the poles or convexity, or also involving insula or temporal lobe, occurred in 110 (78%) in those with IDHmut, 58 (84%) of those with 1p/19q codel, but only 6 (46%) IDHwt. Conversely, tumor restricted to the temporal lobe almost never occurs in 1p/19q codel patients but occurred in 7 (50%) of those IDHwt. Parieto-occipital lobes were involved primarily in 18 (13%) of IDHmut cases and less frequently (n = 1, 7%) in IDHwt cases. Despite these differences, in our fairly uniform neurosurgical practice inclined to maximally safe resection, there was no statistically significant difference in extent of resection achieved between any of the molecular groups. Specific pairwise comparisons of tumor locations and extent of resection by location between groups was not possible because of small numbers in some groups.
The median PFS and the median overall survival (OS) for the collective cohort is 5 years and 15.3 years, respectively (Figure 3A, 3B). There was no difference in either PFS or OS among the three histologies by Kaplan-Meier method, adjusted for patient and treatment characteristics (Figure 4A and 4C). There was a significant difference in OS (p = 0.038) but not in PFS (p = 0.067), comparing the four molecular subgroups, adjusting for age, gender, EOR, histologic diagnosis, and treatment (Figure 4B, 4D). Due to insufficient sample size, we lacked statistical power to test for significant differences in pairwise comparison. The IDHmut-codel subgroup had the longest median PFS (5.6 years) and OS (15.3 years); and OS was significantly longer compared to the other three molecular groups combined (p = 0.025). The IDHmut-noncodel-ATRX loss subgroup median PFS was 4.4 years, and median OS was 12.7 years compared to the IDHmut-noncodel-ATRXwt subgroup median PFS of 2.2 years and median OS of 6.9 years. Median PFS and OS for the IDHwt subgroup were not achieved; however, the 5-year estimate for PFS is 51% (95% CI: 29%–93%), and OS is 71% (95% CI: 47%–100%).
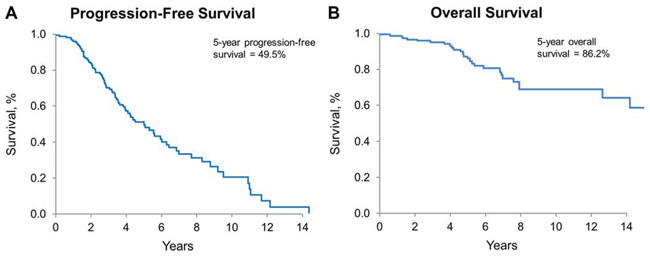
Figure 3: Progression-free and overall survival for all patients. A. PFS for all patients, estimated by Kaplan-Meier method. 5-year PFS: 49.5% (95% CI: 40.6%–60.4%). B. OS for all patients, estimated by Kaplan-Meier method; 5-year OS: 86.2% (95% CI: 79.4%–93.5%)
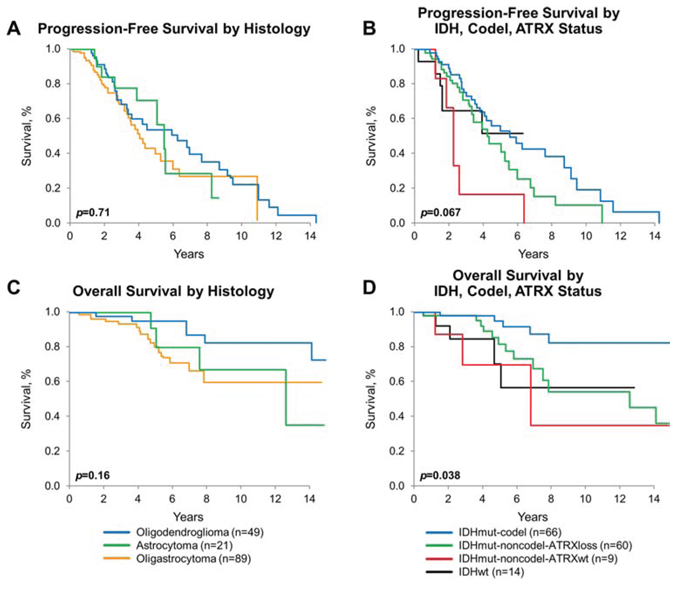
Figure 4: Progression-free and overall survival by diagnosis and molecular subgroups. A. PFS by histologic diagnosis, estimated by Kaplan-Meier method, p = 0.71. B. OS by histologic diagnosis, estimated by Kaplan-Meier method, p = 0.16. C. PFS by molecular subgroup [IDHmut-codel (n = 65), IDHmut-noncodel-ATRXloss (n = 59), IDHmut-noncodel-ATRXwt (n = 9), IDHwt (n = 14)], estimated by Kaplan-Meier method, p = 0.067. D. OS by molecular subgroup [IDHmut-codel (n = 66), IDHmut-noncodel-ATRXloss (n = 60), IDHmut-noncodel-ATRXwt (n = 9), IDHwt (n = 14)], estimated by Kaplan-Meier method, p = 0.038. All p-values adjust for age, sex, histomorphologic diagnosis, extent of resection, and treatment.
