
INTRODUCTION
According to “World Cancer Report 2014” by WHO, in 2012 about 14.1 million new cases of cancer occurred globally and caused 14.6% of all human deaths [1]. It is expected that annual cancer cases will rise from 14.1 to 22 million within the next two decades [1]. Disturbingly, cancer remains the second most common cause of death among children between the ages of 1 to 14 years in the US [1]. Over the past years, with the help of well adapted genomic and proteomic technologies, researchers have made strides in better understanding the biochemical and molecular mechanisms of tumorigenesis. Conventional radio and chemo therapies, coupled with newer targeted cancer therapy are currently utilized for management and treatment of a variety of cancers in the clinic. Although a full understanding of the molecular complexity of cancers is yet to be realized, considerable progress have been made in the treatment of a range of cancers that include chronic myeloid leukemia (CML), acute promyelocytic leukemia (APL), Hodgkin’s lymphoma and testicular cancers [2]. Unfortunately, the successful outcome of many anti-cancer treatments and disease-free survival of the patients is often short lived due in part to limiting toxic side effects of therapies as well as emergence of drug-resistant cancers. Therefore, it is necessary to discover new targets and molecular pathways that regulate processes of carcinogenesis in order to improve diagnoses and therapeutic efficacy of current drugs as well as to permit design and development of novel therapeutic strategies to effectively combat cancers including their drug-resistant variants.
Discovery of CARP-1/CCAR1
By utilizing a functional gene knockout technology, CARP-1/CCAR1 was originally identified as a peri-nuclear phospho-protein that was required for apoptosis signaling by a retinoid (CD437) as well as chemotherapeutics Adriamycin (ADR) and Etoposide in the Human Breast Cancer (HBC) cells [3]. The current GenBank database indicates nucleotide sequences encoding CARP-1/CCAR1 proteins of various species including Mus musculus (mouse), Canis lupus (dog), Rattus norvegicus (rat), Pan troglodytes (chimpanzee), Gallus gallus (domestic chicken), Danio rerio (zebrafish), Xenopus laevis, Caenorhabditis elegans (worm) and Ornithorhynchus anatinus (platypus). Human CARP-1/CCAR1 gene is located at the long arm of chromosome 10 (10q21–10q22), and encodes a transcript of ~3.5 kilobase (kb) size. The CARP-1/CCAR1 protein has 1150 amino acid residues with approximate molecular mass of 130 kiloDaltons (kDa). A number of studies to date have revealed CARP-1/CCAR1 involvement in cell proliferation as well as apoptosis signaling in a variety of cell types including different cancer cells [4, 5].
CARP-1/CCAR1 as a regulator of steroid/thyroid receptor signaling
Nuclear receptors (NRs) are highly conserved family of transcription factors that regulate transcription of target genes in ligand–dependent manner. NRs include receptors for estrogen (ER, NR3A), progesterone (PR, NR3C3), vitamin A (RAR, NR1B), vitamin D (VDR, NR1I1), androgen (AR, NR3C4), and thyroid hormones (TR, NR1A). NRs bind to specific DNA sequences in response to their ligand binding, which in turn recruits other co-activators that remodels the chromatin in regulating RNA polymerase II associated basal transcriptional mechanism [6]. For example, P160 family of coactivators that include Steroid receptor coactivator 1 (SRC-1; also known as nuclear receptor coactivator-1; NCoA-1), SRC-2 (also known as Glutamate receptor interacting protein 1; GRIP1, Transcriptional intermediary factor 2; TIF2 or NcoA-2), and SRC-3 (also known as ACTR, Amplified in breast 1; AIB1, pCIP, RAC3, or Thyroid hormone receptor activator molecule 1; TRAM1) harbor multiple leucine rich repeat (LXXLL) motifs, which promote ligand-dependent interactions with the activation function 2 (AF2) domain of the NRs [6]. The p160 coactivators also recruit multiple acteyltransferases including CBP/p300 and P300-CBP associated protein (PCAF) and methyltransferase coactivators such as coactivator-associated arginine methyl transferase 1 (CARM1) and Protein arginine methyl transferase 1 and 2 (PRMT1 & 2) as well as other downstream targets including coiled-coil coactivator (CoCoA) [7, 8]. Some nuclear hormone receptors such as ERα regulate activator protein (AP)-1 dependent gene expression by directly binding with c-Jun and JunB proteins but not with c-fos proteins [9].
In 2008, Kim et al. reported that CARP-1/CCAR1 functions as co-activator of NRs. CARP-1/CCAR1 associates with components of Mediator and p160 co-activator complexes and is recruited to endogenous NR target genes in the presence of appropriate hormone. CARP-1/CCAR1 interacts with and co-operates with CoCoA and GRIP1 co-activators for estrogen receptor (ER) signaling, and promotes growth of MCF-7 HBC cells in response to estradiol (E2) treatment [10]. CARP-1/CCAR1 also physically and functionally associated with mediator complex including NUT2/MED10 and MED23 [11]. CCAR1/CARP-1 stimulated the basal and hormone dependent activities of glucocorticoid (GR) and thyroid hormone receptor β1 (TR) reporter genes in CV-1 cells [10]. Besides co-activation of GR, CARP-1/CCAR1 also positively regulated adipogenesis through GR signaling pathway [11]. A recent investigation also revealed that carboxyl terminal region of the transcription factor GATA1 interacts with CARP-1/CCAR1, CoCoA, and MED1. CARP-1/CCAR1 and CoCoA in fact synergistically enhanced GATA1-mediated transcription from the γ-globin promoter during erythroid differentiation [12].
Additionally, CARP-1/CCAR1 binds directly with ER and associates with p160 coactivators and in turn recruits mediator MED1/TRAP220 (Thyroid hormone receptor associated Protein 220) to ER target gene pS2 in E2-dependent manner. A recent investigation emphasized that CARP-1/CCAR1 is required for growth of prostate cancer cells in part by functioning as a coactivator of AR transcription. CARP-1/CCAR1 plays a crucial role in recruiting GATA2, a key mediator of transcription by AR, in androgen dependent signaling [13]. Knockdown of endogenous CARP-1/CCAR1, compromised the hormone dependent recruitment of mediator complexes to NRs, and consequently impacted growth of hormone dependent cancer cells [10]. Another recent study revealed CARP-1/CCAR1 is a binding partner of β-catenin and enhances the transcriptional activation of β-catenin-Wnt target genes, and mediates anchorage independent growth of colon cancer cells. Depletion of CARP-1/CCAR1 or β-catenin attenuated ability of the colon cancer cells to form colonies in soft agar [14].
Thus, significant investigations suggest that CARP-1/CCAR1 plays important roles in regulating cancer cell growth in part by recruiting multiple mediators and confers optimal conformation for transcriptional functions of NRs. Additionally CARP-1/CCAR1 co-activates the pro-apoptotic tumor suppressor p53 in chemotherapy (ADR)-treated cells. Co-activation of p53 and NRs by CARP-1/CCAR1 highlights its broader biphasic roles in regulating cell growth and apoptosis signaling [10] that are discussed below.
