
Introduction
The mTOR (Target of Rapamycin) signaling pathway is activated by nutrients (glucose, amino and fatty acids), growth factors, cytokines, oxygen, hormones and many other signals [1-4]. In turn, mTOR stimulates cellular size growth and metabolism as well as differentiation-specific functions [3-19]. In cycling cells, mTOR drives mass growth. If the cell cycle is arrested, then mTOR drives “futile growth” or geroconversion, converting reversible arrest to irreversible senescence [5, 20-22]. Senescence is not just cell cycle arrest: arrested cells can be either quiescent or senescent [21-25]. In quiescent cells, mTOR is deactivated [20, 26-33]. For example, serum withdrawal deactivates mTOR and MEK/MAPK pathways, causing reversible quiescence in normal cells [20, 26, 34-36]. In contrast, in senescent cells, mTOR is active [26, 29, 30, 33, 37- 40] Senescent cells are characterized by a large flat morphology (hypertrophy), active metabolism, differentiation-specific hyper-functions, and irreversible loss of proliferative potential [21, 23, 39, 41-58]. A senescent program includes 2 steps: (a) cell cycle arrest and (b) conversion from arrest to senescence [22]. For example, p21 can arrest cell cycle but does not inhibit mTOR. Therefore, mTOR drives geroconversion from p21-induced arrest to senescence. Since mTOR is fully active in cell culture (high levels of mitogens, nutrients and oxygen), it is usually sufficient for a cell to get arrested, in order to become senescent [22]. Rapamycin (and other rapalogs), certain tumor suppressors, including p53, serum-withdrawal, hypoxia and contact inhibition all suppress geroconversion by deactivating mTOR [19, 28, 59-71], thus maintaining quiescence instead. And vice verse, growth factor receptors, Ras, Raf, MEK, PI3K and Akt, which all activate the mTOR/S6K/S6 pathway, are involved in cellular senescence and cancer [72-76]. They are gerogenes, driving gerogenic conversion and oncogenic transformation [21, 64]. We can predict that activators of these pathways will promote both cancer and aging. Phorbol ester is the most well known tumor promoter, which activates MEK/ERK and mTOR/S6K signaling pathways [77-85].
Depending on the cellular context, PMA can cause either cell cycle progression or cell cycle arrest by inducing both cyclin D1 and p21 via the MEK/ERK pathway [43, 86-88]. Cell cycle arrest by itself can lead to senescence, if mTOR is not inhibited. Furthermore, the ability to activate mTOR predicts that PMA may be gero-promoter (promote geroconversion). Accordingly, it can cause cellular senescence, first by arresting cell cycle and then by converting this arrest to senescence (geroconversion). Cell cycle arrest caused by PMA is well studied. For example in SKBR3 cells, PMA over-activates MEK/ERK/MAPK, which in turn induces p21 and cell cycle arrest [86]. Here we show that cells become senescent, because mTOR is constantly active in SKBR3 cells. By blocking geroconversion, rapamycin rendered PMA-treated cells quiescent but not senescent. We also investigated cell lines that are completely resistant to PMA-induced arrest. In these cell lines, arrest was caused by either ectopic p21 or by serum starvation. In these cases, PMA increased geroconversion. Use of three cellular models demonstrated that, regardless of its ability to provoke senescence by arresting cell cycle (first step), PMA also empowers a second step of a senescent program: geroconversion.
Results
PMA-induced senescence in SKBR3 cells
As it was investigated in detail in SKBR3 cells [86], PMA activates the MEK/ERK pathway, which in turn induces both p21 and cyclin D1, causing G1 and G2 cell cycle arrest. As it was shown later, hyper-accumulation of cyclin D1 in arrested cells is a marker of senescence [39, 88]. Therefore we checked whether PMA-arrested cells acquire senescent morphology (Fig. 1A). We found that PMA caused a large flat morphology with nucleoli enlargement and beta-Gal positivity (Fig. 1 A). Next, we treated cells with PMA in the absence of serum, expecting that serum withdrawal might inhibit the mTOR pathway and prevent senescence. However, PMA caused senescence both in the presence and absence of the serum. In agreement with previous report [86], PMA rapidly activated ERK1/2 followed by p21 and cyclin D1 induction (Fig. 1 B). We also measured phosphorylation of S6 at S235/236 and S240/244 sites, as markers of mTOR activity. Noteworthy, S235/236 sites are phosphorylated by S6K (a substrate of mTOR) and by RSK (MEK-dependent), whereas S240/244 sites are presumably phosphorylated by S6K only [77-85]. Levels of p-S6 were high in both proliferating cells and serum-starved cells and become even higher after PMA treatment (Fig. 1B). First, this explains why arrested SKBR3 cells become senescent and, second, why they become senescent both in the presence and absence of serum.