For the entire cohort, OS, but not PFS, was significantly improved for those who had gross total resection (GTR) when compared to those who had only biopsy (p = 0.042). Outcome for subtotal resection was not significantly different from those with biopsy only (Figure 5A and 5B). Findings were similar when the analysis was limited to any IDH mutant tumor (data not shown). Sample size precluded testing outcome for each molecular group stratified by EOR.
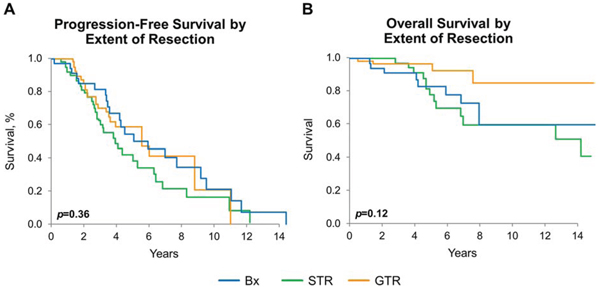
Figure 5: Progression-free and overall survival by extent of resection. A. PFS by extent of resection estimated by Kaplan-Meier method, p = 0.36. B. OS by extent of resection, estimated by Kaplan-Meier method: entire cohort p = 0.12, pairwise GTR vs. biopsy-only: p = 0.042. p-values adjust for age, sex, histomorphologic diagnosis, and treatment.
DISCUSSION
The WHO classification of diffuse gliomas is based on a wealth of clinicopathologic data; however, subjectivity complicates its uniform application. With long-term outcome data from EORTC 26951 and RTOG 9402, the predictive and prognostic significance of 1p19q codel as well as IDH mutation is established in anaplastic gliomas [16, 17]. Recently, ATRX, involved in the regulation of telomeres, was also found to be prognostically important in anaplastic gliomas [19]. Overexpression of p53 is commonly used in clinical practice, although its correlation with TP53 mutation status is far from perfect [21]. The value of these three markers in strictly defined grade II gliomas has yet to be fully established [22, 23]. Towards this end, by using techniques commonly available in clinical laboratories, we analyzed the frequency of alterations in IDH, 1p19q, ATRX, and p53 expression in a uniformly managed cohort of Mayo Clinic patients with strictly defined WHO grade II diffuse gliomas and compared clinical outcomes adjusted for patient and treatment factors.
We chose to test for IDH mutation first by IHC, followed by pyrosequencing in IHC-negative cases as this test sequence is very efficient in clinical practice. IDH1R132H IHC not only allows rapid detection of the most common mutation of IDH, accounting for approximately 90% of the IDH mutations, but in addition can detect even single tumor cells below the density threshold necessary for IDH1-IDH2 pyrosequencing [24]. In this series, all tumors were tested for 1p19q codeletion by FISH, which has been, up to now, widely popular in most clinical laboratories. Evidence has accumulated that FISH is an imperfect technique to test for 1p19q codeletion with a risk of false-positive results as it may not distinguish between partial/interstitial deletions and whole-arm deletions as the length of DNA covered by FISH probes is limited [25], a phenomenon which, in our experience, is uncommon and most frequently seen in high-grade gliomas. In the future, it is likely that laboratories will use techniques for 1p/19q analysis that more extensively ‘scan’ for presence/absence of loss of the relevant chromosome arms. In our own clinical practice, we are in the process of converting the assessment of 1p19q from FISH to aCGH.
