
INTRODUCTION
Ubiquitin/proteasome-mediated protein degradation represents a critical post-translational mechanism for the regulation of protein levels. This process primarily involves polyubiquitination of substrate proteins and subsequent proteolytic degradation by the macromolecular 26S proteasome complex. It has been assumed that inhibition of the proteasome causes accumulation of certain proteins deleterious to the survival of cancer cells, allowing restoration of cell cycle arrest and/or apoptotic cell death [1, 2]. Thus targeting the proteasome has emerged as a promising cancer therapeutic strategy. The successful development of bortezomib, a first-generation proteasome inhibitor, as an FDA-approved anticancer drug has spurred efforts to identify and develop second-generation proteasome inhibitors with improved selectivity and therapeutic efficacy [2, 3].
Carfilzomib (CFZ; also known as PR-171), a second-generation irreversible proteasome inhibitor, is approved by the FDA for the treatment of multiple myeloma. It is a cell-permeable tetrapeptide epoxyketone analog of epoxomicin (Figure 1A) that is structurally distinct from bortezomib and irreversibly binds to and inhibits the chymotrypsin-like site of the proteasome [4, 5]. In addition to hematologic malignancies, CFZ is also being evaluated in clinical trials against solid tumors including small-cell lung cancer, non-small cell lung cancer, refractory renal cell cancer, and metastatic prostate cancer [2].
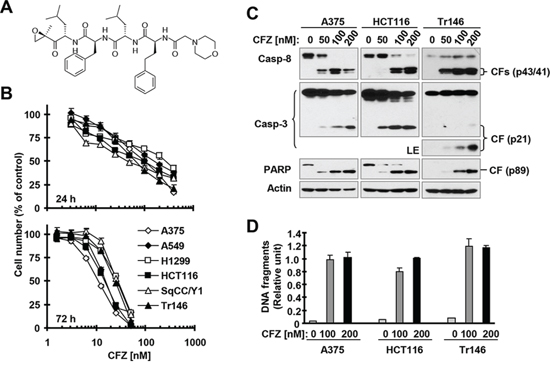
Figure 1: CFZ (A) effectively decreases the survival of cancer cells (B) and induces apoptosis (C and D) A, Chemical structure of CFZ. B, The indicated cancer cell lines were treated with different concentrations of CFZ for 24 or 72 h and then subjected to estimation of cell number with the SRB assay. The data are means ± SDs of four replicate determinations. C and D, The indicated cell lines were treated with the given concentrations of CFZ for 24 h and then subjected to preparation of whole-cell protein lysates and subsequent Western blot analysis (C) or ELISA assay to detect DNA fragments (D). The data are means ± SDs of triplicate determinations. CF, cleaved fragment; LE, long exposure.
Preclinical studies have shown that CFZ induces apoptosis in cell lines derived from multiple myeloma [6], Waldenstromʼs macroglobulinemia [7], chronic lymphocytic leukemia [8], lymphoma [9, 10], head and neck cancer [11] and lung cancer [12]. CFZ-induced apoptosis in different cancer cells has been suggested to be associated with c-Jun N-terminal kinase (JNK) activation [6, 7, 10] and Bik or Bak upregulation [9, 11]. Otherwise, the mechanisms by which CFZ induces apoptosis are largely unknown.
Apoptosis can occur through two pathways: the extrinsic apoptotic pathway that primarily involves signals transduced through death receptors and the intrinsic apoptotic pathway that largely relies on signals from the mitochondria. Both pathways involve the activation of caspase cascades, which in turn cause cleavage of cellular substrates and result in the characteristic morphological and biochemical changes constituting the process of apoptosis [13, 14]. The extrinsic pathway is characterized by the trimerization of cell surface death receptors and activation of caspase-8, while the intrinsic pathway involves the disruption of mitochondrial membranes, release of cytochrome c from the mitochondria, and the activation of caspase-9. One well-known death ligand is tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), which initiates apoptosis upon ligation with two death receptors: death receptor 4 (DR4) and 5 (DR5). TRAIL preferentially induces apoptosis in transformed or malignant cells while sparing most normal cells and thus is a tumor-selective apoptosis-inducing cytokine with cancer therapeutic potential [15].
In this study, we determined the effects of CFZ on the induction of apoptosis in different solid tumor cell lines and were particularly interested in elucidating the mechanisms by which CFZ induces apoptosis in these cancer cells. We demonstrated that CFZ activates and enhances extrinsic apoptosis primarily through DR5 upregulation, in part due to delaying DR5 protein degradation.