DBC1, a paralog of CARP-1/CCAR1
Deleted in breast cancer 1 (DBC1), also known as CCAR2, is a nuclear protein encoded by the KIAA1967 gene and is considered as an important paralog of CARP-1/CCAR1 gene. DBC1 was originally discovered and cloned from short arm of chromosome 8 (8p21), which was homozygously deleted in a subset of breast cancers [15]. DBC1 interacts with and negatively regulates SIRT1 deacetylase activity [16, 17]. Subsequent studies revealed that SIRT1 inhibition by DBC1 results in increased acetylation of p53 that, in turn, mediates p53-dependent apoptosis following DNA damage [16, 17]. Inhibition of SIRT1 by DBC1in the DNA damage response (DDR) depends on ATM-dependent Chk2 activation [18]. This Chk2 activation in turn promotes phosphorylation of the 11S proteasome activator REGγ, which increases REGγ–DBC1 interaction and SIRT1 inhibition [18]. Caspase-dependent processing of DBC1 has also been implicated in its pro-apoptotic activity during tumor necrosis factor alpha (TNFα)-mediated apoptosis [19]. However DBC1 loss also promotes death of breast cancer cells [20] while its expression is associated with distant metastatic relapse in patients after receiving endocrine therapy, and thus is an indicator of poor prognosis in a subset of patients [21, 22]. A broader function of DBC1 in metabolism, aging and cancer has recently been highlighted in an elegant review by Chini et al. [23]. Overall, studies thus far suggest that like CARP-1/CCAR1, DBC1 could also play biphasic roles in proliferation and apoptosis signaling.
Although CARP-1/CCAR1 and DBC1 proteins are paralogs, a recent report revealed that both the DBC1 and CARP-1/CCAR1 are largely disordered proteins that evolved from a single common ancestor, the nematode Caenorhabditis elegans protein lateral signaling target (LST)-3 [24]. DBC1 emerged later in evolution than CARP-1/CCAR1 and first appeared in Danio rerio (zebrafish). Interestingly, the DBC1 gene is not present in insects, amphibians, or birds [24]. Nevertheless, a synergistic co-regulation of CARP-1/CCAR1 by DBC1 in mediating ERα target gene transcription in MCF7 and T47D HBC cells was noted in a recent study [25]. DBC1 and CARP-1/CCAR1 cooperatively enhanced and co-activated the ligand-dependent transcription functions of multiple NRs including AR, GR and TR. Depletion of either DBC1 or CARP-1/CCAR1 or both attenuated transcription of ERα target genes [25]. With reference to the co-activator functions, a genome-wide comparative analysis following knock-down of CARP-1/CCAR1 or DBC1 revealed that each co-regulator regulates only a select subset of physiological pathways controlled by glucocorticoids, and the gene-specific actions of these co-activators correspond to specific physiological pathways [26]
Role of CARP-1/CCAR1 in physiological development
Besides regulating cancer cell growth by co-activating ligand-dependent transcriptional functions of NRs, a number of reports indicate that CARP-1/CCAR1 also co-activates signaling by transcription factors for the normal physiological development. Noggin (Ngn)3 is a helix-loop-helix transcription factor that plays a key role in mediating endocrine differentiation. Notably, Ngn3 regulates pancreatic cell proliferation and differentiation processes and thus serves as a key determinant in pancreatic cell fate during development. CARP-1/CCAR1 binds with Ngn3 in nucleus and co-activates its downstream target gene NeuroD to regulate endocrine differentiation in pancreas during development [27]. Therefore, a broader set of transcriptional co-activation roles of CARP-1/CCAR1 in cell growth, metastasis, and differentiation processes can be deduced on the basis of its ability to co-activate NRs, β-catenin, and Ngn3 [8, 15, 27].
Another recent study revealed modulation of CARP-1/CCAR1 expression and distribution by parathyroid hormone (PTH) in osteoblast differentiation [28]. PTH regulates both anabolic and catabolic process in osteoblast growth and differentiation, and regulates translocation of CARP-1/CCAR1 from nucleus to cytoplasm in remodeling and development of bone tissue. It is well established that Wnt-β-catenin signaling is required for bone physiology [29–31]. Moreover, PTH induces β-catenin activation in multiple cells including UMR, MC3T3-E1 and SAOS [32–35]. Considering the ability of CARP-1/CCAR1 to co-activate Wnt-β-catenin target gene transcription, it is conceivable that CARP-1/CCAR1 could serve as a docking protein to finely regulate β-catenin signaling and apoptosis in the presence of PTH in remodeling bone tissue and development.
Necdin, a member of the MAGE (Melanoma Antigen) protein family, binds with CARP-1/CCAR1, and promotes proteosomal degradation of pro-apoptotic CARP-1/CCAR1 in turn enhancing myoblast differentiation and survival [36]. Interestingly, Necdin also functions as a transcriptional repressor of p53 and inhibits apoptosis [37] regulated by TNFα signaling in preventing muscle atrophy [38]. In light of the fact that CARP-1/CCAR1 co-activates pro-apoptotic transcriptional function of p53, it appears that by negatively regulating CARP-1/CCAR1 Necdin counter-acts the pro-apoptotic activities of p53 and thus contributes to myoblast survival. Additionally, CARP-1/CCAR1 serves as a binding partner of AKAP350A, a multifunctional scaffolding protein, present in Golgi apparatus and centrosomes, along with RAS-GAP SH3 domain binding protein (G3BP), a stress granule marker, and Caprin-1 (Cytoplasmic activation/proliferation-associated protein 1). The oxidative stress following arsenite treatment results in translocation of CARP-1/CCAR1-AKAP350A-G3BP-Caprin-1 complex to RNA stress granules [39]. Since G3BP is known to protect cellular mRNA under stress conditions [40], and CARP-1/CCAR1 protein harbors a cold-shock RNA-binding domain [3], it appears that CARP-1/CCAR1 association with AKAP350A/G3BP/Caprin-1 complex in stress granules likely functions to sequester RNAs to maintain stability and integrity of cellular RNAs in a microtubule-dependent manner during the conditions of cellular stress [39].
CARP-1/CCAR1 as a regulator of stress-dependent apoptosis
Apoptosis, a process of programmed cell death, plays a pivotal role in maintaining proper tissue homeostasis. Deregulation of apoptosis contributes to various pathological conditions including inflammation and tumorigenesis. Apoptosis is considered as an important attribute of the chemotherapy drugs and serves to eliminate the damaged cancer cells [41, 42]. However, resistance to therapy-induced apoptosis is often encountered in the clinic and contributes to failure in therapy response in cancer patients and results in poor prognosis. Therefore, urgent efforts are warranted to identify additional perhaps new druggable targets to improve therapy efficacy and overcome the resistance mechanisms.
