
CANONICAL HEDGEHOG SIGNALING IN CANCER
Canonical HH signaling engages PTCH, SMO and the GLI family of transcription factors (Figure 1), and in normal cellular processes is involved in embryogenesis, tissue patterning, stem cell function, and differentiation[1, 2]. Several types of human cancers have demonstrated aberrant activation of the HH pathway by ligand-independent signaling such as, amplification of GLI1 or GLI2, mutations in PTCH or SMO, or dysregulated gene expression[1, 3]. In colon cancer, aberrant HH signaling progresses during carcinogenesis and in metastatic disease[4-6], and is also activated in human colon carcinoma cell lines[7-9] and xenograft models[4], by ligand-dependent activation, that occurs in GI cancers[1, 10]. However, the role of HH signaling and its importance in driving cellular survival in colon cancer are not well defined. Small molecule inhibitors of SMO have been studied in preclinical models, and applied to the treatment of various types of cancers in humans[4, 9, 11-14]. Those tumors sensitive to SMO inhibitors, which include basal cell carcinoma[15, 16] and medulloblastoma[11, 17], rely on canonical HH signaling for cellular survival. In other cancer types, SMO inhibitors including GDC-0449, IPI-926 or LDE225, have demonstrated limited clinical activity (reviewed in [11, 12]). Intrinsic resistance to SMO inhibitors is frequent[11-14, 18, 19], and acquired resistance to GDC-0449 following initial response has been reported in medulloblastoma (heterozygous mutation, Asp->His at aa 473 in SMO)[20]. Thus targeting the GLI genes downstream of SMO, that constitute the core of HH-dependent gene regulation, may provide a significant advantage in eliminating HH signaling.
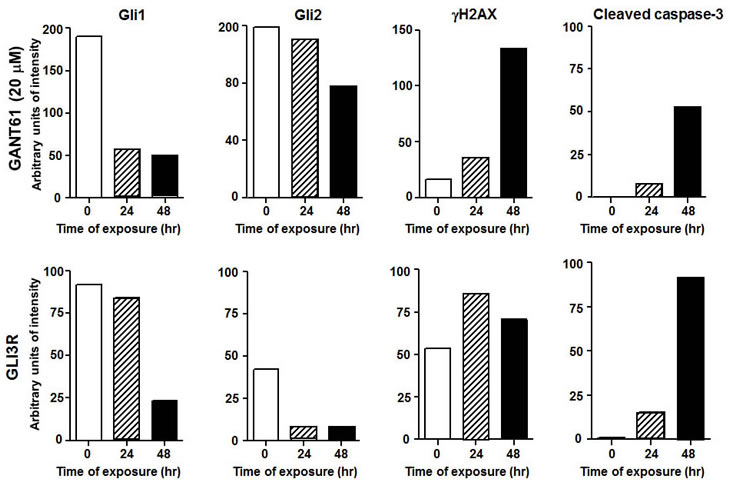
Figure 1: Canonical HH signaling and non-canonical GLI gene activation.
Activation of GLI by oncogenic, non-canonical signaling pathways
Non-canonical, oncogene-driven signaling pathways converge on the activation of GLI genes and further converge on their specific downstream targets[3, 18, 21, 22] (see Figure 1). The RAS/RAF/MEK/ERK pathway, with activating mutations in K-RAS or B-RAF that occur in high frequency in colon cancers[23-25], activates GLI function[18, 19, 21]. In HT29 cells (mutated B-RAF V600E[25]), we demonstrated inhibition of GLI-luciferase reporter activity, reduced expression of GLI1 mRNA and protein, and of p-ERK in response to the MEK/ERK and RAS/RAF signaling inhibitor U0126[26, 27] (Figure 2). While loss-of-function mutations in PTCH and gain-of-function mutations in SMO activate HH signaling[1], acquired mutations in SMO or non-canonical GLI activation render cancer cells resistant to SMO antagonists. These observations emphasize the importance of targeting the GLI genes downstream of SMO for terminating HH-dependent survival and inducing cell death in colon carcinoma cells. It therefore follows that termination of HH signaling at the level of GLI may constitute a molecular switch that determines the balance between cell survival or cell death.