RESULTS
CFZ effectively decreases the survival of different cancer cell lines and induces apoptosis
To determine the effective concentration range of CFZ that suppresses the growth of different cancer cell lines, we treated 6 cancer cell lines with varied concentrations of CFZ for 24 and 72 h, and then estimated cell numbers. We found that CFZ effectively decreased the survival of these cells with IC50s ranging from 50 nM to 300 nM (24 h exposure) and from 10 to 30 nM (72 h treatment) (Figure 1B). Clearly, prolonged treatment enhances the efficacy of CFZ in inhibiting cancer cell growth. We further determined the effects of CFZ on hallmarks of apoptosis in three cancer cell lines, specifically, cleavage (or activation) of caspases and PARP and induction of DNA fragmentation. We detected cleavage of caspase-8, caspase-3 and PARP as indicated by the appearance of cleaved forms of these proteins in A375, HCT116 and Tr146 cells exposed to CFZ for 24 h, demonstrating that CFZ activates these caspases (Figure 1C). In agreement, we detected increased levels of DNA fragments in cells treated with CFZ (Figure 1D), indicating that CFZ induces DNA fragmentation. Hence, CFZ clearly induces apoptosis in cancer cells, constituting an important mechanism accounting for its cell-killing effect.
CFZ induces caspase-dependent apoptosis in part through activation of the extrinsic apoptotic pathway
The above data clearly show that CFZ strongly induces cleavage of, and thus activates, caspase-8, a well-known initiator caspase in the extrinsic apoptotic pathway. To examine whether activation of caspases, particularly caspase-8, is required for CFZ-induced apoptosis, we compared PARP cleavage in the absence and presence of the pan-caspase inhibitor, Z-VAD-FMK, and found that PARP cleavage by CFZ was substantially inhibited by Z-VAD-FMK (Figure 2A). When caspase-8 was silenced with caspase-8 small interfering RNA (siRNA), CFZ-induced cleavage of both caspase-3 and PARP were blocked (Figure 2B). These results indicate that CFZ induces caspase-8-dependent apoptosis. Moreover, we determined whether CFZ induces apoptosis through activation of the extrinsic apoptotic pathway. We compared the effects of CFZ on decreasing cell survival and inducing apoptosis in isogenic HCT116 cell lines with wild-type (WT) FADD and with FADD knocked out (FADD-KO). In the survival assay, both FADD-KO HCT116 cell lines were significantly more resistant than WT HCT116 cells to CFZ, although they were not completely resistant (Figure 2C). In agreement, CFZ induced strong cleavage of caspase-8, caspase-3 and PARP in WT HCT116 cells, but only weakly in FADD-KO cells (Figure 2D). Hence, it is clear that deficiency of FADD, an essential component in mediating extrinsic apoptotic signaling, partially protects cancer cells from induction of apoptosis by CFZ, indicating that activation of the extrinsic apoptotic pathway contributes to CFZ-induced apoptosis.
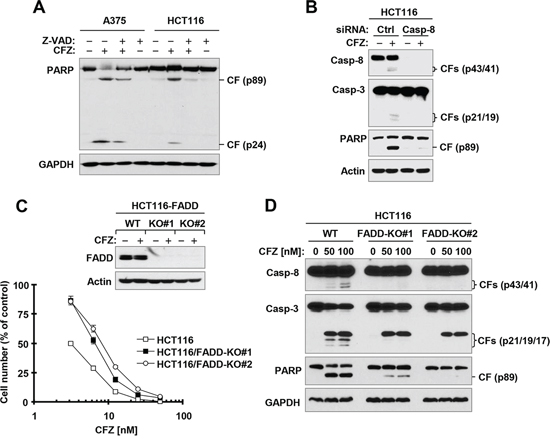
Figure 2: Caspase inhibition (A) caspase-8 knockdown (B) or FADD deficiency (C and D) protects cancer cells from CFZ-induced apoptosis. A, The indicated cell lines were treated with 100 nM CFZ in the absence or presence of 20 μM Z-VAD-FMK for 24 h and then harvested for preparation of whole-cell protein lysates and subsequent Western blot analysis. B, HCT116 cells were transfected with the indicated siRNAs and then after 36 h, were exposed to 100 nM CFZ for an additional 24 h. The cells were harvested for preparation of whole-cell protein lysates and subsequent Western blot analysis. C, The indicated WT and FADD-KO cell lines were treated with 100 nM CFZ for 8 h and then harvested for preparation of whole-cell protein lysates and subsequent Western blot analysis to confirm FADD expression. The indicated cell lines were also treated with different concentrations of CFZ for 48 h and then subjected to estimation of cell number with the SRB assay. The data are means ± SDs of four replicate determinations. D, The given cell lines were treated with the indicated concentrations of CFZ for 17 h and then subjected to preparation of whole-cell protein lysates and subsequent Western blot analysis. CF, cleaved fragment.
CFZ upregulates the expression of TRAIL death receptors, particularly DR5, in cancer cells
To understand the mechanism by which CFZ activates extrinsic apoptosis, we examined the effects of CFZ on the expression of DR4 and DR5, which are important cell surface death receptors that activate extrinsic apoptotic signaling though recruitment of FADD. At a concentration range of 25–100 nM, CFZ increased the levels of DR5 in a concentration-dependent manner in all of the tested cancer cell lines (Figure 3A). CFZ also increased DR4 levels somewhat in these cell lines, but the effects were either very weak (HCT116 and SqCC/Y1) or not concentration-dependent (Tr146 and SqCC/Y1) (Figure 3A). The increase in DR5 and DR4 expression in CFZ-treated cells occurred by 4 h, peaked at 8 h, and was maintained for up to 24 h (Figure 3B), indicating that CFZ-induced upregulation of DR5 and DR4 is an early and persistent event. Consistent with the greater increase in expression of DR5 compared with that of DR4 in CFZ-treated cells (Figure 3A), cell surface levels of DR5 were substantially increased by CFZ treatment while surface DR4 levels were only weakly increased (Figure 3C). Taken together, we conclude that CFZ strongly increases total and cell surface DR5 expression in cancer cells.