As mentioned earlier, CARP-1/CCAR1 was originally indentified and characterized as perinuclear phosphoprotein that was required for apoptosis signaling by chemotherapy drugs ADR and Etoposide independent of the p53 status of the HBC cells [3]. As summarized in figure 1, apoptosis induction by CARP-1/CCAR1 involves its binding with 14–3-3 protein, stimulation of CDKI p21WAF1CIP1 levels and down-regulation of cell growth and cell cycle regulators c-Myc, topoisomerase IIα, cyclin B and p21 Rac1 [3]. Genetic studies revealed that C. elegans Lst3, an ortholog of human CARP-1/CCAR1, is a transducer of Notch signaling. Lst3 also functions as an inhibitor of the EGFR-MAPK pathway [43]. These findings support our earlier studies that demonstrated CARP-1/CCAR1 requirement for apoptosis signaling following EGFR blockage in HBC cells [4]. Apoptosis signaling following EGFR inhibition invloves CARP-1/CCAR1 phosphorylation at tyrosine (Y)192, activation of the stress-activated protein kinase (SAPK) p38α/β and caspase 9 [4]. Interestingly, phosphorylation of Y189 of murine CARP-1/CCAR1 that corresponds with the Y192 of the human homolog has been noted in a global screening of p-tyrosine profile of Src-transformed MEFs [44]. It is likely that a context-dependent tyrosine phosphorylation of CARP-1/CCAR1 plays an important role in signaling for tumor growth or apoptosis, and a thorough functional characterization is needed for further understanding of the mechanism(s) involved. Pharmcologic inhibition of Protein kinase A (PKA) resulted in suppression of HBC cell growth in part by targeting CARP-1/CCAR1 threonine (T)667-dependent reduced c-Myc transcription [5]. A number of previous studies have revealed serine (S), and/or threonine (T) as well as tyrosine (Y) phosphorylation of CARP-1/CCAR1 by a variety of different signaling pathways [45–47]. CARP-1/CCAR1 was also found to be a nuclear protein in HeLa cells where it was phosphorylated at its Carboxyl terminus S1149, although the precise signaling context was not elucidated [45]. A compilation of the CARP-1/CCAR1 residues modified by phosphorylation and ubiquitination culminating from various proteomic and signaling studies thus far is available at http://www.phosphosite.org. Of note is that signaling by RIP3 kinase, oncogenic phosphatase CDC25B, Aurora and Polo-like kinases phosphorylate CARP-1/CCAR1 T627 [48–51], however, no kinase has been identified that directly phosphorylates CARP-1/CCAR1 Y192, T627, T667, or S1149.
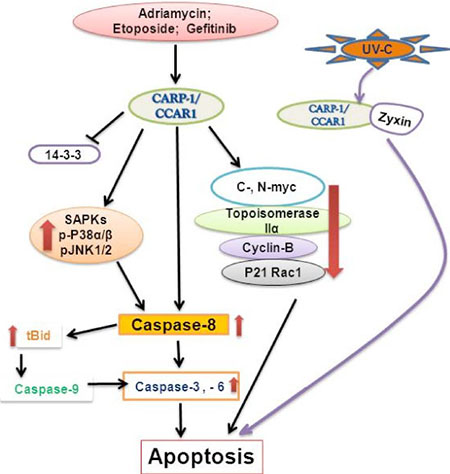
Figure 1: A Schematic of CARP-1/CCAR1 Apoptosis Signaling.
Epigenetic modifications such as CpG island methylation, histone modification, and deregulation of DNA binding proteins are as important as gene mutation in the process of tumorigenesis. Such epigenetic alterations could either silence key tumor suppressor(s) or promote activation of oncogene(s). In this context our prior reports revealed that CARP-1/CCAR1 expression correlated inversely with the grades of human tumors of breast and lymphoma origins [52, 53]. Although a limited number of specimens were analyzed, diminished expression of CARP-1/CCAR1 in human breast cancers involved methylation-dependent gene silencing that was associated with hypermethylation of CARP-1/CCAR1 promoter [52]. On the other hand, HBC cells stably overexpressing CARP-1/CCAR1 formed reduced sized subcutaneous tumors compared with their wild-type counterparts when xenografted in the immunocompromised SCID mice [52].
A recent study reported that Par-4 (prostate apoptosis response factor -4) and THAP1, a sequence specific DNA binding factor, synergestically cooperate to enhance CARP-1/CCAR1 expression, and promote T-cell acute lymphoblastic leukemia cells (T-ALL) apoptosis [54]. It is known that deregulation of Notch signaling contributes to generation of T-ALL [55]. Here, Par-4/THAP1 complex and Notch 3 competitively regulate alternative splicing of CARP-1/CCAR1 mRNA and impact the T-ALL cell survival. Thus exogenous expression of Notch-ICD (intracellular domain) results in production of truncated form of CARP-1/CCAR1 protein of ~90 kDa along with the full length CARP-1/CCAR1 protein of ~130 kDa, and inhibits Par-4/THAP1 induced CARP-1/CCAR1 expression and apoptosis. Silencing of Notch resulted in the disappearance of truncated ~90 kDa form of CARP-1/CCAR1 and restoration of apoptosis [54]. Although a truncated variant of CARP-1/CCAR1 appears to function as an inhibitor of apoptosis signaling by wild-type CARP-1/CCAR1 in T-ALL cells, ectopic expression of retroviral TAT-domain fusions of a number of non-overlapping CARP-1/CCAR1 peptides that accumulated in the cytosolic compartment, inhibited growth of HBC and lymphoma cells in vitro, and suppressed growth of the lymphoma as well as the breast cancer cell-derived xenografts in SCID mice [52, 53]. Another recent report demonstrated that CARP-1/CCAR1 directly binds with LIM-domain protein Zyxin and promotes UV-induced apoptosis in mouse embryonic fibroblasts (MEFs) [56]. Zyxin regulates actin assembly and shuttles between focal adhesions and cell nuclei in respose to mechanical stress. Further, in response to UV-C irradiation, CARP-1/CCAR1 binds to LIM domain of zyxin and promotes its pro-apoptotic activity while expression of zyxin mutant lacking LIM domain failed to bind with CARP-1/CCAR1 and interfered with apoptosis [56]. Consistent with the property of CARP-1/CCAR1 to regulate apoptosis signaling, a couple of studies revealed that CARP-1/CCAR1 is downregulated in a gene expression profiles of tumors derived from rasv12/E1A-transformed MEFs [57], and in peripheral blood cells from neurodegenerative disease, Friedreich’s Ataxia patients [58]. Overall, it is increasingly becoming evident that CARP-1/CCAR1 also functions as a tumor suppressor and/or mediator of apoptosis in diverse cancers [3–5, 10, 45, 52–54]. The pathways and interactions involved in pleiotropic signal transduction by CARP-1/CCAR1 that are known to date are summarized in Table 1.