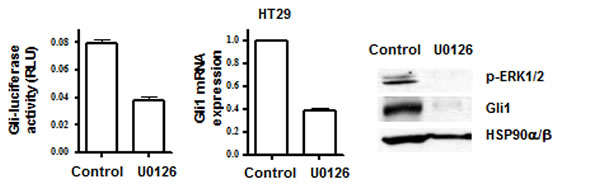
Figure 2: Inhibition of the RAS/RAF pathway by UO126 (20 µM) decreases GLI-luc activity (left), GLI1 mRNA (center), GLI1 and p-ERK1/2 proteins (right).
TARGETING GLI1 AND GLI2 WITH GANT61
One of the gaps in our knowledge is that the impact of terminating HH signaling at the level of the GLI genes is unclear. The GLI family of transcription factors regulates target gene expression that determines HH-dependent survival. GLI1 and GLI2 are the primary activators of HH signaling; further, the cooperative roles of GLI1 and GLI2 are critical in the transcriptional regulation of HH target genes[1, 28-30]. While SMO has been extensively investigated as a therapeutic target[11-15, 17], few agents are available that target the GLI genes[31]. GANT61 was identified in a cell-based screen for small molecule inhibitors of GLI1-mediated transcription. In the original study[31], GANT61 1) functions in the nucleus to abrogate GLI function; 2) blocks both GLI1- and GLI2- mediated transcription; 3) in SUFU-null MEFS with constitutively active HH signaling, reduces expression of GLI1 and HIP1 mRNA in contrast to cyclopamine; 4) inhibits GLI1 DNA binding activity (EMSA analysis). We further demonstrated the specificity of GANT61 in targeting GLI1 and GLI2 from reduced GLI1 and GLI2 protein expression[8], inhibition of the binding of GLI1 and GLI2 to the promoter regions of HH target genes, specificity of reduction in GLI-luciferase reporter activity, and rapid inhibition of the transcriptional regulation of the GLI target gene BCL-2, within 1 hr of GANT61 exposure[7].
Inhibition of GLI induces greater cytotoxicity than targeting SMO
GANT61, and the classic SMO inhibitor cyclopamine (for comparison with other model systems), were evaluated in a panel of human colon carcinoma cell lines to determine the impact of inhibiting GLI1/GLI2 vs SMO. Inhibition of GLI1 and GLI2 by GANT61 induced > 80% cell death in 5/7 human colon carcinoma cell lines following 72 hr exposure (20 µM). In contrast, cyclopamine induced < 30% cell death in 6/7 cell lines, at equimolar concentrations (20 µM; Figure 3). In time course studies, 24 hr exposure to GANT61 or 72 hr exposure to cyclopamine was required to initiate cell death[8]. In addition the SMO inhibitor GDC-0449, like cyclopamine, demonstrated limited cytotoxic activity in HT29 cells at equimolar concentrations (unpublished data). Collectively, the data demonstrate increased sensitivity of human colon cancer cells to inhibition of GLI genes compared to that of SMO. This is consistent with the presence of a non-canonical mode of GLI activation in human colon cancer cells. These concentrations and time frames for the induction of cellular effects are similar to those determined in other model systems for inhibitors of HH signaling[4, 19, 33].
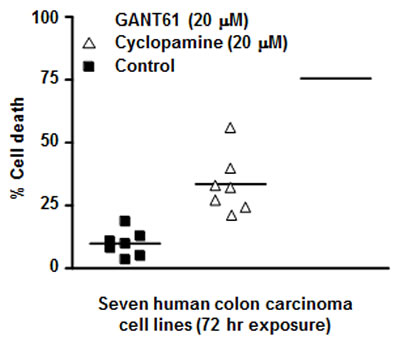
Figure 3: GANT61- and Cyclopamine- induced cell death.