These senescent cells lost the reversibility or regenerative potential (RP). In fact, PMA-treated SKBR3 cells poorly proliferated after PMA was washed out (Fig. 1 C, D) (Note: PMA is known to be poorly washable. Yet, even without washing, PMA-induced p21 disappears by day 3 [86] and this is a functional equivalent of washing PMA out).
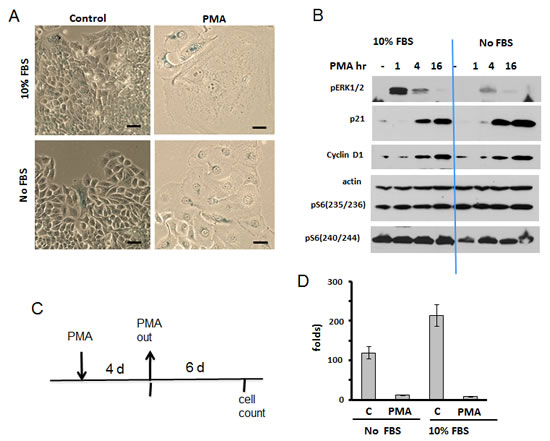
Figure 1: PMA-induced senescence in SKBr3 cells. A. Beta-gal staining. SKBR3 cells were treated with 100 nM PMA either in serum-free or in complete (10% FBS) medium. After 4 days drug was washed out and cells were cultured in drug-free medium and stained for beta-gal. Bar – 100 µm. B. Immunoblot analysis. SKBR3 cells were treated with 100 nM PMA in either serum-free or complete medium for times indicated and lysed. Results shown were obtained from 2 separate gels. C-D. RP (reversibility potential) of SKBR3 treated with PMA. C – Schema of experiment; D – RP: SKBR3 cells were plated at low density and treated with 100 nM PMA either in serum-free medium or in complete medium (10% FBS). After 4 days drug was washed out and cells were incubated in drug-free complete medium (10% FBS) for 6 days and counted. Fold increase in cell number was calculated relative to initially plated numbers. Data presented as mean ±SD from triplicates.
Rapamycin suppresses geroconversion in PMA-arrested cells
As shown in Fig. 2A, PMA caused typical senescent morphology in 30% of SKBR3 cells. Rapamycin by itself slightly inhibited proliferation but did not cause senescent morphology (Fig 2A and Fig. S1). Importantly, rapamycin abrogated PMA-induced senescent morphology (Fig. 2A). We also determined the reversibility potential by the ability of PMA-treated cells to form colonies in drug-free medium. The ability to restart proliferation or RP was decreased in PMA-pretreated cells, measured when PMA was washed out (Fig. 2 B). Rapamycin increased the number of colonies approximately 5-fold (Fig. 2B). We also excluded that rapamycin forced PMA-treated cells to proliferate in the presence of PMA. (Fig. S1A). As expected, co-addition of rapamycin and PMA inhibited proliferation (Fig. S1A). Also, rapamycin alone inhibited proliferation (Fig. S1), whereas inhibiting proliferation, rapamycin prevented PMA-induced loss of the potential to proliferate or RP. As emphasized previously, “proliferation” and “potential to proliferate” should not be confused. Rapamyin never induces proliferation but preserves the potential. Perhaps terms “regenerative potential (RP)”, “reversibility potential (RP)”, “the potential”, “reversibility” should be used to distinguish “proliferative potential” from “proliferation”. So rapamyin suppressed senescent morphology, hypertrophy and maintained reversibility potential (RP). In other words, rapamycin suppressed conversion from reversible arrest to senescence (geroconversion) (Fig. 2C).