Previous investigations of the frequencies of molecular and genetic alterations in low-grade gliomas disclosed IDH mutations in as many as 85% [14], 1p19q codeletion in up to 80% of O [26], and ATRX loss as high as 70% to 72% in IDH mut-noncodel tumors [12, 18]. We identified in 21 A, 49 O, and 89 OA alteration rates of 91% in IDH, 46% in 1p19q loss, and among IDHmut-noncodel tumors, 85% prevalence of ATRX loss. No instance of ATRX loss was found in the 12 of 66 IDHmut-codel tumors tested, and only 7 of the 64 ATRX loss tumors were O, consistent with prior correlations of ATRX loss with astrocytic and oligoastrocytic morphology [20, 21]. The frequency of ATRX loss in 3 (of 14) IDHwt tumors (21%), 2% of the entire cohort, is similar to previously reported ATRX loss in tumors which are not IDHmut-noncodel [19].
Our study confirms that ATRX loss is highly correlated with IDH mutation. The high rate of ATRX alteration in our strictly defined grade II IDHmut-noncodel tumor cohort supports the use of ATRX as a discriminating molecular marker in clinical practice, together with IDH testing.
We demonstrate that IDH mutation, 1p19q codeletion, and ATRX loss, considered together, may better define the clinical course than histologic diagnosis alone, after adjusting for age, EOR, and treatment. While better prognosis of patients with IDH mutations and 1p19q codeletion is well-established [3, 12, 16, 17], ATRX loss has only recently been of clinical research interest. Several studies have reported ATRX loss in grade II and III IDHmut-noncodel tumors to be prognostically favorable [18, 19]. Conversely, in this series of strictly defined grade II gliomas, IDHwt and IDHmut-non-codel-ATRXwt subgroups do somewhat more poorly, particularly in terms of relatively short PFS, but OS is much better than glioblastoma, as might be surmised from combined grade II and grade III glioma cohorts [27]. It is likely that additional studies will define markers more capable of identifying the most aggressive grade II lesions yet, even within the four groups we have identified.
There was no difference in frequency of seizure presentation between the groups. There was no difference in EOR achieved between any of the molecular groups. There are differences in the location frequencies between the groups [27], but location did not influence the extent of resection. In this cohort of relatively young patients with minimal comorbidities, the majority of whom were operated in a surgical practice inclined toward maximally safe resections incorporating image-guided techniques and awake craniotomy, extent of resection can be confidently assumed to be related to tumor volume and/or involvement in eloquent brain. Realizing these confounders, we confirm observations from other series that GTR (in this series, meaning >90% of all MRI defined T2 signal abnormality) resulted in improved survival, at least when compared to biopsy alone [28, 29]. However, having shown that extent of resection was not significantly different between the molecular groups, yet showing OS differences between these groups, our results from a relatively uniformly managed group of patients clearly suggest that the biology of the groups is important, independent of initial surgical management.
IDHwt is relatively uncommon in strictly defined grade II glioma, 9%(14 patients) in this series. Here, classic astrocytoma morphology predominates, but it remains for a much larger, well-annotated study of these tumors to demonstrate whether or not prognostically different molecular subtypes can be defined within this group and whether or not clinical and treatment factors influence their outcomes.
Despite the retrospective nature of the study, our cohort has the unique advantage of treatment uniformity and long-term follow-up. The majority of patients with >90% EOR were observed closely and received radiation therapy at first recurrence, while most patients with subtotal resection had adjuvant radiation. In this strictly defined grade II glioma cohort, we demonstrate that IDHmut, 1p19q codel, and ATRX loss, considered together, are prognostically useful. Molecular groups defined by combinations of these markers should be incorporated in the neuropathologic evaluation.