Genetic downregulation of GLI1 and GLI2 by GLI3R parallels cellular effects mediated by pharmacologic inhibition of GLI1 and GLI2 by GANT61
A third member of the GLI family is GLI3. A cleaved C-terminally truncated form of GLI3 (GLI3R) demonstrates repressor activity for GLI1 and GLI2 transcriptional regulation, and thereby silences HH-GLI target genes[4, 34]. We confirmed the critical role of GLI1 and GLI2 in cell survival by genetic downregulation of GLI1 and GLI2 following transient transfection of GLI3R[4, 34] in HT29 cells. Transient transfection and expression of GLI3R (GLI3R-pCS2-MT, N-terminal Myc-epitope tag) over a period of 72 hr paralleled the effects of GANT61 by inducing significant changes in cellular morphology, reduced expression of GLI1 and GLI2, reduced proliferation, and induction of cell death. GLI3R-myc expression was detected by 24 hr, with strong expression by 48 hr[32]. Under these conditions of GLI3R transient transfection, GLI1 and GLI2 expression was decreased, with significant cleavage of caspase-3 (Figure 4). To begin to explore the cellular mechanisms downstream of GLI1/GLI2 inhibition that lead to cell death, we demonstrated that γH2AX, a marker of DNA DSBs[35], was expressed in GANT61-treated cells (Figure 4). Further, γH2AX nuclear foci were detected in the cells expressing GLI3R-myc[32]. Thus, both genetic and pharmacologic downregulation of GLI1 and GLI2 induce parallel changes in events leading to cell death, including the induction of γH2AX nuclear foci. One of the critical regulators of DNA damage response is p53, which is stabilized upon DNA damage and modulates multiple components of the DNA damage response pathway[36, 37]. Expression of Gli3R-myc in human colon carcinoma cell lines harboring mutant p53 (functionally inactive) demonstrated DNA damage response suggesting a p53 independent mechanism[[32]]. Further, GANT61 treatment of HT29 cells demonstrated a p21Cip1-independent mechanism of cell death[32].
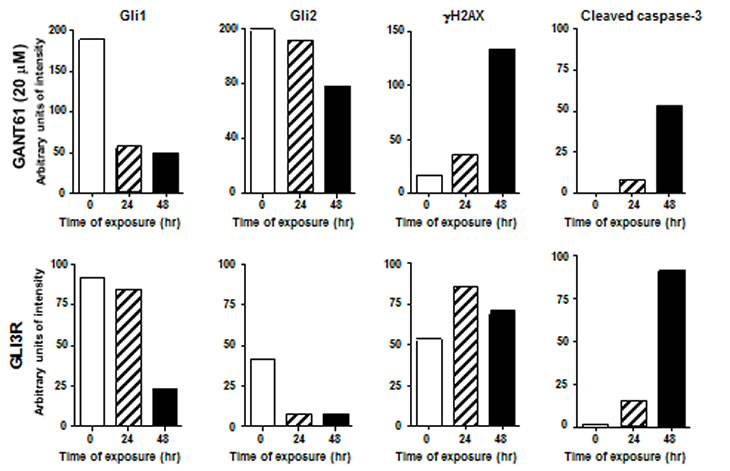
Figure 4: Treatment of HT29 cells with vehicle alone (0.2% DMSO) or GANT61 (20 µM; upper panel), or transient transfection of vector alone, or GLI3R-pCS2-MT[4, 34](lower panel) for 72 hr. Expression of proteins was by western analysis and analyzed by densitometry.
INHIBITION OF GLI1/GLI2 BY GANT61 INDUCES DNA DAMAGE VIA ATM/CHK2 SIGNALING
To further investigate the molecular mechanisms underlying DNA damage signaling downstream of GLI1/GLI2 inhibition, HT29 cells treated with GANT61 (20 µM) were examined for expression of the phosphorylated (active) forms of ATM, ATR, Chk1 and Chk2 by Western analysis or by confocal microscopy. p-ATM and p-Chk2 were detected as early as 4 hr, and their expression was sustained for at least 24 hr. In contrast, p-ATR and p-Chk1 expression remained undetectable[32]. p-Chk2 nuclear foci co-localized with γH2AX nuclear foci at the sites of DNA DSBs in GANT61-treated cells (Figure 5). An ATM/Chk2 axis was therefore activated in GANT61-treated human colon carcinoma cells as an early event in the response to DNA damage.
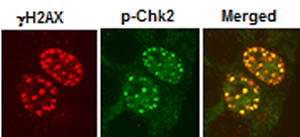
Figure 5: Co-localization of γH2AX and p-Chk2 nuclear foci following GANT61 (20 µM) for 4 hr in HT29 cells.