We further determined whether CFZ alters the levels of other proteins including c-FLIP, Mcl-1, Bcl-2 and Bcl-XL that are known to inhibit apoptosis [14]. As presented in Figure 3D, CFZ clearly increased the levels of FLIPS and Mcl-1 in a concentration-dependent manner in both A549 and HCT116 cells with limited effects on changing the levels of Bcl-2 and Bcl-XL.
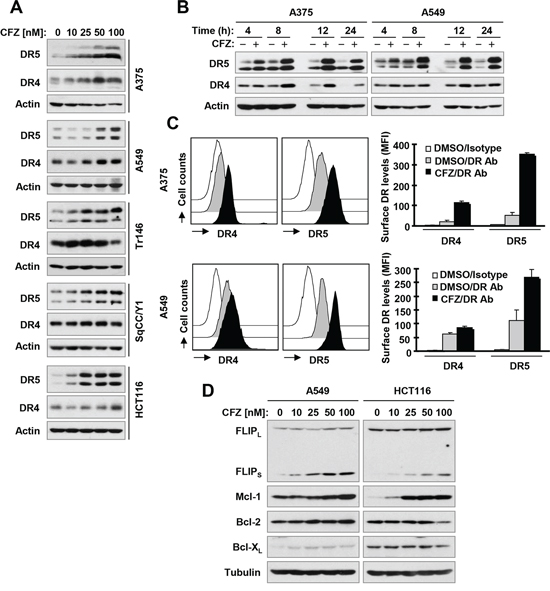
Figure 3: CFZ increases total (A and B) and cell surface (C) levels of DR5 and DR4 accompanied with elevated c-FLIP and Mcl-1 (D) A, B and D, The indicated cell lines were treated with the given concentrations of CFZ for 8 h (A and D) or with 100 nM CFZ for different times (B) and then subjected to preparation of whole-cell protein lysates. The given proteins were detected using Western blot analysis. C, The indicated cell lines were treated with 100 nM CFZ for 15 h and then harvested for analysis of cell surface DR5 and DR4 by immunofluorescence staining and subsequent flow cytometry. The open peak represents DMSO-treated cells stained with a matched control PE-conjugated IgG isotype antibody. The filled grey peaks show DMSO-treated cells stained with PE-conjugated anti-DR5 or DR4 antibody. The filled black peaks represent CFZ-treated cells stained with PE-conjugated anti-DR5 or DR4 antibody.
CFZ enhances TRAIL-induced apoptosis
Given that CFZ increases cell surface levels of both DR5 and DR4, which are well known death receptors for the death ligand TRAIL, we speculated that CFZ would sensitize cancer cells to TRAIL-induced apoptosis. To test this hypothesis, we examined the effects of CFZ in combination with TRAIL on cell survival and apoptosis in several cancer cell lines that are relatively less sensitive to TRAIL. TRAIL alone or CFZ alone at the tested concentration ranges weakly decreased the survival of the tested cancer cell lines; however, the combination of CFZ and TRAIL was much more active than either single agent in decreasing cell survival (Figure 4A). The combination indexes (CIs) for most combinations were far lower than 1, indicating strongly synergistic effects on cancer cell killing. Consistently, the combination of CFZ and TRAIL was more potent than each single agent in inducing cleavage of caspase-8, caspase-3 and PARP, evidenced by higher levels of cleaved forms in cells treated with the combination than in cells treated with either CFZ or TRAIL alone (Figure 4B). These data further support that the combination of CFZ and TRAIL enhances apoptosis in cancer cells.
It is well known that TRAIL induces apoptosis exclusively through a FADD-dependent mechanism. To determine whether the enhanced cell-killing effect seen with the CFZ and TRAIL combination is simply due to enhancement of TRAIL-induced apoptosis, we compared the effects of the combination on cell death and caspase cleavage in WT and FADD-KO HCT116 cell lines. Indeed, the combination of CFZ and TRAIL enhanced cell death and cleavage of caspase-8, caspase-3 and PARP in WT HCT116, but not in FADD-KO cells (Figure 5). These data convincingly demonstrate that CFZ enhances TRAIL-induced apoptosis and cell-killing.