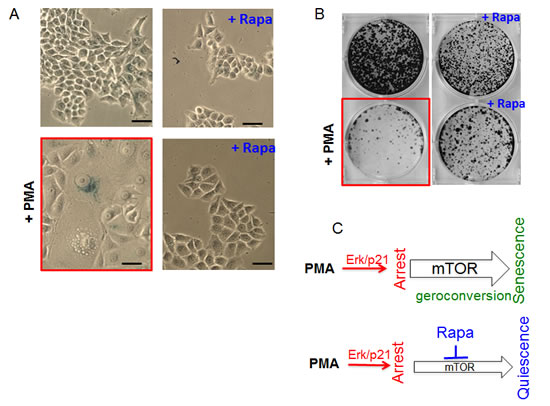
Figure 2: Suppression of PMA-induced senescence by rapamycin in SkBR3 cells. A. Beta-gal staining. SkBR3 cells were treated with PMA +/- rapamycin (20 nM) for 5 days, then drugs were washed out and cells were cultured for another 3 days and stained for beta-gal. Bar – 100 µm. B. RP (reversibility potential). SkBR3 cells were plated at low density and treated with 100 nM PMA -/+ rapamycin (20 nM). After 4 days cells were washed and colonies were allowed to regrow in drug-free medium and stained with Crystal Violet after 13 days in culture. C. Schema: PMA-induced senescence and its suppression by rapamycin (Rapa).
Rapamycin partially abrogates PMA-induced hyper-activation of mTOR targets
PMA induced p-ERK1/2 and p-S6K in both isoforms p70 and p85 (T412) (Fig. 3A). As expected, rapamycin did not affect PMA-induced phosphorylation of ERK1/2. Rapamycin abrogated p-S6K at T389 and p-S6 at S240/244 both in the absence and presence of PMA (Fig. 3A). Also, rapamycin completely eliminated phospho-T421/S424-S6K and phospho-S235/236 -S6 in the absence of PMA. However, rapamycin only marginally affected levels of phospho-T421/S424-S6K and phospho-S235/236 -S6 in the presence of PMA. In other words, PMA caused phosphorylation of S6K and S6 at these sites, even in the presence of rapamycin. This indicates that PMA activates S6K and S6 phosphorylation in part independent of mTOR.
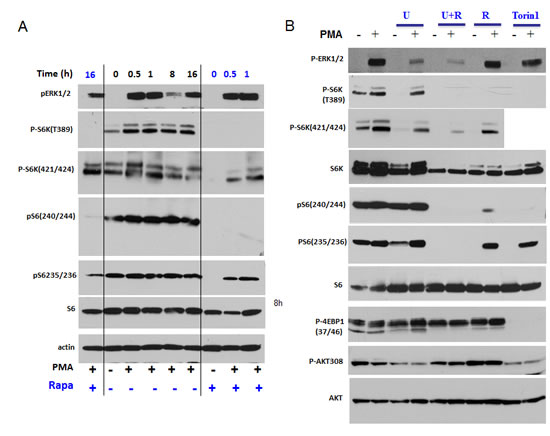
Figure 3: PMA-induced activation of the mTOR pathway in SkBr3 cells. A. Immunoblot analysis. SkBR3 cells were treated with 100 nM PMA for the times indicated and lysed. One set was pre-treated (and co-treated) with 100 nM rapamycin for 16 h before adding PMA, as indicated at the bottom (+ Rapa). B. Immunoblot analysis. SkBR3 cells were pre-treated and co-treated with either 10 µMU0126 (U), rapamycin 100 nM (R) or their combination, or with Torin 1 (100 nM) for 24 h. Then 100 nM PMA was added for 1 h and cells were lysed.