MATERIALS AND METHODS
The Mayo Clinic Brain SPORE (Specialized Program of Research Excellence) Registry was queried to identify patients aged 16 years or older with a tissue diagnosis of WHO grade II O, OA, or A between January 1, 2003, and December 31, 2012. A total of 159 patients who received care at Mayo Clinic were identified with tissue available for review and appropriate consent. Seven patients had resection at Mayo Clinic Rochester for a WHO grade II glioma prior to 2003, dating 1992 at the earliest. Thirty-two patients had previous resection elsewhere dating to 1990 at the earliest. Slides from these previous resections were reviewed by a Mayo neuropathologist and diagnosed as WHO grade II glioma. Additional studies (IHC, 1p19q FISH) were obtained at Mayo as needed. Their pertinent clinical data was abstracted from the electronic medical record including: 1) extent of resection (EOR) as determined by surgeon and corroborated by treating oncologist and interpreting neuroradiologist, 2) dates of tumor progression as determined by treating oncologist, 3) and dates of most recent MRI documenting tumor stability. Institutional Review Board approval was obtained. Data from 37 patients have been previously published [4].
All cases were primarily diagnosed by a neuropathologist, applying strict uniform diagnostic criteria, including: low to moderate cellularity defined as cellularity of tumors in which cell density is such that background infiltrated parenchyma is easily recognizable even in low-intermediate power (x10–x20), in contrast to high cellularity in which cell density is such that tumor cells are tightly packed touching each other (intervening parenchyma is present but its recognition requires intermediate-high power resolution x20–x40; e.g. to recognize entrapped neurons); low mitotic index, which differs between astrocytic and oligodendroglial tumors. In WHO grade II astrocytoma, no mitotic activity should be seen, although a single mitosis was not considered sufficient for a grade III a designation [30]. In WHO grade II oligodendroglioma and oligoastrocytoma, mitotic index should be <6 per 10 high-power fields (HPF), a threshold we established and [31] has been widely used in the neuropathology community and referenced in the WHO 2007. Grade II astrocytoma (WHO) were typically low to moderately cellular tumors with no or, at most, a rare mitotic figure. Grade II oligodendroglioma and oligoastrocytoma (WHO) were low to moderately cellular tumors with <6 mitoses per 10 HPF. Neither microvascular proliferation nor necrosis were present [31]. With only three neuropathologists on service over the time period involved, one of which trained under the other two, diagnostic criteria and their application were very uniform, as shown by the high intraclass correlation coefficient (“substantial to almost perfect”) observed among two (of three) neuropathologists on all grading criteria in oligodendroglioma [31]. At the time of this study, histology review was conducted by a single neuropathologist (CG) to confirm the diagnosis and grade as well as to assess tumoral tissue markers. The following markers were evaluated in each case: p53 protein and ATRX expression by immunohistochemistry (IHC); IDH mutation status by IDH1 R132H immunohistochemistry followed by IDH1-IDH2 pyrosequencing in all IHC-negative cases, 1p19q status by FISH. Clinical testing of codel by FISH has been in use since 2003, while IDH1R132H IHC was implemented in 2011, and IDH1-IDH2 pyrosequencing in 2012. Immunohistochemistry for p53 protein has been routinely used for many years. Thus, within our grade II glioma cohort, 1p19q status was known in 128 cases, IDH mutation status in 37, and p53 expression had been tested in 71. Analysis was completed in all cases lacking one or more of the tissue markers (p53 expression, IDH status, and 1p19q status) if sufficient tissue was available from the primary tumor resection (p53 expression) and/or from subsequent tumor resections (IDH, ATRX and 1p19q status). Since 1p19q codeletion, IDH and ATRX mutation are early genomic alterations, typically shared between primary and recurrent tumors, testing of recurrent tumors should not have significantly affected the results of our series. 1p19q status was assessed in the primary tumor resection in 142 (of 151) cases (94%) and IDH status in 128 (of 155) cases (82%) while in remaining cases was assessed on tumor from subsequent resections. Mutations of IDH were evaluated by immunohistochemistry using the monoclonal antibody to IDH1R132H H09 and by pyrosequencing in immunonegative cases as recently described [32]. IDH1R132H IHC was scored as positive (consistent with IDH1 R132H mutation) or negative. IDH1-IDH2 pyrosequencing is designed to detect IDH1 R132C, R132G, R132H, R132L, R132P, and R132S and IDH2 R172G, R172K, and R172M mutations, while IDH1 R132V and R100Q and IDH2 R172S, R172T and R172W, which occur at very low frequency, are not detected. In 20 cases, testing was performed using a recently developed PCR assay for one-step detection of 12 IDH1/IDH2 mutations, which allows for identification of 12 IDH1-IDH2 mutations, including IDH1 R100 mut, three among which are specifically identified (IDH1 R132H, IDH1 R132C and IDH2 R172K), while the others are reported as “unknown” [33]. FISH for 1p19q was performed according to previously described methods [15]. IHC stain for p53 protein was performed according to standard techniques. Positive p53 staining was used as a surrogate for altered TP53 function as previously described [34]. Percentage of cells displaying strong nuclear staining was scored semi-quantitatively as 0 to 10% of tumor cell, 11 to 50%, 51 to 80%, or >80%.