CARP-1/CCAR1 signaling as a basis for development of novel approaches for tumor suppression
High throughput proteomic studies revealed that CARP-1/CCAR1 interacts with SAPK p38 [59] and NF-κB upstream kinase subunit NEMO/IKKγ [60]. Considering the fact that TAT-mediated transduction of various non-overlapping peptides of CARP-1/CCAR1 suppressed the growth of HBC and lymphoma cells in vitro and in vivo [52, 53], and CARP-1/CCAR1 co-activates p53 to transduce ADR-dependent apoptosis in breast cancer cells [10], we speculated that CARP-1/CCAR1 likely regulates cell growth and apoptosis signaling by associating with additional key components of cell growth and cell cycle signal transduction pathways. To test this possibility, we performed a yeast two-hybrid screening assay to identify additional binding partners of CARP-1/CCAR1. We found that CARP-1/CCAR1 specifically interacts with Anaphase Promoting Complex/Cyclosome (APC/C) subunit APC-2 protein [61]. Co-immunoprecipitation (co-IP)-western blot (WB) experiments revealed that CARP-1/CCAR1 (896–978) peptide harbored the epitope which interacts with APC-2. Additional co-IP-WB experiments revealed that CARP-1/CCAR1 also interacts with Cdc20 and Cdh1, co-activators of APC/C. The CARP-1/CCAR1 epitope that interacts with Cdc20 or Cdh1 is distinct from its APC-2-interacting epitope [61]. These findings indicate a broader CARP-1/CCAR1 association with APC/C proteome [61]. In light of the fact that APC/C plays a distinct role in various cell cycle checkpoints [62, 63] and deregulation of APC/C and its regulators and substrates has been implicated in tumor progression [64], the components of the APC/C proteome including its co-activator CARP-1/CCAR1 therefore represent attractive targets for design of cell cycle inhibitory strategies with potential for therapeutic use [65–67].
Table 1: List of CARP-1/CCAR1 Functions and Interacting Proteins
CARP-1/CCAR-1 functions and interacting partners |
References |
Transducer of apoptosis signaling |
Rishi et al., 2003 [3] |
Negative regulator of EGFR signaling |
|
Binds and co-activates p53 |
Kim et al., 2008 [10] |
Regulator of steroid/thyroid receptor signaling |
Kim et al., 2008 [10] |
Positively regulates adipocyte differentiation |
Ou et al., 2014 [11] |
Binding partner of β-catenin |
Ou et al., 2009 [14] |
Associated with DBC-1 in NRs transcription |
Yu et al., 2011 [25] |
Binds to Ngn3 in nucleus |
Lu et al., 2012 [27] |
Negatively regulated by Necdin |
François et al., 2012 [36] |
Binding partner of AKAP350A |
Kolobova et al., 2009 [39] |
Regulates Notch signaling in association with Par-4/THAP1 |
Lu et al., 2013 [54] |
Binding partner of LIM protein Zyxin |
Hervy et al., 2010 [56] |
Interacts with SAPK p38 |
Papin et al., 2005 [59] |
Interacts with NEMO/IKKγ |
Bouwmeester et al., 2004 [60] |
Binds and co-activates APC-2 protein |
Puliyappadamba et al., 2011 [61] |
Interacts with CDC20 and Cdh1 |
Puliyappadamba et al., 2011 [61] |
Involved in apoptosis signaling in Mesothelioma, Medulloblastoma, and Neuroblastoma cells |
Ashour et al., 2013 [68] |
Binding partner of DEDD2, FADD, RIPK1 and p43/41 fragment of cleaved Caspase 8 |
Muthu et al., 2015 [71] |
CARP-1/CCAR1 Functional Mimetics (CFMs)
Based on the knowledge of CARP-1/CCAR1 pro-apoptotic signaling in diverse cancer cells, and the fact that CARP-1/CCAR1 binds with APC-2 and co-activates APC/C, while APC/C is a crucial regulator of cell cycle, led us to speculate that disruption of APC/C co-activation by CARP-1/CCAR1 could impact cell cycle progression and ultimately the growth and survival of the cells. To test this possibility, we conducted a high-throughput chemical biology experiment to identify small molecule inhibitors (SMIs) of CARP-1/CCAR1-APC-2 interaction. This experiment yielded multiple, novel SMIs of CARP-1/CCAR1-APC-2 binding, termed CARP-1/CCAR1 Functional Mimetics (CFMs) [61]. CFM-1, CFM-4, and CFM-5 compounds inhibited CARP-1/CCAR1-APC-2 interaction with IC50 values of 4, 1, and 0.75 μM, respectively, while CFM-4 and CFM-5 compounds also bind with CARP-1/CCAR1. Although all the three compounds inhibited growth of a variety of cancer cells including HBC cells in vitro, CFM-4 and CFM-5 compounds induced G2M cell cycle arrest, promoted loss of cyclin B1, p21WAF1/CIP1, p27KIP1. CFMs cause increase in CARP-1/CCAR1 levels, and apoptosis in part by activation of caspases-9, -8, and -3 [61]. Moreover, pharmacologic blockage of caspase-8, but not caspase-9, prevented apoptosis by CFM-4 indicating that caspase-8 activation is necessary for apoptosis signaling by CFM-4 [61; Figure 2]. Importantly, knock-down of CARP-1/CCAR1 or APC/C co-activator Cdh1 interferes with apoptosis by CFM-4, suggesting that CARP-1/CCAR1 and Cdh1 play a crucial role in regulating apoptosis by CFMs [61]. Moreover, CFM-4 attenuates proliferation of the ADR or Tamoxifen (TAM)-resistant MCF7 HBC cells without impacting the growth of immortalized, non-tumorigenic human MCF-10A cells [61]. Thus, our discoveries of CARP-1/CCAR1 and CFMs provide a proof-of-concept that CARP-1/CCAR1 signaling could be exploited to identify agents to inhibit breast cancer cells and their drug-resistant variants.
Given the fact that CFMs inhibited growth of breast cancer cells, we further investigated their potential in inhibiting other cancers such as malignant pleural mesothelioma (MPM), medulloblastoma (MB), neuroblastoma (NB) and non-small cell lung cancer (NSCLC) that often have poor prognosis and are known to develop resistance to therapeutic modalities in clinic. Consistent with our previous observations with HBC cells, CFM-4 and CFM-5 compounds inhibited the growth of MPM, MB, NB, and NSCLC cells [68–71] in part by activating apoptosis signaling and diminishing the levels of key cell cycle regulatory proteins including cyclin-B1, c- and N-myc. Both CFM-4 and CFM-5 compounds enhanced expression of CARP-1/CCAR1 along with activation of pro-apoptotic SAPKs (p38α/β and JNK1/2) [68–71]. CFMs, particularly CFM-4, also activated NF-κB signaling by depleting its inhibitors ABIN1 and inhibitory κB (IκB) α and β proteins [68–70], while increasing the expression of pro-apoptotic death receptor (DR) 4 protein. CFM-4 also enhanced expression of serine-phosphorylated cell surface sialo-glycoprotein podoplanin that regulates cell motility and integrity, while promoting cleavage of vimentin in MPM cells [69]. In light of the fact that serine phosphorylation in the short cytoplasmic domain of podoplanin interferes with the processes of cellular motility [72], podoplanin phosphorylation and cleavage of vimentin in the CFM-4-treated MPM cells would suggest for motility and metastasis-inhibitory properties of CFMs [69]. Our gene-array based analysis in MB cells revealed that CFM-4 enhanced the expression of neurotrophin (NTF3), while depletion of NTF3 expression inhibited MB cell apoptosis by CFM-4, suggesting a potential therapeutic window with CFM-4 mediated apoptosis through NTF3 signaling in MB cells [68].