Inhibition of GLI1/GLI2 BY GANT61 induces arrest in early S
Using HT29 cells we have demonstrated that cells treated with GANT61 (20 µM) accumulate in early S-phase and fail to progress further through or beyond early S before becoming subG1[7, 32]. By cell cycle analysis, cells were observed to move from G1 into early S by 32 hr where they remain, detected as two discrete peaks (G1/S, early S; Figure 6A). Analysis by FACS/BrdU incorporation at 32 hr after GANT61 exposure demonstrated a shift in G1/S-phase cells from 8.0% BrdU incorporation in the control to 52.3% at 32 hr, and S-phase cells from 25.0% to 33.6%[32]. In contrast 5-fluorouracil (FUra) combined with leucovorin (1 µM), which targets thymidylate synthase in the inhibition of DNA replication and induction of DNA damage, arrested HT29 cells in mid S-phase[38] (67.0% by FACS/PI staining; Figure 6A), thereby demonstrating the difference in S-phase target location of the mechanism of GANT61-induced inhibition of DNA replication. The cell cycle checkpoints constitute a regulatory mechanism to arrest the cell cycle in response to DNA damage so that cell cycle progression and repair may be temporally coordinated[39-42]. In contrast to the checkpoints at the G1/S and G2/M transitions, the S-phase checkpoint can only delay the progression of S-phase. ATM is the master transducer of the S-phase checkpoint, responds to DNA DSBs, and together with its effector kinase Chk2, is activated in response to GLI1/GLI2 inhibition by GANT61. In activation of the intra-S-phase checkpoint, Cdc25A is phosphorylated on Ser123, which targets this phosphatase for degradation by the proteasome. Activation of Cdk2 and the loading of Cdc45 onto replication origins are inhibited, DNA replication is inhibited, the intra-S-phase checkpoint is activated, and cyclin E accumulates[43-45], as demonstrated in GANT61-treated HT29 cells at 24 hr (Figure 6B). Accumulation of HT29 cells in early S[9](Figure 6A), and the data of Figure 6B, suggest activation of an intra-S-phase checkpoint, which is not sustained.
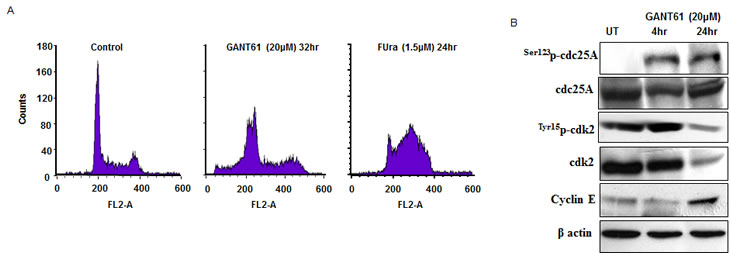
Figure 6. A: Flow cytometric analysis of propidium iodide-stained HT29 cells at 0 hr, or after GANT61 (20 µM) for 32 hr[7], or FUra (1.5 µM/leucovorin 1 µM) for 24 hr. B: Expression profile of proteins involved in activation of the intra-S-phase checkpoint (western).
GANT61 MODULATES GENES DOWNSTREAM OF GLI1/GLI2 INHIBITION INVOLVED IN THE DNA DAMAGE RESPONSE
To identify unique downstream targets of the GLI genes, analysis with cDNA microarray gene profiling of 18,401 genes identified Differentially Expressed Genes (DEGs) in HT29 (and GC3/c1)[7]. Analyses using GenomeStudio (statistics), Matlab (heat map), Ingenuity (canonical pathway analysis), or by qRT-PCR, identified decreased expression of mRNA for genes involved in DNA replication (including thymidylate synthase, thymidine kinase, topoisomerase2, DNA polymerases, E2F, replication factor C); DNA damage (including H2AX, MDC1, BRCA1, FANCD2, GADD45G, GADD153, REDD1, PCNA); and DNA repair (including RAD51, RAD54, FEN1, MSH6, Exonuclease 1), 24 hr after GANT61 exposure (20 µM; Table 1).