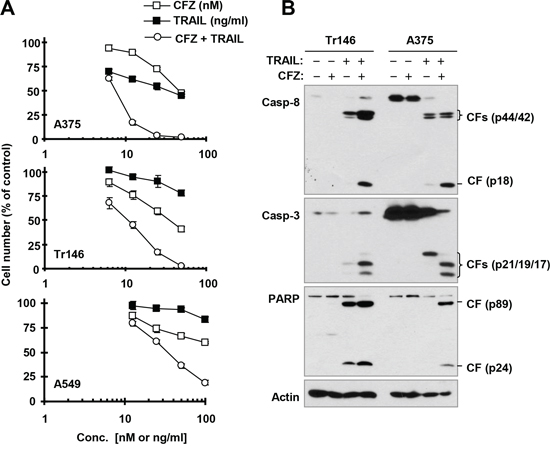
Figure 4: The combination of CFZ and TRAIL augments killing of cancer cells (A) with enhanced activation of caspases (B) A, The indicated cancer cell lines were treated with different concentrations of CFZ alone, TRAIL alone, and the combination of CFZ and TRAIL for 24 h and then subjected to estimation of cell number with the SRB assay. The data are means ± SDs of four replicate determinations. B, The indicated cell lines were treated with 50 nM CFZ alone, 25 ng/ml TRAIL alone or their combination for 24 h and then subjected to preparation of whole-cell protein lysates and subsequent Western blot analysis. CF, cleaved fragment.
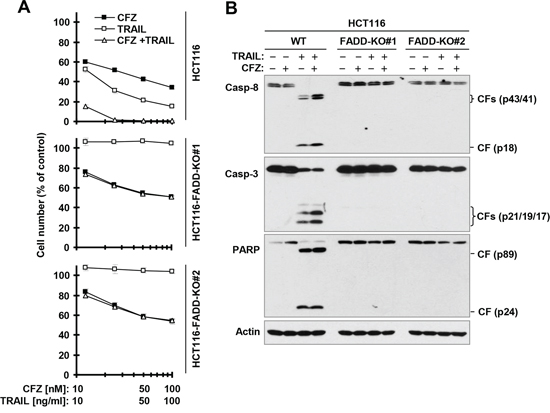
Figure 5: FADD deficiency abolishes augmented induction of apoptosis by the CFZ and TRAIL combination. A, The indicated cancer cell lines were treated with different concentrations of CFZ alone, TRAIL alone, and the combination of CFZ and TRAIL for 24 h and then subjected to estimation of cell number with the SRB assay. The data are means ± SDs of four replicate determinations. B, The indicated cell lines were treated with 50 nM CFZ alone, 10 ng/ml TRAIL alone or their combination for 8 h and then subjected to preparation of whole-cell protein lysates and subsequent Western blot analysis. CF, cleaved fragment.
DR5 upregulation contributes to CFZ-induced apoptosis
To determine whether DR5 upregulation is involved in CFZ-induced apoptosis in human cancer cells, we compared the effects of CFZ on cell death and caspase activation in WT and DR5-KO HCT116 cells (Figure 6A). We found that DR5-KO cells were significantly less sensitive than WT cells to CFZ in the cell survival assay (Figure 6B). Moreover, CFZ strongly induced cleavage of caspase-8, caspase-3 and PARP in WT HCT116 cells, but did so only minimally in DR5-KO cells (Figure 6C). Hence these results clearly show that DR5 upregulation is a critical event for CFZ to induce apoptosis.
DR5 upregulation contributes to CFZ-mediated enhancement of TRAIL-induced apoptosis
We further determined whether CFZ enhances TRAIL-induced apoptosis via upregulation of DR5 using the same isogenic cell lines. The combination of CFZ and TRAIL enhanced cell death and cleavage of caspase-8, caspase-3 and PARP, as indicated by the elevated levels of cleaved forms of these proteins in WT cells, but not in DR5-KO cells (Figure 6D and 6E). This effect was consistent with the results observed with TRAIL alone, i.e., TRAIL induced cell death and cleavage of caspase-8, caspase-3 and PARP with less effect in DR5-KO cells than in WT cells (Figure 6D and 6E). Together, these results clearly indicate that CFZ upregulates DR5 expression, leading to enhancement of TRAIL-induced apoptosis.
CFZ increases DR5 expression through enhancing protein stability and gene transcription
Since DR5 upregulation is strongly induced by CFZ across the tested cancer cell lines and plays a critical role in mediating CFZ-induced apoptosis and enhancement of TRAIL-induced apoptosis, we were interested in the mechanism by which CFZ upregulates DR5 expression. To this end, we examined the effect of CFZ on DR5 protein stability, since CFZ is a novel proteasome inhibitor. Hence, we conducted a cycloheximide (CHX) chase assay to compare degradation rates or half-lives of DR5 protein in the absence and presence of CFZ in 3 different cancer cell lines. The half-lives of DR5 in CFZ-treated cells were 4–6 h, compared with 1–2 h in DMSO-treated cells (Figure 7A and 7B), indicating that DR5 was degraded more slowly in CFZ-treated cells than in DMSO-treated cells. Hence, it is apparent that CFZ stabilizes DR5 protein.