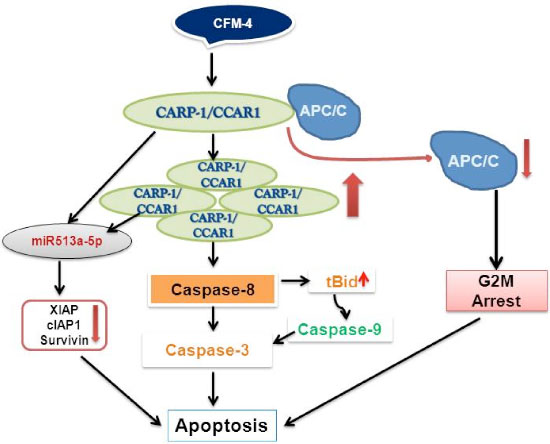
Figure 2: A Schematic of CFM-4-CARP-1/CCAR1-APC/C Signaling.
Interestingly, a high-throughput miRNA profiling of malignant pediatric tumor NB cells revealed up-regulation of XIAP-targeting miR513a-5p in response to CFM-4 treatment [70]. Both CFM-4 and CFM-5 compounds significantly abrogated the expression of cell survival associated XIAP1, cIAP1 and survivin proteins in NB as well as in HBC cells. Moreover, expression of anti-miR513a-5p or miR513a-5p mimic interfered with or enhanced, respectively, the apoptosis by CFM-4 in HBC cells [70], suggesting involvement of miR-dependent post-transcriptional targeting of XIAP family of proteins in inhibiting both NB and HBC cell growth in the presence of CFM-4 [70; Figure 2]. Although CFM compounds target podoplanin and vimentin signaling, further in vitro studies indicate that CFMs impact ability of the MB, MPM, NB, NSCLC, and HBC cells to grow in soft agar, migrate to close a wound, and invade through the matrigel-coated membranes [68–71].
In an attempt to develop CFMs as potential anti-cancer agent, we performed structure activity relationship (SAR) studies and identified six additional compounds, termed as CFM-4.1 – CFM-4.6, that were structurally similar to our lead compound CFM-4. Among all, CFM-4.6 was very effective in inhibiting the growth of NSCLC (A549, H1299) and MDA-MB-231 triple negative breast cancer (TNBC) cells in vitro. Consistent with our earlier studies, both CFM-4 and CFM-4.6 enhanced the CARP-1/CCAR1 expression and activated the pro-apoptotic SAPKs p38α/β and JNK1/2. Additionally, we found that these compounds stimulated the expression of death effector domain containing protein DEDD2. Co-IP-WB studies revealed that CARP-1/CCAR1 directly interacts with DEDD2, FADD, RIPK1 and p43/41 fragment of cleaved caspase 8 [71]. Since activation of caspase 8 is required for apoptosis by CFM-4 [61], and the fact that activation of caspase 8 is often regulated by TNFα receptor (TNFR)/DR family of proteins [73–75], it is likely that CFMs promote formation of an apical, pro-apoptotic sub-complex of CARP-1/CCAR1-DEDD2-FADD-RIPK1-p43/41 caspase 8 proteins. Moreover, since CARP-1/CCAR1 and DEDD2 are known to shuttle between cytosol and nucleus [3, 76, 77], it is possible that CFMs induce apoptosis by stimulating levels of CARP-1/CCAR1 and DEDD2 proteins and their intracellular shuttling to transduce apoptosis [71]. CFM-4, CFM-5, and CFM-4 analog CFM-4.6 disrupt formation of tubules by human vascular endothelial cells (HUVECs) in vitro, suggesting that these compounds likely also possess anti-angiogenic properties [71].
CFM-4 nano lipid formulations (CFM-4 NLFs)
To demonstrate the therapeutic potential of CFMs, preclinical studies in xenograft models of MB, MPM, and breast cancers were carried out. Direct, intravenous administration of CFM-4 cocktail failed to inhibit growth of subcutaneously xenografted tumor cells in immunocompromised mice [71]. Since, nanolipid based formulations often increase the physicochemical stability of both the drug molecules and particulate system, and have higher drug-loading capacity, we next investigated whether a nanolipid based formulation of CFM-4 could be generated and utilized to further test the therapeutic potential of CFM-4 compound. A natural, biocompatible cationic polysaccharide Chitosan-based nanolipid formulation of CFM-4 (CFM-4 NLF) was prepared as described before [71, 78]. CFM-4 NLF had superior bioavailability and pharmacokinetics when compared with the CFM-4 free drug [71], and inhibited growth of xenografted TNBC and NSCLC tumors in nude mice, when administered orally [71]. Biochemical analyses of representative NSCLC and TNBC tumor xenografts from placebo (control) or CFM-4 NLF-treated animals revealed accumulation of CFM-4 NLF in tumors of treated animals. Additionally, Immuno-histological analyses showed elevated levels of CARP-1/CCAR1 and fragmented DNA in tumors of CFM-4 NLF-treated animals when compared with those of placebo-treated controls [71]. These findings provide a proof-of-concept that CFMs possess therapeutic potential, and function in vivo in part by stimulating CARP-1/CCAR1 levels and apoptosis.