Both mTOR and MEK pathways are sufficient to phosphorylate S6
Since PMA activates the MEK/ERK pathway, we investigated whether inhibition of MEK can prevent PMA-induced phosphorylation of S6K and S6. PMA alone stimulated phosphorylation of ERK1/2 (dramatically), S6K (moderately) and only marginally S6, because S6 has been already near-maximally phosphorylated in proliferating untreated SKBR3 cells (Fig. 3B). Also, SKBR3 cells were treated with PMA in the presence of either U0126 (U), rapamycin (R), U+R or Torin 1 (an inhibitor of both TORC1 and TORC2). As expected, U0126, an inhibitor of MEK, abrogated PMA induced p-ERK1/2 but did not affect p-S6K and p-S6 at all sites tested (Fig. 3B). In contrast, rapamycin abrogated p-S6K (at both sites) and p-S6 (at both sites) in the absence of PMA-stimulation. Yet in the presence of PMA, while abrogating phospho-T389-S6K and phospho-S240/244-S6, rapamycin did not abrogate PMA-induced rapamycin-resistant (RR sites, for brevity) sites: namely, phospho-T421/S424-S6K and phospho-S235/236-S6 sites. Interestingly, Torin 1, which inhibited both complexes of mTOR and prevented phosphorylation of all substrates of mTORC1, as shown for phospho-4EBP1 (T37/46), failed to affect RR sites in the presence of PMA (Fig. 3B). In other words, PMA caused phosphorylation of RR sites in the presence of either rapamycin, U0126 or Torin 1 (Fig. 3B). Only a combination of U+R prevented PMA-induced RR sites (phospho-T421/S424-S6K and phospho-S235/236-S6). We conclude that, in the presence of PMA, S6K can be fully phosphorylated at T421/S424 via mTORC1 (rapamycin sensitive) and via MEK (rapamycin-insensitive) pathways (Fig. 4). Similarly, S6 can be fully phosphorylated on S235/236 via mTORC1 (rapamycin sensitive) and via MEK (rapamycin-insensitive) pathways in the presence of PMA (Fig. 4). Noteworthy, PMA did not cause AKT (T308) phosphorylation. Rapamycin increased Akt phosphorylation (Fig. 3B). This suggests that Akt itself (unlike mTORC1) does not empower geroconversion.
We emphasize that rapamycin only partly suppressed geroconversion. We investigated whether PMA-induced phospho-T421/S424-S6K and phospho-S235/236 S6 contribute to geroconversion in the presence of rapamycin. As we discussed, addition of U0126 to rapamycin eliminated PMA-induced RR phosphorylation (Fig. 3B). However, this combination did not further suppress senescent morphology in comparison to rapamycin alone (Fig. 5).
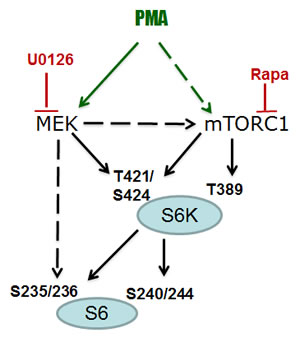
Figure 4: PMA-activated pathways.
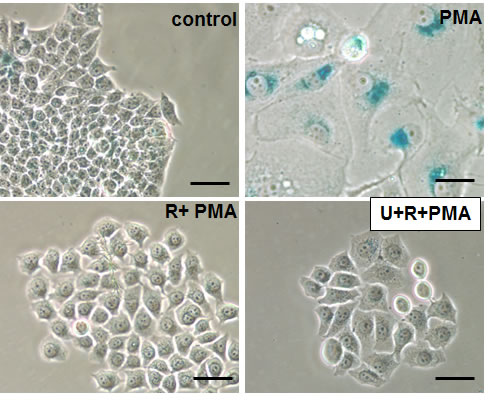
Figure 5: Effects of rapamycin plus U0126 on senescent morphology. Beta-gal staining. SkBR3 cells were pre-treated with rapamycin (100 nM) or its combination with U126 (10 µM) for 24 h before adding 100 nM PMA. After 3-day treatment with PMA drugs were washed out and cells were cultured for another 3 days in drug-free medium and stained for beta-gal. Bar – 100 µm.
PMA increased geroconversion in HT-p21 cells arrested by p21
We next investigated whether PMA contributes to senescence independently of cell cycle arrest. In HT-p21 model, cell cycle arrest was induced not by PMA but by IPTG-inducible p21 (Fig. 6A). In agreement with previous reports [89], a transient (for 4 days) induction of p21 led to cellular senescence, as evident by senescent morphology (Fig. 6 B). Rapamycin partially suppressed geroconversion to senescent morphology; rapamycin decreased cell size and beta-Gal-staining (Fig. 6 B). In addition, rapamycin preserved the reversibility potential (RP) measured by the ability to form colonies after removal of IPTG. (Note once again: The potential to proliferate (the reversibility potential) should not be confused with proliferation. Thus, rapamycin did not abrogate IPTG-induced arrest but instead preserved the potential to proliferate, when IPTG was washed out).