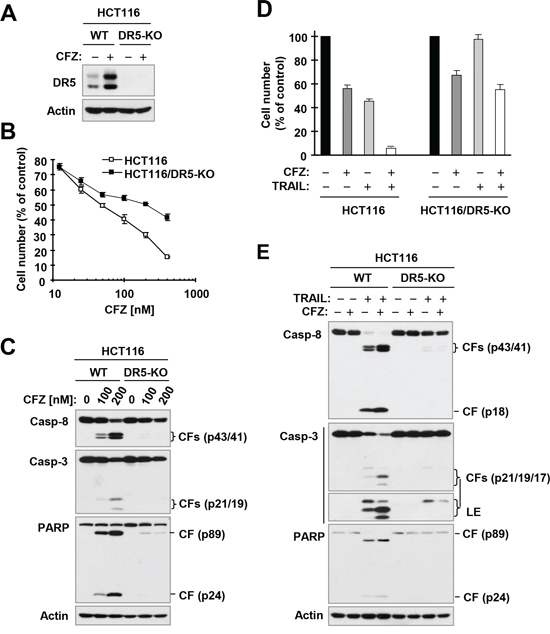
Figure 6: DR5 deficiency (A) protects cancer cells from undergoing apoptosis induced by CFZ (A and C) or CFZ and TRAIL combination (D and E). A, Detection of DR5 expression by Western blotting. B and D, The indicated cancer cell lines were treated with different concentrations of CFZ (B) or with 25 nM CFZ, 25 ng/ml TRAIL or the combination of CFZ and TRAIL (D). After 24 h, the cells were subjected to estimation of cell number with the SRB assay. The data are means ± SDs of four replicate determinations. C and E, The indicated cell lines were treated with different concentrations of CFZ (C) or with 50 nM CFZ alone, 15 ng/ml TRAIL alone or their combination (E) for 24 h (C) or 8 h (E) and then subjected to preparation of whole-cell protein lysates and subsequent Western blot analysis. CF, cleaved fragment. LE, longer exposure.
In addition we found that CFZ also increased DR5 mRNA levels as evaluated with reverse transcription PCR (RT-PCR) in both A375 and A549 cells (Figure 7C). Moreover, the presence of the transcription inhibitor, actinomycin D, inhibited DR5 upregulation by CFZ (Figure 7D). Therefore, CFZ also increases DR5 transcription.
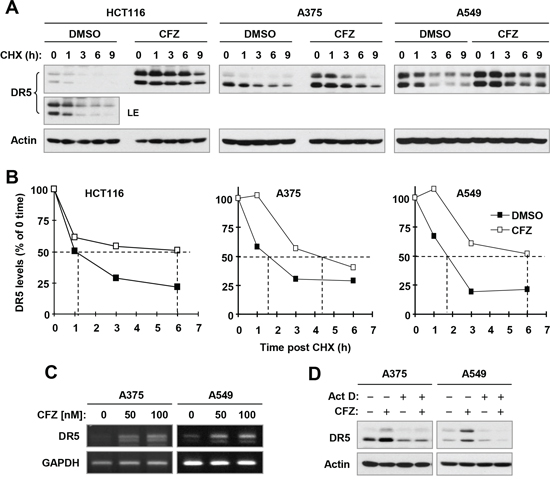
Figure 7: CFZ stabilizes DR5 protein (A and B) and increases DR5 transcription (C and D) in cancer cells. A and B, The indicated cancer cell lines were exposed to 100 nM CFZ for 6 h. The cells were then washed with PBS 3 times and re-fed with fresh medium containing 10 μg/ml CHX. At the indicated times post-CHX, the cells were harvested for preparation of whole-cell protein lysates and subsequent Western blot analysis (A). Protein levels were quantified with NIH Image J Software and were normalized to actin (B). C, The indicated cell lines were treated with the given concentrations of CFZ for 8 h and then harvested for preparation of total cellular RNA and subsequent RT-PCR. D, The indicated cell lines were pre-treated with 2.5 μM actinomycin D (Act D) for 30 minutes and then co-treated with 100 nM CFZ for an additional 8 h. The cells were then harvested for preparation of whole-cell protein lysates and subsequent Western blot analysis. LE, longer exposure.
DISCUSSION
In the present study, we have shown that CFZ effectively induces apoptosis and decreases the survival of different cancer cell lines. The induction of apoptosis by CFZ is at least in part due to activation of the extrinsic apoptotic pathway because deficiency of FADD, an essential component of the extrinsic apoptotic pathway, protected cells from CFZ-induced cell death and cleavage of caspases and PARP. Moreover, we have demonstrated that CFZ substantially enhances TRAIL-induced apoptosis, as evidenced by the synergistic induction of FADD-dependent apoptosis by the CFZ and TRAIL combination. To the best of our knowledge, this is the first study to indicate that CFZ induces and enhances extrinsic apoptosis in cancer cells.
Both DR5 and DR4 are death receptors through which TRAIL initiates extrinsic apoptosis. In this study, we found that total and cell surface levels of DR5 were strongly elevated by CFZ across the tested cancer cell lines, in comparison with a relatively modest increase in DR4. Hence, this study primarily focused on demonstrating the role of DR5 in mediating CFZ-induced apoptosis and enhancement of TRAIL-induced apoptosis and the mechanism accounting for CFZ-induced DR5 upregulation. Using genetically-manipulated DR5-deficient cells, we have robustly demonstrated for the first time that DR5 upregulation is a critical event that mediates CFZ-induced apoptosis and enhancement of TRAIL-induced apoptosis, since DR5 deficiency substantially protected cells from undergoing apoptosis induced by either CFZ or the CFZ and TRAIL combination. However, we cannot rule out that DR4 upregulation also contributes to CFZ-induced apoptosis and enhancement of TRAIL-induced apoptosis in a given cell line (e.g., A375).