CONCLUSION AND FUTURE PERSPECTIVE
Overall, it is becoming increasingly evident that CARP-1/CCAR1 regulates signaling ranging from co-activation of physiological responses to steroids, processes of cellular differentiation and homeostasis in different tissues, to the chemotherapy-dependent apoptosis signaling with or without co-activation of tumor suppressor p53. Identification of CFMs through the chemical biology strategies provides a further proof-of-concept that CARP-1/CCAR1 and an aspect of its signaling could be exploited to inhibit cancer cells. The lead compound CFM-4 inhibits growth of a variety of cancer cells including the chemo-resistant breast cancer cells without impacting growth of immortalized, non-tumorigenic breast epithelial cells. Although CFM-4 and CFM-5 compounds function in part by binding and elevating cellular levels of CARP-1/CCAR1, apoptosis by radiation, chemotherapy (such as ADR, Etoposide, or Gefitinib), the CFM-4 and CFM-5 compounds, and the physiological responses of various steroids nevertheless require CARP-1/CCAR1. Thus targeting of CARP-1/CCAR1 could allow for physiological fine tuning of steroid responses, the discovery of CFMs and their future analogs on the other hand offer novel tools for design of anti-cancer strategies for combating a variety of cancers including the TNBCs and their drug-resistant variants. The promising in vitro and in vivo pre-clinical studies thus far provide a compelling rationale for further exploration of the pleiotropic cell growth and apoptosis signaling by CARP-1/CCAR1, and potential of CFMs as novel agents for optimization of anti-cancer therapeutic strategies. Since current treatment options for TNBCs as well as drug-resistant cancers of breast, lung, MPM, and neurological origins are limited, discovery and development of molecules based on CARP-1/CCAR1 signaling could yield novel tools for management and treatment of cancers and thus potentially serve an urgent and unmet need.
REFERENCES
1. Stewart B, Wild CP. (Eds) World Cancer Report 2014. Chapter 1: Cancer Worldwide. International Agency for Research on Cancer, World Health Organization.
2. Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, Smith T, Cooper D, Gansler T, Lerro C, Fedewa S, Lin C, Leach C, Cannady RS, Cho H, Scoppa S, Hachey M, Kirch R, Jemal A, Ward E. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012; 62:220–41.
3. Rishi AK, Zhang L, Boyanapalli M, Wali A, Mohammad RM, Yu Y, Fontana JA, Hatfield JS, Dawson MI, Majumdar AP, Reichert U. Identification and characterization of a cell cycle and apoptosis regulatory protein-1 as a novel mediator of apoptosis signaling by retinoid CD437. J Biol Chem. 2003; 278:33422–35.
4. Rishi AK, Zhang L, Yu Y, Jiang Y, Nautiyal J, Wali A, Fontana JA, Levi E, Majumdar APN. Cell cycle and apoptosis regulatory protein [CARP]-1 is involved in apoptosis signaling by epidermal growth factor receptor. J Biol Chem. 2006; 281:13188–98.
5. Jiang Y, Puliyappadamba VT, Zhang L, Wu W, Wali A, Yaffe MB, Fontana JA, Rishi AK. A novel mechanism of cell growth regulation by cell cycle and apoptosis regulatory protein (CARP)-1. J Mol Signaling. 2010; 5:7.
6. Sladek FM. What are nuclear receptor ligands? Mol Cell Endocrinol. 2011; 334:3–13.
7. Stallcup MR, Kim JH, Teyssier C, Lee YH, Ma H, Chen D. The roles of protein-protein interactions and protein methylation in transcriptional activation by nuclear receptors and their coactivators. J Steroid Biochem Mol Biol. 2003; 85:139–45.
8. Kim JH, Li H, Stallcup MR. CoCoA, a nuclear receptor coactivator which acts through an N-terminal activation domain of p160 coactivators. Mol Cell. 2003; 12:1537–49.
9. Teyssier C, Belguise K, Galtier F, Chalbos D. Characterization of the physical interaction between-estrogen receptor α and JUN proteins. J. Biol. Chem. 2001; 276:36361–36369.
10. Kim JH, Yang CK, Heo K, Roeder RG, An W, Stallcup MR. CCAR1, a key regulator of mediator complex recruitment to nuclear receptor transcription complexes. Mol Cell. 2008; 31:510–9.
11. Ou CY, Chen TC, Lee JV, Wang JC, Stallcup MR. Coregulator cell cycle and apoptosis regulator 1 (CCAR1) positively regulates adipocyte differentiation through the glucocorticoid signaling pathway. J Biol Chem. 2014; 289:17078–86.
12. Mizuta S, Minami T, Fujita H, Kaminaga C, Matsui K, Ishino R, Fujita A, Oda K, Kawai A, Hasegawa N, Urahama N, Roeder RG, Ito M. CCAR1/CoCoA pair-mediated recruitment of the Mediator defines a novel pathway for GATA1 function. Genes Cells. 2014; 19:28–51.
13. Seo WY, Jeong BC, Yu EJ, Kim HJ, Kim SH, Lim JE, Kwon GY, Lee HM, Kim JH. CCAR1 promotes chromatin loading of androgen receptor (AR) transcription complex by stabilizing the association between AR and GATA2. Nucleic Acids Res. 2013; 41:8526–36.
14. Ou CY, Kim JH, Yang CK, Stallcup MR. Requirement of cell cycle and apoptosis regulator 1 for target gene activation by Wnt and beta-catenin and for anchorage-independent growth of human colon carcinoma cells. J Biol Chem. 2009; 284:20629–37.
15. Hamaguchi M, Meth JL, von Klitzing C, Wei W, Esposito D, Rodgers L, Walsh T, Welcsh P, King MC, Wigler MH. DBC2, a candidate for a tumor suppressor gene involved in breast cancer. Proc Natl Acad Sci U S A. 2002; 99:13647–52.
16. Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008; 451:583–6.
17. Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. 2008; 451:587–90.
18. Magni M, Ruscica V, Buscemi G, Kim JE, Nachimuthu BT, Fontanella E, Delia D, Zannini L. Chk2 and REGγ-dependent DBC1 regulation in DNA damage induced apoptosis. Nucleic Acids Res. 2014; 42:13150–60.
19. Sundararajan R, Chen G, Mukherjee C, White E. Caspase-dependent processing activates the proapoptotic activity of deleted in breast cancer-1 during tumor necrosis factor-alpha-mediated death signaling. Oncogene. 2005; 24:4908–20.
20. Trauernicht AM, Kim SJ, Kim NH, Clarke R, Boyer TG. DBC-1 mediates endocrine resistant breast cancer cell survival. Cell Cycle. 2010; 9:1218–1219.
21. Lee H, Kim KR, Noh SJ, Park HS, Kwon KS, Park BH, Jung SH, Youn HJ, Lee BK, Chung MJ, Koh DH, Moon WS, Jang KY. Expression of DBC1 and SIRT1 is associated with poor prognosis for breast carcinoma. Hum Pathol. 2011; 42:204–13.
22. Radvanyi L, Singh-Sandhu D, Gallichan S, Lovitt C, Pedyczak A, Mallo G, Gish K, Kwok K, Hanna W, Zubovits J, Armes J, Venter D, Hakimi J, Shortreed J, Donovan M, Parrington M, Dunn P, Oomen R, Tartaglia J, Berinstein NL. The gene associated with trichorhinophalan-geal syndrome in humans is overexpressed in breast cancer. Proc Natl Acad Sci U S A. 2005; 102:11005–10.
23. Chini EN, Chini CC, Nin V, Escande C. Deleted in breast cancer -1 (DBC-1) in the interface between metabolism, aging and cancer. Biosci Rep. 2013; 33:637–643.