ATRX expression was evaluated in 93 tumors, including all IDHmut-noncodel tumors with sufficient tumor tissue from either the original and/or a subsequent tumor surgery and a small number (n = 12) of 1p19q codel tumors. ATRX expression was assessed in the primary tumor resection in 75 (of 93) cases (81%) and in tumor from subsequent resections in remaining cases [35]. IHC was performed using the monoclonal antibody anti-ATRX (clone D5) 1:1000 dilution of primary antibody (Santa Cruz, catalog # sc-55584), performed on the Ventana BenchMark XT stainer with the following conditions: CC1 for 32 min as pretreatment, primary antibody incubation time of 16 minutes at 37°C, Optiview detection, followed by Ventana DAB, 8 minutes hematoxylin and 4 minutes Bluing Reagent. ATRX expression was either retained or lost in tumor cells (Figure 1). In a few cases (n = 6), the number of ATRX-negative cells seemed to be focally less than the number of atypical tumor cells identified morphologically and by IDH1 R132H IHC stain (5 cases were IDH1R132H IHC positive). These were areas in which vascular cells and neurons stained positively, excluding that the lack of stain could be artifactual. We originally tracked these cases as “partial ATRX loss,” since when we reviewed the ATRX stain we were blind to the 1p19q results. We wanted to exclude that these cases may represent possible rare examples of “true mixed gliomas with molecular features of composite oligodendroglioma and astrocytoma [36, 37].” The cases were 1p19q intact and were grouped with the ATRX loss cases.
Statistical analyses
Group comparisons of patient, surgical, and tumor characteristics were made using the chi-square test for categorical variables and the Kruskal-Wallis test for continuous variables. Cumulative survival probabilities were estimated using the Kaplan-Meier method. Cox proportional hazards regression was used to assess the association of patient, surgical, and tumor characteristics with overall and progression-free survival (PFS). All analyses included age, sex, histomorphologic diagnosis, extent of resection, radiation therapy, and chemotherapy as covariates. In all cases, p-values < 0.05 were considered statistically significant.
ACKNOWLEDGMENTS
We thank Helene Peyro-Saint-Paul from QIAGEN for providing a kit to complete IDH mutation analysis in this patient cohort. We thank Denise Chase (Mayo Clinic) for the manuscript preparation.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
GRANT SUPPORT
CTSA Grant (UL1 TR000135) from the National Center for Advancing Translational Sciences (NCATS), a component of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NIH. Work at the Mayo Clinic was supported, in part, by the National Institutes of Health (P50CA108961).
REFERENCES
1. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007; 114:97–109.
2. van den Bent MJ. Interobserver variation of the histopathological diagnosis in clinical trials on glioma: a clinician’s perspective. Acta Neuropathol. 2010; 120:297–304.
3. Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F, El Hallani S, Boisselier B, Mokhtari K, Hoang-Xuan K, Delattre JY. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol. 2009; 27:4150–4.
4. Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, Pekmezci M, Rice T, Kosel ML, Smirnov IV, Sarkar G, Caron AA, Kollmeyer TM, et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N Engl J Med. 2015; 372:2499–508.
5. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med. 2015; 372:2481–98.
6. Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA, Jr., Friedman AH, Friedman H, Gallia GL, Giovanella BC, Grollman AP, He TC, He Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013; 110:6021–6.
7. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009; 360:765–73.
8. Watanabe T, Nobusawa S, Kleihues P, Ohgaki H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol. 2009; 174:1149–53.
9. Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, Felsberg J, Wolter M, Mawrin C, Wick W, Weller M, Herold-Mende C, Unterberg A, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 2009; 118:469–74.
10. Duncan CG, Barwick BG, Jin G, Rago C, Kapoor-Vazirani P, Powell DR, Chi JT, Bigner DD, Vertino PM, Yan H. A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genome Res. 2012; 22:2339–55.
11. Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010; 17:510–22.
12. Cairncross JG, Wang M, Jenkins RB, Shaw EG, Giannini C, Brachman DG, Buckner JC, Fink KL, Souhami L, Laperriere NJ, Huse JT, Mehta MP, Curran WJ, Jr. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J Clin Oncol. 2014; 32:783–90.
13. Erdem-Eraslan L, Gravendeel LA, de Rooi J, Eilers PH, Idbaih A, Spliet WG, den Dunnen WF, Teepen JL, Wesseling P, Sillevis Smitt PA, Kros JM, Gorlia T, van den Bent MJ, et al. Intrinsic molecular subtypes of glioma are prognostic and predict benefit from adjuvant procarbazine, lomustine, and vincristine chemotherapy in combination with other prognostic factors in anaplastic oligodendroglial brain tumors: a report from EORTC study 26951. J Clin Oncol. 2013; 31:328–36.
14. Kim YH, Nobusawa S, Mittelbronn M, Paulus W, Brokinkel B, Keyvani K, Sure U, Wrede K, Nakazato Y, Tanaka Y, Vital A, Mariani L, Stawski R, et al. Molecular classification of low-grade diffuse gliomas. Am J Pathol. 2010; 177:2708–14.
15. Jenkins RB, Blair H, Ballman KV, Giannini C, Arusell RM, Law M, Flynn H, Passe S, Felten S, Brown PD, Shaw EG, Buckner JC. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. 2006; 66:9852–61.
16. van den Bent MJ, Brandes AA, Taphoorn MJ, Kros JM, Kouwenhoven MC, Delattre JY, Bernsen HJ, Frenay M, Tijssen CC, Grisold W, Sipos L, Enting RH, French PJ, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol. 2013; 31:344–50.
17. Cairncross G, Wang M, Shaw E, Jenkins R, Brachman D, Buckner J, Fink K, Souhami L, Laperriere N, Curran W, Mehta M. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol. 2013; 31:337–43.
18. Kannan K, Inagaki A, Silber J, Gorovets D, Zhang J, Kastenhuber ER, Heguy A, Petrini JH, Chan TA, Huse JT. Whole-exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget. 2012; 3:1194–203.
19. Wiestler B, Capper D, Holland-Letz T, Korshunov A, von Deimling A, Pfister SM, Platten M, Weller M, Wick W. ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol. 2013; 126:443–51.
20. Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF, Rodriguez FJ, Rosemberg S, Oba-Shinjo SM, Nagahashi Marie SK, Bettegowda C, Agrawal N, Lipp E, et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget. 2012; 3:709–22.
21. James CD, Galanis E, Frederick L, Kimmel DW, Cunningham JM, Atherton-Skaff PJ, O’Fallon JR, Jenkins RB, Buckner JC, Hunter SB, Olson JJ, Scheithauer BW. Tumor suppressor gene alterations in malignant gliomas: histopathological associations and prognostic evaluation. Int J Oncol. 1999; 15:547–53.
22. Buckner JC, Pugh SL, Shaw EG, Gilbert MR, Barger G, Coons S, Ricci P, Bullard D, Brown PD, Stelzer K, Brachman D, Suh JH, Schultz CJ, et al. Phase III study of radiation therapy (RT) with or without procarbazine, CCNU, and vincristine (PCV) in low-grade glioma: RTOG 9802 with Alliance, ECOG, and SWOG (Abstract 2000). ASCO MEETING ABSTRACTS. 2014.