Ubiquitination and proteasomal degradation is a key post-translational mechanism that regulates protein levels and biological functions including apoptosis [16]. It has been suggested that DR5 is regulated by this mechanism [17]. Since CFZ primarily functions as a proteasome inhibitor, it is reasonable to determine whether CFZ elevates DR5 levels by suppressing its degradation. Indeed, we demonstrated that CFZ treatment delayed the degradation of DR5 in a CHX chase assay (Figure 7). Therefore we conclude that stabilization of DR5 protein is an important mechanism by which CFZ upregulates DR5 expression. DR5 induction can also occur at transcriptional level [18–21]. In this study, we observed that CFZ also increased DR5 mRNA levels (Figure 7C) and inhibition of transcription with actinomycin D abolished the ability of CFZ to increased DR5 levels. Therefore, transcriptional regulation of DR5 expression is another mechanism contributing to the CFZ-induced upregulation of DR5. Clearly CFZ increases DR5 expression through enhancing both protein stabilization and gene transcription.
It is known that suppression of c-FLIP, a key negative regulator of the extrinsic apoptotic pathway, and/or Bcl-1 family members such as Mcl-1, Bcl-1 and Bcl-XL, which are negative regulators of the intrinsic apoptotic pathway, induces apoptosis or enhances TRAIL-induced apoptosis [22–24]. In this study, CFZ clearly increased the levels of both FLIPS and Mcl-1 (Figure 3D). These findings are similar to previous observations that proteasome inhibition increased FLIPS and Mcl-1 levels [25, 26]. Hence, elevation of these protein levels is likely to be a survival mechanism, which, however, can be overridden by upregulation of DR5 (and DR4) in cells exposed to CFZ, eventually resulting in apoptosis.
It is known that TRAIL rapidly induces apoptosis in a wide variety of transformed cells but is not cytotoxic in normal cells in vitro and in vivo [15, 27]. Thus, TRAIL is considered to be a tumor-selective, apoptosis-inducing cytokine and a promising new candidate for cancer therapy. In addition, agonistic anti-DR5 antibodies can induce DR5 trimerization, which triggers the extrinsic apoptotic pathway, and thus have great cancer therapeutic potential [28]. Unfortunately, certain cancer cells and tumors are resistant to apoptosis induced by TRAIL or DR5 agonistic antibody. Therefore, CFZ may be useful in combination with TRAIL or an agonistic anti-DR5 antibody to enhance the induction of apoptosis or overcome TRAIL resistance in human cancer cells.
MATERIALS AND METHODS
Reagents
CFZ was purchased from Selleck Chemicals (Houston, TX) and was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 1 mM, and aliquots were stored at −80°C. Stock solutions were diluted to the desired final concentrations with growth medium just before use. CHX and actinoimycin D were purchased from Sigma Chemical Co. (St. Louis, MO). Human recombinant TRAIL was purchased from PeproTech, Inc. (Rocky Hill, NJ). Rabbit polyclonal anti-DR5 antibody was purchased from ProSci, Inc. (Poway, CA). Mouse monoclonal anti-DR4 antibody (B-N28) was purchased from Diaclone (Stamford, CT). Mouse monoclonal anti-caspase-3 was purchased from Imgenex (San Diego, CA). Rabbit anti-caspase-8 and anti-PARP antibodies were purchased from Cell Signaling Technology, Inc. (Beverly, MA). Mouse monoclonal anti-FLIP antibody (NF6) was purchased from Alexis Biochemicals (San Diego, CA). Rabbit polyclonal Mcl-1 and Bcl-XL and mouse monoclonal Bcl-2 antibody were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Rabbit polyclonal anti-β-actin antibody was purchased from Sigma Chemical Co.
Cell lines and cell culture
A549 (lung cancer), H1299 (lung cancer), Tr146 (head and neck cancer) and SqCC/Y1 (head and neck cancer) cell lines were described previously [29, 30]. A375 (melanoma) cells were provided by Dr. J. Arbiser (Emory University, Atlanta, GA). HCT116 (colon cancer) and its isogenic FADD-KO and DR5-KO cell lines were provided by Dr. J. Yu (University of Pittsburgh, Pittsburgh, PA) and Dr. L. Zhang (University of Pittsburgh, Pittsburgh, PA), respectively. Except for A549 cells, which were authenticated by Genetica DNA Laboratories, Inc. (Cincinnati, OH) through analyzing short tandem repeat DNA profile, other cell lines were not authenticated. Cells were grown in monolayer culture in RPMI 1640 with glutamine, DMEM/F12 or McCoy’s 5A modified medium supplemented with 5% fetal bovine serum at 37°C in a humidified atmosphere consisting of 5% CO2 and 95% air.