As shown in Figure S1B, PMA transiently phosphorylated RR sites in the presence of rapamycin. In agreement with the appearance of phospho-S6, this treatment affected geroconversion, increasing the number of senescent cells (morphology) and decreased RP (Fig. 6 B, C).
We next investigated induction of p-S6(S235/236) by PMA in detail. Basal levels of phospho-S6K (T389), phospho-S6(S235/236) and phospho-ERK1/2 were high and, therefore, the effect of PMA was marginal (Fig. 7 A). Inhibitor of MEK U0126, inhibitor of TOR rapamycin and inhibitor of TOR kinase Torin 1 all eliminated basal level of phospho-S6K(T389)/ phospho-S6(S235/236). This is in agreement with data obtained in SKBR3 cells. Impressively, none of these inhibitors prevented the induction of phospho-S6 (S235/236) by PMA. Only a combination of U0126 and Rapamycin prevented induction of phospho-S6(S235/236) by PMA. These data support the model shown in figure 4.
In HT-p21 cells, both rapamycin and U0126 suppressed geroconversion, as evidenced by preservation of RP (Fig. 7B). Yet, rapamycin and U0126 did not have any additive effect (Fig. 7B). This indicates that PMA-induced phosphorylation of S6 at S235/236 sites is not sufficient by itself to promote geroconversion. Importantly, PMA increased geroconversion both in the absence or presence of rapamycin (Fig. 6 C), suggesting that mTORC1-dependent and -independent pathways are involved in geroconversion.
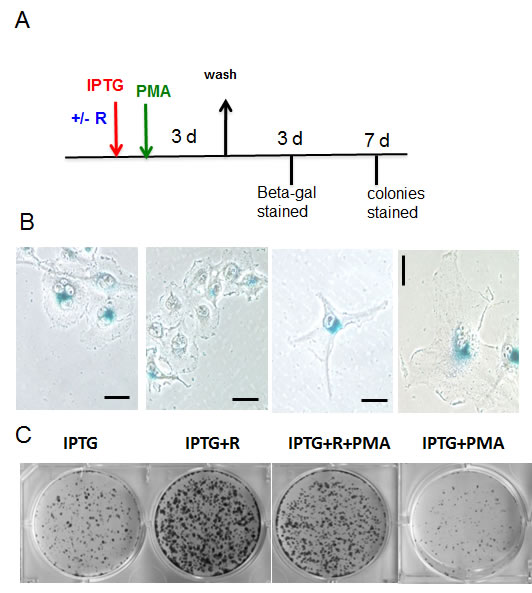
Figure 6: Effects of PMA on IPTG-induced senescence in HT-p21 cells. A. Schema of experiment. Rapamycin – R. B-C. HT-p21 cells were plated at low density and treated with IPTG, rapamycin (R) (500 nM) and PMA (100 nM) as indicated in Schema (A). After 3 days drugs were washed out, cells were incubated in drug-free medium for another 3 days and stained for beta-gal (B) (bar – 100 µm) and colonies were stained 7 days after wash (C). As indicated in the Schema, rapamycin was added 1 h before PMA.
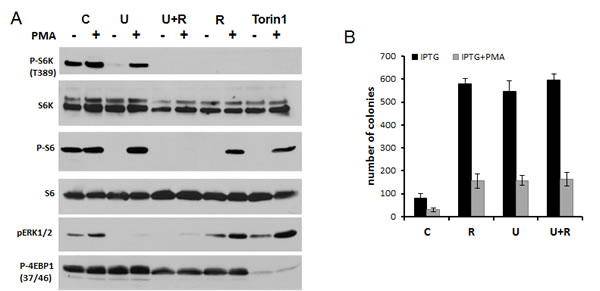
Figure 7: PMA induced rapamycin-insensitive p-S6 in HT-p21 cells A. Immunoblot analysis. HT-p21 cells were pre-treated with IPTG in the presence of either rapamycin (500 nM), U126 (10 µM) or their combination or torin 1 (100 nM) for 24 h, then 100 nM PMA was added for 1 h and cells were lysed. All treatments were performed in the presence of IPTG to match conditions shown in fig. 6 and panel 7B. B. RP: HT-p21 cells were pre-treated with IPTG in the presence of different drugs as in panel A for 24 h, then 100 nM PMA was added. After 3 day-treatment with PMA (4 days with IPTG and other drugs), drugs were washed out and colonies were allowed to grow and stained with Crystal violet after 9 days in culture and counted in triplicates. Data are presented as mean ± SD. C – cells treated with IPTG alone; R – treated with IPTG in the presence of rapamycin; U – treated with IPTG in the presence of U126; U+R – treated with IPTG in the presence of combination of rapamycin and U126.