SUMMARY AND FUTURE DIRECTIONS
We have demonstrated in human colon carcinoma cells that the GLI genes, the transcriptional regulators of the HH signaling response, are activated by both canonical signaling (via SMO) and by non-canonical activation (via the RAS/RAF pathway), which is activated in high frequency in colon cancers. Both pharmacologic and genetic inhibition of GLI function reduced proliferation, GLI1 and GLI2 expression, induced γH2AX expression, cleavage of caspase-3, and cell death. In contrast inhibition of SMO was significantly less effective at inducing cell death in contrast to targeting GLI. DNA damage signaling involved an ATM/Chk2 axis, with Chk2 co-localizing with γH2AX at the sites of DNA DSBs. Further, cells accumulated in early S-phase following GANT61 exposure but did not progress before becoming subG1, suggesting an intra-S-phase checkpoint that could not be sustained. From cDNA microarray gene profiling, genes involved in the inhibition of DNA replication, DNA damage response, and DNA repair were identified downstream of GANT61-induced GLI1/GLI2 inhibition. Chromosome instability is found in the majority of colon cancers, resulting primarily from deregulation of the DNA replication and mitotic spindle checkpoints (reviewed in [45]). Genes involved in canonical HH signaling have been linked to genomic instability, involving inactivation of DNA repair mechanisms, defects in checkpoint activation, and predisposition to development of cancers[46-49]. It therefore follows that termination of HH signaling at the level of GLI may constitute a critical event in determining the balance between cell survival and cell death. The combination of molecular and cellular approaches that are being employed will 1) provide critical new insight into the role of GLI1/GLI2 in driving cellular survival, which has never been addressed in any type of human cancer, and will answer a fundamental question in HH biology, 2) address how dysregulation of the GLI1/GLI2 axis induces DNA damage upstream of cell death or DNA repair upstream of cell survival and 3) provide a framework for the design of strategies and therapeutics specifically targeted at HH signaling via GLI1 and GLI2 in human colon carcinoma.
Table 1: Decreased expression of genes downstream of GLI1/GLI2 inhibition following GANT61 (20 μM), 24 hr, HT29 cells; cDNA microarray gene profiling.
| Function | Fold change | Genes |
| DNA replication | -2.0 to -4.2 | TYMS, TK1, TOP2A, RRM1, RRM2, PRPS2, POLE, POLE2, POLA1, POLA2, POLQ, E2F2, CDT1, PRIM1, GMNN, RFC2, RFC3, RFC4, RFC5 |
| DNA damage response | -1.9 to -4.9 | H2AFX, MDC1, BRCA1, FANCD2, BARD1, CDC45L, DDIT2, DDIT3, DDIT4, PPP1R15A, PCNA, ATF3 |
| DNA repair | -1.9 to -4.2 | RAD51, RAD51C, RAD54B, RAD54L, FEN1, MSH6, KIAA0101, UNG, LIG1, EXO1, HELLS |
REFERENCES
1. Katoh Y, Katoh M: Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr Mol Med 2009, 9(7):873-886.
2. Yu M, Gipp J, Yoon JW, Iannaccone P, Walterhouse D, Bushman W: Sonic hedgehog-responsive genes in the fetal prostate. J Biol Chem 2009, 284(9):5620-5629.
3. Ruiz i Altaba A, Mas C, Stecca B: The Gli code: an information nexus regulating cell fate, stemness and cancer. Trends Cell Biol 2007, 17(9):438-447.
4. Varnat F, Duquet A, Malerba M, Zbinden M, Mas C, Gervaz P, Ruiz i Altaba A: Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol Med 2009, 1(6-7):338-351.
5. Yoshikawa K, Shimada M, Miyamoto H, Higashijima J, Miyatani T, Nishioka M, Kurita N, Iwata T, Uehara H: Sonic hedgehog relates to colorectal carcinogenesis. J Gastroenterol 2009, 44(11):1113-1117.
6. Bian YH, Huang SH, Yang L, Ma XL, Xie JW, Zhang HW: Sonic hedgehog-Gli1 pathway in colorectal adenocarcinomas. World J Gastroenterol 2007, 13(11):1659-1665.
7. Shi T, Mazumdar T, Devecchio J, Duan ZH, Agyeman A, Aziz M, Houghton JA: cDNA microarray gene expression profiling of hedgehog signaling pathway inhibition in human colon cancer cells. PLoS One 2010, 5(10):e13054.