24. Brunquell J, Yuan J, Erwin A, Westerheide SD, Xue B. DBC1/CCAR2 and CCAR1 are largely disordered proteins that have evolved from one common ancestor. BioMed Res Int. 2014; 1–13.
25. Yu EJ, Kim SH, Heo K, Ou CY, Stallcup MR, Kim JH. Reciprocal roles of DBC1 and SIRT1 in regulating estrogen receptor α activity and co-activator synergy. Nucleic Acids Res. 2011; 39:6932–43.
26. Wu DY, Ou CY, Chodankar R, Siegmund KD, Stallcup MR. Distinct, genome-wide, gene-specific selectivity patterns of four glucocorticoid receptor coregulators. Nucl Recept Signal. 2014; 12:e003.
27. Lu CK, Lai YC, Lin YF, Chen HR, Chiang MK. CCAR1 is required for Ngn3-mediated endocrine differentiation. Biochem Biophys Res Commun. 2012; 418:307–12.
28. Sharma S, Mahalingam CD, Das V, Jamal S, Levi E, Rishi AK, Datta NS. Cell cycle and apoptosis regulatory protein (CARP)-1 is expressed in osteoblasts and regulated by PTH. Biochem Biophys Res Commun. 2013; 436:607–12.
29. Rawadi G, Roman-Roman S. Wnt signalling pathway: a new target for the treatment of osteoporosis. Expert Opin Ther Targets. 2005;9:1063–77.
30. Krishnan V, Bryant HU, Macdougald OA. Regulation of bone mass by Wnt signaling. J Clin Invest. 2006; 116:1202–9.
31. Almeida M, Han L, Bellido T, Manolagas SC, Kousteni S. Wnt proteins prevent apoptosis of both uncommitted osteoblast progenitors and differentiated osteoblasts by beta-catenin-dependent and -independent signaling cascades involving Src/ERK and phosphatidylinositol 3-kinase/AKT. J Biol Chem. 2005; 280:41342–51.
32. Kulkarni NH, Halladay DL, Miles RR, Gilbert LM, Frolik CA, Galvin RJ, Martin TJ, Gillespie MT, Onyia JE. Effects of parathyroid hormone on Wnt signaling pathway in bone. J Cell Biochem. 2005; 95:1178–90.
33. Tobimatsu T, Kaji H, Sowa H, Naito J, Canaff L, Hendy GN, Sugimoto T, Chihara K. Parathyroid hormone increases beta-catenin levels through Smad3 in mouse osteoblastic cells. Endocrinology. 2006; 147:2583–90 Epub 2006.
34. Wan M, Yang C, Li J, Wu X, Yuan H, Ma H, He X, Nie S, Chang C, Cao X. Parathyroid hormone signaling through low-density lipoprotein-related protein 6. Genes Dev. 2008; 22:2968–79. doi: 10.1101/gad.1702708.
35. Suzuki A, Ozono K, Kubota T, Kondou H, Tachikawa K, Michigami T. PTH/cAMP/PKA signaling facilitates canonical Wnt signaling via inactivation of glycogen synthase kinase-3beta in osteoblastic Saos-2 cells. J Cell Biochem. 2008; 104:304–17.
36. François S, D’Orlando C, Fatone T, Touvier T, Pessina P, Meneveri R, Brunelli S. Necdin enhances myoblasts survival by facilitating the degradation of the mediator of apoptosis CCAR1/CARP1. PLoS One. 2012; 7:e43335. doi: 10.1371/journal.pone.0043335 Epub 14..
37. Taniura H, Matsumoto K, Yoshikawa K. Physical and functional interactions of neuronal growth suppressor necdin with p53. J Biol Chem. 1999; 274:16242–8.
38. Sciorati C, Touvier T, Buono R, Pessina P, François S, Perrotta C, Meneveri R, Clementi E, Brunelli S. Necdin is expressed in cachectic skeletal muscle to protect fibers from tumor-induced wasting. J Cell Sci. 2009; 122:1119–25.
39. Kolobova E, Efimov A, Kaverina I, Rishi AK, Schrader JW, Ham AJ, Larocca MC, Goldenring JR. Microtubule-dependent association of AKAP350A and CCAR1 with RNA stress granules. Exp Cell Res. 2009; 315:542–55.
40. Tourrière H, Chebli K, Zekri L, Courselaud B, Blanchard JM, Bertrand E, Tazi J. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J Cell Biol. 2003; 160:823–31.
41. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972; 26:239–257.
42. Johnstone RW, Rufeli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002; 108:153–164.
43. Yoo AS, Bais C, Greenwald I. Crosstalk between the EGFR and LIN-12/Notch pathways in C. elegans vulval development. Science. 2004; 303:663–666.
44. Luo W, Slebos RJ, Hill S, Li M, Brábek J, Amanchy R, Chaerkady R, Pandey A, Ham AJ, Hanks SK. Global impact of oncogenic Src on a phosphotyrosine proteome. J Proteome Res. 2008; 7:3447–60.
45. Jiang Y, Puliyappadamba VT, Zhang L, Wu W, Wali A, Yaffe MB, Fontana JA, Rishi AK. A novel mechanism of cell growth regulation by Cell Cycle and Apoptosis Regulatory Protein (CARP)-1. J Mol Signal. 2010; 1:5.
46. Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villen J, Li J, Cohn MA, Cantley LC, Gygi SP. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc Natl Acad Sci USA. 2004; 101:12130–12135.
47. Blagoev B, Kratchmarova I, Ong SE, Nielsen M, Foster LJ, Mann M. A proteomics strategy to elucidate functional protein-protein interactions applied to EGF signaling. Nat Biotechnol. 2003; 21:315–318.
48. Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ. ATM, and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007; 316:1160–1166.
49. Wu X, Tian L, Li J, Zhang Y, Han V, Li Y, Xu X, Li H, Chen X, Chen J, Jin W, Xie Y, Han J, Zhong CQ. Investigation of receptor interacting protein (RIP3)-dependent protein phosphorylation by quantitative phosphoproteomics. Mol Cell Proteomics. 2012; 11:1640–51.
50. Kettenbach AN, Schweppe DK, Faherty BK, Pechenick D, Pletnev AA, Gerber SA. Quantitative phosphoproteomics identifies substrates and functional modules of aurora and polo-like kinase activities in mitotic cells. Sci Signal. 2011; 4:rs5.
51. Chen RQ, Yang QK, Lu BW, Yi W, Cantin G, Chen YL, Fearns C, Yates JR 3rd, Lee JD. CDC25B mediates rapamycin-induced oncogenic responses in cancer cells. Can Res. 2009; 69:2663–8.
52. Zhang L, Levi E, Majumder P, Yu Y, Aboukameel A, Du J, Xu H, Mohammad R, Hatfield JS, Wali A, Adsay V, Majumdar AP, Rishi AK. Transactivator of transcription-tagged cell cycle and apoptosis regulatory protein-1 peptides suppress the growth of human breast cancer cells in vitro and in vivo. Mol Cancer Ther. 2007; 6:1661–72.