PMA-induced geroconversion in quiescent RPE cells
Next, we investigated PMA-induced geroconversion in normal human retinal pigment epithelial (RPE) cells, arrested by serum starvation (Fig. 8). In RPE cells, serum withdrawal causes reversible quiescence, characterized by low levels of p-S6 [26, 34]. In quiescent cells arrested by serum starvation, PMA transiently induced phospho-S6 (Fig. 8 A). PMA did not induce proliferation but instead induced “futile growth” or geroconversion”. This geroconversion is manifested by hypertrophy and beta-Gal staining in approximately 20% of cells, observed after re-addition of serum (Fig. 8 B).
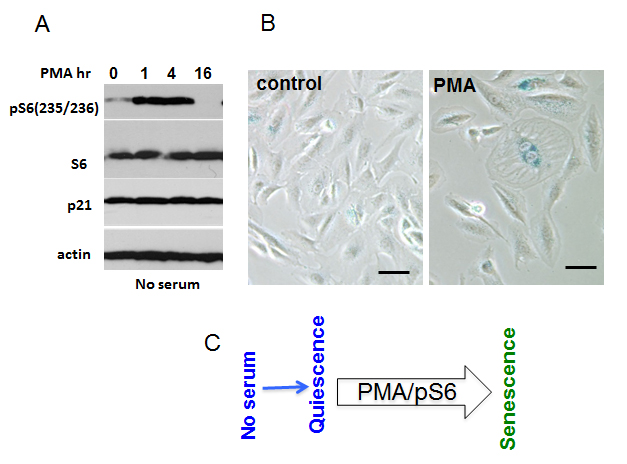
Figure 8: mTOR-dependent geroconversion in RPE cells by PMA. A. Immunoblot analysis. RPE cells were incubated in serum-free MEM overnight and then treated with 100 nM PMA for the times indicated. B. Beta-gal staining. RPE cells were pre-incubated in serum free medium before being treated with 100 nM PMA. After 2 day-treatment PMA was washed out, cells were incubated in drug-free medium for another 2 days and stained for beta-gal. Bar – 100 µm. C. Mechanism.
Discussion
Growth-promoting pathways such as the PI-3K/mTOR pathway are involved in both cancer and aging [21, 64, 90, 91]. Rapamycin prevents age-related diseases and cancer in mammals, including humans [92-116]. Therefore, inhibitors of mTOR are both tumor suppressors and gero-suppressors. To study aging in accelerated fashion, it would be useful to identify “antipode for rapamycin”, an agent that promotes geroconversion. Such agent is expected to (a) activate mTOR and related pathway, (b) be a tumor-promoter. It is known that PMA, a classic tumor-promoter, activates mTOR and MAPK pathways. Importantly, rapamycin can suppress tumor-promotion caused by PMA [117-119].
Given that PMA activates the mTOR pathway, we predicted that PMA can accelerate geroconversion in cell culture. Here we showed that phorbol ester indeed displayed the gero-converting activity. This activity can be obscured by the ability of PMA to initiate senescence, simply by inducing cell cycle arrest. In SKBR3 cells, strong activation of MEK/ERK pathway by PMA causes induction of p21 and cell cycle arrest. When the cell cycle was arrested, still active mTOR pathway drove geroconversion from arrest to senescence. So in SKBR3 cells, PMA caused cell cycle arrest, which was sufficient to cause senescence in the presence of active mTOR. This is consistent with the model of two-step senescence program: cell cycle arrest by PMA plus geroconversion by active mTOR. The mTOR pathway was constitutively activated in cancer SKBR3 cells even in the absence of serum. Rapamycin decreased geroconversion, indicating that mTOR indeed is involved in PMA-induced senescence.