23. Louis DN, Perry A, Burger P, Ellison DW, Reifenberger G, von Deimling A, Aldape K, Brat D, Collins VP, Eberhart C, Figarella-Branger D, Fuller GN, Giangaspero F, et al. International Society Of Neuropathology—Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol. 2014; 24:429–35.
24. Horbinski C. What do we know about IDH1/2 mutations so far, and how do we use it? Acta Neuropathol. 2013; 125:621–36.
25. Horbinski C, Nikiforova MN, Hobbs J, Bortoluzzi S, Cieply K, Dacic S, Hamilton RL. The importance of 10q status in an outcomes-based comparison between 1p/19q fluorescence in situ hybridization and polymerase chain reaction-based microsatellite loss of heterozygosity analysis of oligodendrogliomas. J Neuropathol Exp Neurol. 2012; 71:73–82.
26. Smith JS, Perry A, Borell TJ, Lee HK, O’Fallon J, Hosek SM, Kimmel D, Yates A, Burger PC, Scheithauer BW, Jenkins RB. Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. J Clin Oncol. 2000; 18:636–45.
27. Gorovets D, Kannan K, Shen R, Kastenhuber ER, Islamdoust N, Campos C, Pentsova E, Heguy A, Jhanwar SC, Mellinghoff IK, Chan TA, Huse JT. IDH mutation and neuroglial developmental features define clinically distinct subclasses of lower grade diffuse astrocytic glioma. Clin Cancer Res. 2012; 18:2490–501.
28. Schomas DA, Laack NN, Rao RD, Meyer FB, Shaw EG, O’Neill BP, Giannini C, Brown PD. Intracranial low-grade gliomas in adults: 30-year experience with long-term follow-up at Mayo Clinic. Neuro Oncol. 2009; 11:437–45.
29. Smith JS, Chang EF, Lamborn KR, Chang SM, Prados MD, Cha S, Tihan T, Vandenberg S, McDermott MW, Berger MS. Role of extent of resection in the long-term outcome of low-grade hemispheric gliomas. J Clin Oncol. 2008; 26:1338–45.
30. Giannini C, Scheithauer BW, Burger PC, Christensen MR, Wollan PC, Sebo TJ, Forsyth PA, Hayostek CJ. Cellular proliferation in pilocytic and diffuse astrocytomas. J Neuropathol Exp Neurol. 1999; 58:46–53.
31. Giannini C, Scheithauer BW, Weaver AL, Burger PC, Kros JM, Mork S, Graeber MB, Bauserman S, Buckner JC, Burton J, Riepe R, Tazelaar HD, Nascimento AG, et al. Oligodendrogliomas: reproducibility and prognostic value of histologic diagnosis and grading. J Neuropathol Exp Neurol. 2001; 60:248–62.
32. van den Bent MJ, Hartmann C, Preusser M, Strobel T, Dubbink HJ, Kros JM, von Deimling A, Boisselier B, Sanson M, Halling KC, Diefes KL, Aldape K, Giannini C. Interlaboratory comparison of IDH mutation detection. J Neurooncol. 2013; 112:173–8.
33. Catteau A, Girardi H, Monville F, Poggionovo C, Carpentier S, Frayssinet V, Voss J, Jenkins R, Boisselier B, Mokhtari K, Sanson M, Peyro-Saint-Paul H, Giannini C. A new sensitive PCR assay for one-step detection of 12 IDH1/2 mutations in glioma. Acta Neuropathol Commun. 2014; 2:58.
34. Giannini C, Hebrink D, Scheithauer BW, Dei Tos AP, James CD. Analysis of p53 mutation and expression in pleomorphic xanthoastrocytoma. Neurogenetics. 2001; 3:159–62.
35. Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K, Asthana S, Jalbert LE, Nelson SJ, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014; 343:189–93.
36. Huse JT, Diamond EL, Wang L, Rosenblum MK. Mixed glioma with molecular features of composite oligodendroglioma and astrocytoma: a true “oligoastrocytoma”? Acta Neuropathol. 2015; 129:151–3.
37. Wilcox P, Li CC, Lee M, Shivalingam B, Brennan J, Suter CM, Kaufman K, Lum T, Buckland ME. Oligoastrocytomas: throwing the baby out with the bathwater? Acta Neuropathol. 2015; 129:147–9.