8. Mazumdar T, DeVecchio J, Shi T, Jones J, Agyeman A, Houghton JA: Hedgehog signaling drives cellular survival in human colon carcinoma cells. Cancer Res 2011, 71(3):1092-1102.
9. Tremblay MR, McGovern K, Read MA, Castro AC: New developments in the discovery of small molecule Hedgehog pathway antagonists. Curr Opin Chem Biol 2010, 14(3):428-435.
10. Low JA, de Sauvage FJ: Clinical experience with Hedgehog pathway inhibitors. J Clin Oncol 2010, 28(36):5321-5326.
11. Mas C, Ruiz i Altaba A: Small molecule modulation of HH-GLI signaling: current leads, trials and tribulations. Biochem Pharmacol 2010, 80(5):712-723.
12. De Smaele E, Ferretti E, Gulino A: Vismodegib, a small-molecule inhibitor of the hedgehog pathway for the treatment of advanced cancers. Curr Opin Investig Drugs 2010, 11(6):707-718.
13. Lorusso PM, Rudin CM, Reddy JC, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, Chang I, Darbonne WC et al: Phase I Trial of Hedgehog Pathway Inhibitor Vismodegib (GDC-0449) in Patients with Refractory, Locally Advanced or Metastatic Solid Tumors. Clin Cancer Res 2011, 17(8):2502-2511.
14. Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR et al: Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med 2009, 361(12):1164-1172.
15. Dierks C: GDC-0449--targeting the hedgehog signaling pathway. Recent Results Cancer Res 2010, 184:235-238.
16. Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, Holcomb T, Stinson J, Gould SE, Coleman B et al: Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med 2009, 361(12):1173-1178.
17. Ji Z, Mei FC, Xie J, Cheng X: Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J Biol Chem 2007, 282(19):14048-14055.
18. Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, Beermann F, Ruiz IAA: Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc Natl Acad Sci U S A 2007, 104(14):5895-5900.
19. Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T, Pujara K, Stinson J, Callahan CA, Tang T et al: Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326(5952):572-574.
20. Schnidar H, Eberl M, Klingler S, Mangelberger D, Kasper M, Hauser-Kronberger C, Regl G, Kroismayr R, Moriggl R, Sibilia M et al: Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res 2009, 69(4):1284-1292.
21. Yoon JW, Kita Y, Frank DJ, Majewski RR, Konicek BA, Nobrega MA, Jacob H, Walterhouse D, Iannaccone P: Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1-induced cell transformation. J Biol Chem 2002, 277(7):5548-5555.
22. Allegra CJ: National Institutes of Health State-of-the-Science Conference Statement: Diagnosis and Management of Ductal Carcinoma In Situ September 22-24, 2009. J Natl Cancer Inst. 2010, 102(3):161-9.
23. Mercer KE, Pritchard CA: Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim Biophys Acta 2003, 1653(1):25-40.
24. Hinoue T, Weisenberger DJ, Pan F, Campan M, Kim M, Young J, Whitehall VL, Leggett BA, Laird PW: Analysis of the association between CIMP and BRAF in colorectal cancer by DNA methylation profiling. PLoS One 2009, 4(12):e8357.
25. Marampon F, Bossi G, Ciccarelli C, Di Rocco A, Sacchi A, Pestell RG, Zani BM: MEK/ERK inhibitor U0126 affects in vitro and in vivo growth of embryonal rhabdomyosarcoma. Mol Cancer Ther 2009, 8(3):543-551.
26. Bedogni B, Welford SM, Kwan AC, Ranger-Moore J, Saboda K, Powell MB: Inhibition of phosphatidylinositol-3-kinase and mitogen-activated protein kinase kinase 1/2 prevents melanoma development and promotes melanoma regression in the transgenic TPRas mouse model. Mol Cancer Ther 2006, 5(12):3071-3077.
27. Thiyagarajan S, Bhatia N, Reagan-Shaw S, Cozma D, Thomas-Tikhonenko A, Ahmad N, Spiegelman VS: Role of GLI2 transcription factor in growth and tumorigenicity of prostate cells. Cancer Res 2007, 67(22):10642-10646.