Cell survival and apoptosis assays
Cells were seeded in 96-well cell culture plates and treated the next day with the tested agents. Viable cell numbers were determined using sulforhodamine B (SRB) assay as described previously [29]. CI was calculated using the CompuSyn software (ComboSyn, Inc.; Paramus, NJ) to indicate drug interaction (e.g., synergy). Apoptosis was evaluated primarily by detecting caspase and PARP cleavage with Western blot analysis as described below. We also used a Cell Death Detection ELISAPlus kit (Roche Molecular Biochemicals, Indianapolis, IN) to detect DNA fragments according to the manufacturerʼs instructions as an additional indicator of apoptosis.
Western blot analysis
Preparation of whole-cell protein lysates and the procedures for Western blotting were described previously [31].
Detection of cell surface death receptors
The procedure for direct antibody staining and subsequent flow cytometric analysis of cell surface proteins was described previously [25]. Mean fluorescence intensity (MFI), which represents antigenic density on a per cell basis, was used to represent the cell surface death receptor expression level.
Gene silencing with siRNA
The control and caspase-8 siRNA were the same as described previously [31]. siRNA transfection was performed as previously described [19, 32].
Detection of DR5 mRNA expression
DR5 mRNA was detected with RT-PCR as described previously [33].
ACKNOWLEDGMENTS
We are grateful to Drs. L. Zhang and J. Yu for providing their valuable FADD-KO and DR5-KO cell lines and to Dr. A. Hammond in our department for editing the manuscript.
This study was supported by Emory Winship Cancer Institute Halpern Research Scholar award (to SYS) and Melanoma Research Fund (to JD).
FRK and SYS are Georgia Research Alliance Distinguished Cancer Scientists. SYS is a Halpern Research Scholar.
REFERENCES
1. Shen M, Schmitt S, Buac D, Dou QP. Targeting the ubiquitin-proteasome system for cancer therapy. Expert opinion on therapeutic targets. 2013; 17:1091–1108.
2. Johnson DE. The ubiquitin-proteasome system: opportunities for therapeutic intervention in solid tumors. Endocr Relat Cancer. 2015; 22:T1–T17.
3. Pellom ST Jr, Shanker A. Development of Proteasome Inhibitors as Therapeutic Drugs. Journal of clinical & cellular immunology. 2012; S5:5.
4. Kortuem KM, Stewart AK. Carfilzomib. Blood. 2013; 121:893–897.
5. Herndon TM, Deisseroth A, Kaminskas E, Kane RC, Koti KM, Rothmann MD, Habtemariam B, Bullock J, Bray JD, Hawes J, Palmby TR, Jee J, Adams W, Mahayni H, Brown J, Dorantes A, et al. U.s. Food and Drug Administration approval: carfilzomib for the treatment of multiple myeloma. Clin Cancer Res. 2013; 19:4559–4563.
6. Kuhn DJ, Chen Q, Voorhees PM, Strader JS, Shenk KD, Sun CM, Demo SD, Bennett MK, van Leeuwen FW, Chanan-Khan AA, Orlowski RZ. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood. 2007; 110:3281–3290.
7. Sacco A, Aujay M, Morgan B, Azab AK, Maiso P, Liu Y, Zhang Y, Azab F, Ngo HT, Issa GC, Quang P, Roccaro AM, Ghobrial IM. Carfilzomib-dependent selective inhibition of the chymotrypsin-like activity of the proteasome leads to antitumor activity in Waldenstromʼs Macroglobulinemia. Clin Cancer Res. 2011; 17:1753–1764.
8. Gupta SV, Hertlein E, Lu Y, Sass EJ, Lapalombella R, Chen TL, Davis ME, Woyach JA, Lehman A, Jarjoura D, Byrd JC, Lucas DM. The proteasome inhibitor carfilzomib functions independently of p53 to induce cytotoxicity and an atypical NF-kappaB response in chronic lymphocytic leukemia cells. Clin Cancer Res. 2013; 19:2406–2419.
9. Gu JJ, Hernandez-Ilizaliturri FJ, Kaufman GP, Czuczman NM, Mavis C, Skitzki JJ, Czuczman MS. The novel proteasome inhibitor carfilzomib induces cell cycle arrest, apoptosis and potentiates the anti-tumour activity of chemotherapy in rituximab-resistant lymphoma. Br J Haematol. 2013; 162:657–669.
10. Zhang L, Pham LV, Newberry KJ, Ou Z, Liang R, Qian J, Sun L, Blonska M, You Y, Yang J, Lin X, Rollo A, Tamayo AT, Lee J, Ford RJ, Zhao X, et al. In vitro and in vivo therapeutic efficacy of carfilzomib in mantle cell lymphoma: targeting the immunoproteasome. Mol Cancer Ther. 2013; 12:2494–2504.
11. Zang Y, Thomas SM, Chan ET, Kirk CJ, Freilino ML, DeLancey HM, Grandis JR, Li C, Johnson DE. The next generation proteasome inhibitors carfilzomib and oprozomib activate prosurvival autophagy via induction of the unfolded protein response and ATF4. Autophagy. 2012; 8:1873–1874.