53. Levi E, Zhang L, Aboukameel A, Rishi S, Mohammad RM, Polin L, Hatfield JS, Rishi AK. Cell cycle and apoptosis regulatory protein (CARP)-1 is a novel, adriamycin-inducible, diffuse large B-cell lymphoma (DLBL) growth suppressor. Cancer Chemother Pharmacol. 2011; 67:1401–13.
54. Lu C, Li JY, Ge Z, Zhang L, Zhou GP. Par-4/THAP1 complex and Notch3 competitively regulated pre-mRNA splicing of CCAR1 and affected inversely the survival of T-cell acute lymphoblastic leukemia cells. Oncogene. 2013; 32:5602–13.
55. Tzoneva G, Ferrando AA. Recent advances on NOTCH signaling in T-ALL. Curr Top Microbiol Immunol. 2012; 360:163–82.
56. Hervy M, Hoffman LM, Jensen CC, Smith M, Beckerle MC. The LIM Protein Zyxin Binds CARP-1 and Promotes Apoptosis. Genes Cancer. 2010; 1:506–515.
57. Vasseur S, Malicet C, Calvo EL, Dagorn JC, Iovanna JL. Gene expression profiling of tumours derived from rasV12/E1A-transformed mouse embryonic fibroblasts to identify genes required for tumour development. Mol Cancer. 2005; 16;4:4.
58. Haugen AC, Di Prospero NA, Parker JS, Fannin RD, Chou J, Meyer JN, Halweg C, Collins JB, Durr A, Fischbeck K, Van Houten B. Altered gene expression and DNA damage in peripheral blood cells from Friedreich’s ataxia patients: cellular model of pathology. PLoS Genet. 2010; 6:e1000812.
59. Papin JA, Hunter T, Palsson BO, Subramaniam S. Reconstruction of cellular signalling networks and analysis of their properties. Nat Rev Mol Cell Biol. 2005; 6:99–111.
60. Bouwmeester T, Bauch A, Ruffner H, Angrand PO, Bergamini G, Croughton K, Cruciat C, Eberhard D, Gagneur J, Ghidelli S, Hopf C, Huhse B, Mangano R, Michon AM, Schirle M, Schlegl J, Schwab M, Stein MA, Bauer A, Casari G, Drewes G, Gavin AC, Jackson DB, Joberty G, Neubauer G, Rick J, Kuster B, Superti-Furga G. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol. 2004; 6:97–105.
61. Puliyappadamba VT, Wu W, Bevis D, Zhang L, Polin L, Kilkuskie R, Finley RL Jr, Larsen SD, Levi E, Miller FR, Wali A, Rishi AK. Antagonists of anaphase-promoting complex (APC)-2-cell cycle and apoptosis regulatory protein (CARP)-1 interaction are novel regulators of cell growth and apoptosis. J Biol Chem. 2011; 286:38000–17.
62. Zachariae W, Nasmyth K. Whose end is destruction: cell division and the anaphase-promoting complex. Genes Dev. 1999; 13:2039–58.
63. Harper JW, Burton JL, Solomon MJ. The anaphase-promoting complex: it’s not just for mitosis any more. Genes Dev. 2002; 16:2179–206.
64. Lehman NL, Tibshirani R, Hsu JY, Natkunam Y, Harris BT, West RB, Masek MA, Montgomery K, van de Rijn M, Jackson PK. Oncogenic regulators and substrates of the anaphase promoting complex/cyclosome are frequently overexpressed in malignant tumors. Am J Pathol. 2007; 170:1793–805.
65. Bhati R, Gökmen-Polar Y, Sledge GW Jr, Fan C, Nakshatri H, Ketelsen D, Borchers CH, Dial MJ, Patterson C, Klauber-DeMore N. 2-methoxyestradiol inhibits the anaphase-promoting complex and protein translation in human breast cancer cells. Cancer Res. 2007; 67:702–8.
66. Braunstein I, Miniowitz S, Moshe Y, Hershko A. Inhibitory factors associated with anaphase-promoting complex/cylosome in mitotic checkpoint. Proc Natl Acad Sci U S A. 2007; 104:4870–5.
67. Carvajal RD, Tse A, Schwartz GK. Aurora kinases: new targets for cancer therapy. Clin Cancer Res. 2006; 12:6869–75.
68. Ashour AE, Jamal S, Cheryan VT, Muthu M, Zoheir KM, Alafeefy AM, Abd-Allah AR, Levi E, Tarca AL, Polin LA, Rishi AK. CARP-1 functional mimetics: a novel class of small molecule inhibitors of medulloblastoma cell growth. PLoS One. 2013; 8:e66733.
69. Jamal S, Cheriyan VT, Muthu M, Munie S, Levi E, Ashour AE, Pass HI, Wali A, Singh M, Rishi AK. CARP-1 functional mimetics are a novel class of small molecule inhibitors of malignant pleural mesothelioma cells. PLoS One. 2014; 9:e89146.
70. Muthu M, Cheriyan VT, Munie S, Levi E, Frank J, Ashour AE, Singh M, Rishi AK. Mechanisms of neuroblastoma cell growth inhibition by CARP-1 functional mimetics. PLoS One. 2014; 9:e102567.
71. Muthu M, Somagoni JM, Cheriyan VT, Munie S, Levi E, Ashour AE, Alafeefy AM, Sochacki P, Polin LA, Reddy KB, Larsen SD, Singh M, Rishi AK. Identification and testing of novel CARP-1 functional mimetic compounds as inhibitors of non-small cell lung and triple-negative breast cancers. J Biomed Nanotechnol. 2015; 11:1–20.
72. Krishnan H, Ochoa-Alvarez JA, Shen Y, Nevel E, Lakshminarayanan M, Williams MC, Ramirez MI, Miller WT, Goldberg GS. Serines in the intracellular tail of podolplanin (PDPN) regulates cell motility. J Biol Chem. 2013; 288:12215–12221.
73. Biton S, Ashkenazi A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-α feedforward signaling. Cell. 2011; 145:92–103.
74. Bremer E. Targeting of the Tumor Necrosis Factor Receptor Superfamily for Cancer Immunotherapy. ISRN Oncology. 2013; 2013:1–25.
75. Lin Y, Devin A, Rodriguez Y. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Develop. 1999; 13:2514–2526.
76. Roth W, Stenner-Liewen F, Pawlowski K, Godzik A, Reed JC. Identification and characterization of DEDD2, a death effector domain-containing protein. J Biol Chem. 2002; 277:7501–7508.
77. Alcivar A, Hu Shimin, Tang J, Yang X. DEDD and DEDD2 associate with caspase-8/10 and signal cell death. Oncogene. 2003; 22:291–297.
78. Paños I, Acosta N, Heras A. New drug delivery systems based on chitosan. Curr Drug Discov Technol. 2008; 5:333–41.