28. Ikram MS, Neill GW, Regl G, Eichberger T, Frischauf AM, Aberger F, Quinn A, Philpott M: GLI2 is expressed in normal human epidermis and BCC and induces GLI1 expression by binding to its promoter. J Invest Dermatol 2004, 122(6):1503-1509.
29. Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL: Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development 2002, 129(20):4753-4761.
30. Curtin JC, Lorenzi MV: Drug discovery approaches to target Wnt signaling in cancer stem cells. Oncotarget 2010, 1(7):563-577.
31. Lauth M, Bergstrom A, Shimokawa T, Toftgard R: Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci U S A 2007, 104(20):8455-8460.
32. Mazumdar T, Devecchio J, Agyeman A, Shi T, Houghton JA: Blocking Hedgehog survival signaling at the level of the GLI genes induces DNA damage and extensive cell death in human colon carcinoma cells. Cancer Res 2011, [Epub ahead of print].
33. Dormoy V, Danilin S, Lindner V, Thomas L, Rothhut S, Coquard C, Helwig JJ, Jacqmin D, Lang H, Massfelder T: The sonic hedgehog signaling pathway is reactivated in human renal cell carcinoma and plays orchestral role in tumor growth. Mol Cancer 2009, 8:123.
34. Ruiz i Altaba A: Gli proteins encode context-dependent positive and negative functions: implications for development and disease. Development 1999, 126(14):3205-3216.
35. Lobrich M, Shibata A, Beucher A, Fisher A, Ensminger M, Goodarzi AA, Barton O, Jeggo PA: gammaH2AX foci analysis for monitoring DNA double-strand break repair: strengths, limitations and optimization. Cell Cycle 2010, 9(4):662-669.
36. Carson DA, Lois A: Cancer progression and p53. Lancet 1995, 346(8981):1009-1011.
37. Holzel M, Burger K, Muhl B, Orban M, Kellner M, Eick D: The tumor suppressor p53 connects ribosome biogenesis to cell cycle control: a double-edged sword. Oncotarget 2010, 1(1):43-47.
38. Petak I, Tillman DM, Houghton JA: p53 dependence of Fas induction and acute apoptosis in response to 5-fluorouracil-leucovorin in human colon carcinoma cell lines. Clin Cancer Res 2000, 6(11):4432-4441.
39. Nojima H: G1 and S-phase checkpoints, chromosome instability, and cancer. Methods Mol Biol 2004, 280:3-49.
40. Hartwell LH, Weinert TA: Checkpoints: controls that ensure the order of cell cycle events. Science 1989, 246(4930):629-634.
41. Sherr CJ: Principles of tumor suppression. Cell 2004, 116(2):235-246.
42. Falck J, Petrini JH, Williams BR, Lukas J, Bartek J: The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat Genet 2002, 30(3):290-294.
43. Woo RA, Poon RY: Cyclin-dependent kinases and S phase control in mammalian cells. Cell Cycle 2003, 2(4):316-324.
44. Zhu Y, Alvarez C, Doll R, Kurata H, Schebye XM, Parry D, Lees E: Intra-S-phase checkpoint activation by direct CDK2 inhibition. Mol Cell Biol 2004, 24(14):6268-6277.
45. Grady WM: Genomic instability and colon cancer. Cancer Metastasis Rev 2004, 23(1-2):11-27.
46. Frappart PO, Lee Y, Russell HR, Chalhoub N, Wang YD, Orii KE, Zhao J, Kondo N, Baker SJ, McKinnon PJ: Recurrent genomic alterations characterize medulloblastoma arising from DNA double-strand break repair deficiency. Proc Natl Acad Sci U S A 2009, 106(6):1880-1885.
47. Leonard JM, Ye H, Wetmore C, Karnitz LM: Sonic Hedgehog signaling impairs ionizing radiation-induced checkpoint activation and induces genomic instability. J Cell Biol 2008, 183(3):385-391.
48. Snijders AM, Huey B, Connelly ST, Roy R, Jordan RC, Schmidt BL, Albertson DG: Stromal control of oncogenic traits expressed in response to the overexpression of GLI2, a pleiotropic oncogene. Oncogene 2009, 28(5):625-637.