12. Baker AF, Hanke NT, Sands BJ, Carbajal L, Anderl JL, Garland LL. Carfilzomib demonstrates broad anti-tumor activity in pre-clinical non-small cell and small cell lung cancer models. Journal of experimental & clinical cancer research : CR. 2014; 33:778.
13. Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998; 281:1305–1308.
14. Hengartner MO. The biochemistry of apoptosis. Nature. 2000; 407:770–776.
15. Kelley SK, Ashkenazi A. Targeting death receptors in cancer with Apo2L/TRAIL. Curr Opin Pharmacol. 2004; 4:333–339.
16. Vucic D, Dixit VM, Wertz IE. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat Rev Mol Cell Biol. 2011; 12:439–452.
17. Song JJ, Szczepanski MJ, Kim SY, Kim JH, An JY, Kwon YT, Alcala MA Jr, Bartlett DL, Lee YJ. c-Cbl-mediated degradation of TRAIL receptors is responsible for the development of the early phase of TRAIL resistance. Cell Signal. 2010; 22:553–563.
18. Wu GS, Burns TF, McDonald ER 3rd, Jiang W, Meng R, Krantz ID, Kao G, Gan DD, Zhou JY, Muschel R, Hamilton SR, Spinner NB, Markowitz S, Wu G, el-Deiry WS. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet. 1997; 17:141–143.
19. Oh YT, Liu X, Yue P, Kang S, Chen J, Taunton J, Khuri FR, Sun SY. ERK/ribosomal S6 kinase (RSK) signaling positively regulates death receptor 5 expression through co-activation of CHOP and Elk1. J Biol Chem. 2010; 285:41310–41319.
20. Zou W, Liu X, Yue P, Zhou Z, Sporn MB, Lotan R, Khuri FR, Sun SY. c-Jun NH2-terminal kinase-mediated up-regulation of death receptor 5 contributes to induction of apoptosis by the novel synthetic triterpenoid methyl-2-cyano-3, 12-dioxooleana-1, 9-dien-28-oate in human lung cancer cells. Cancer Res. 2004; 64:7570–7578.
21. Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem. 2004; 279:45495–45502.
22. Zhang L, Fang B. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther. 2005; 12:228–237.
23. Chan SL, Yu VC. Proteins of the bcl-2 family in apoptosis signalling: from mechanistic insights to therapeutic opportunities. Clin Exp Pharmacol Physiol. 2004; 31:119–128.
24. Fesik SW. Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer. 2005; 5:876–885.
25. Liu X, Yue P, Chen S, Hu L, Lonial S, Khuri FR, Sun SY. The proteasome inhibitor PS-341 (bortezomib) up-regulates DR5 expression leading to induction of apoptosis and enhancement of TRAIL-induced apoptosis despite up-regulation of c-FLIP and survivin expression in human NSCLC cells. Cancer Res. 2007; 67:4981–4988.
26. Yuan BZ, Chapman J, Ding M, Wang J, Jiang B, Rojanasakul Y, Reynolds SH. TRAIL and proteasome inhibitors combination induces a robust apoptosis in human malignant pleural mesothelioma cells through Mcl-1 and Akt protein cleavages. BMC Cancer. 2013; 13:140.
27. Wang S, El-Deiry WS. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003; 22:8628–8633.
28. Ichikawa K, Liu W, Zhao L, Wang Z, Liu D, Ohtsuka T, Zhang H, Mountz JD, Koopman WJ, Kimberly RP, Zhou T. Tumoricidal activity of a novel anti-human DR5 monoclonal antibody without hepatocyte cytotoxicity. Nat Med. 2001; 7:954–960.
29. Sun SY, Yue P, Dawson MI, Shroot B, Michel S, Lamph WW, Heyman RA, Teng M, Chandraratna RA, Shudo K, Hong WK, Lotan R. Differential effects of synthetic nuclear retinoid receptor-selective retinoids on the growth of human non-small cell lung carcinoma cells. Cancer Res. 1997; 57:4931–4939.
30. Sun SY, Yue P, Mao L, Dawson MI, Shroot B, Lamph WW, Heyman RA, Chandraratna RA, Shudo K, Hong WK, Lotan R. Identification of receptor-selective retinoids that are potent inhibitors of the growth of human head and neck squamous cell carcinoma cells. Clin Cancer Res. 2000; 6:1563–1573.
31. Liu X, Yue P, Zhou Z, Khuri FR, Sun SY. Death receptor regulation and celecoxib-induced apoptosis in human lung cancer cells. J Natl Cancer Inst. 2004; 96:1769–1780.
32. Oh YT, Yue P, Zhou W, Balko JM, Black EP, Owonikoko TK, Khuri FR, Sun SY. Oncogenic Ras and B-Raf proteins positively regulate death receptor 5 expression through co-activation of ERK and JNK signaling. J Biol Chem. 2012; 287:257–267.
33. Sun SY, Liu X, Zou W, Yue P, Marcus AI, Khuri FR. The Farnesyltransferase Inhibitor Lonafarnib Induces CCAAT/Enhancer-binding Protein Homologous Protein-dependent Expression of Death Receptor 5, Leading to Induction of Apoptosis in Human Cancer Cells. J Biol Chem. 2007; 282:18800–18809.