To elucidate the role of PMA in geroconversion, we used the model of IPTG-induced senescence, HT-p21 cells. In this model IPTG, not PMA, caused p21 induction and cell cycle arrest. PMA increased senescence in this cell model, acting as an enhancer of geroconversion. Rapamycin partially decreased geroconversion in the presence and absence of PMA. Yet, PMA still enhanced geroconversion in the presence and absence of rapamycin. This indicates that geroconversion involves some rapamycin-insensitive pathways (in addition to rapamycin-sensitive pathways), which are activated by PMA and are involved in geroconversion. Noteworthy, rapamycin-insensitive phosphorylation of S6K(T421/S424) and S6(S235/236) was also mTOR -independent because Torin 1 (a direct inhibitor of both mTORC1 and mTORC2) did not abrogate-rapamycin insensitive phosphorylation of S6(S235/236) and S6K(T421/S424). We identified pathways that led to rapamycin-insensitive S6K(T421/S424) phosphorylation by PMA (Fig. 4). In agreement with previous reports, PMA induced phosphorylation of S6K and S6 at both rapamycin-sensitive and -insensitive sites in all 3 cell lines tested here. In part, PMA-induced rapamycin-insensitive phosphorylation was dependent on the MEK pathway. Either MEK or mTOR was sufficient to phosphorylate these sites of S6K and S6. Thus, neither rapamycin nor U0126 inhibited phosphorylation of S6K and S6 at T421/S424 and S235/236, respectively, whereas a combination of U0126 and rapamycin eliminated phosphorylation of S6K and S6 on these sites. Yet (and importantly), the addition of U0126 to rapamycin had no effect on geroconversion. This indicates that rapamycin-insensitive phosphorylation of these sites alone is not sufficient to cause geroconversion, when mTORC1 is inhibited. In turn, this indicates that, although rapamycin sensitive-pathways are involved in geroconversion, some unidentified rapamycin-insensitive pathways also contribute to geroconversion. So identification of such pathways remains a challenge.
In conclusion, PMA possesses two senescence-promoting activities: cell cycle arrest (in some cell lines such as SKBR3) and geroconversion. When the cell cycle is arrested by other condition (IPTG-induced p21 or serum starvation), then the geroconverting activity of PMA becomes apparent.
This study further validates the utility of two-step model of senescence for identification of agents which can promote or in contrast suppress aging.
Materials and Methods
Cell lines and reagents
SKBR3, breast adenocarcinoma cell line (ATCC), was cultured in high-glucose DMEM (-pyruvate) with 10% FBS. HT-p21 cells, derived from HT1080 human fibrosarcoma cells (ATCC, Manassas, VA) were previously described [20, 120, 121] and were cultured in high-glucose DMEM without pyruvate plus 10% FC2 serum (HyClone FetalClone II from Thermo Scientific, Logan, Utah). In these cells, p21 can be turned on or off by isopropyl-thio-galactosidase (IPTG) [20, 120, 121]. Normal retinal pigment epithelial RPE cell line (ATCC, Manassas, VA) was maintained in MEM plus 10% FBS. IPTG was purchased from Invitrogen (Grand Island, NY). Rapamycin was obtained from LC Laboratories (MA, USA). U0126 and PMA were from Sigma-Aldrich (St. Louis, MO). Torin 1 was obtained from Selleck chemicals LCC (Houston, TX).
SA-β-Gal staining
Beta-Gal staining was performed using Senescence-galactosidase staining kit (Cell Signaling Technology) according to manufacturer’s protocol. Cells were incubated at 37oC until beta-gal staining becomes visible. Development of color was detected under light microscope.
Immunoblot analysis
Whole cell lysates were prepared using boiling lysis buffer (1% SDS, 10 mM Tris.HCl, pH 74.). Equal amounts of proteins were separated using Criterion or mini gradient polyacrylamide gels (Bio-Rad, Hercules, CA) and transferred to PVDF membranes. The following rabbit antibodies for: phospho-S6 (Ser235/236 and S240/244), phospho-AKT (T308), phospho ERK ½ , AKT, phospho-4EBP1(T37/46) and phospho-S6K(T421/S424)- were from Cell Signaling Biotechnology (Danvers, MA). Mouse anti-phospho-Thr 389 -S6K and anti-S6 antibody were from Cell Signaling Biotechnology. Rabbit anti-actin antibody was from Sigma-Aldrich (St. Louis, MO); mouse antibodies for p21 and cyclin D1 were from from BD Biosciences (San Jose, CA) and Santa Cruz Biotechnology (Paso Robles, CA), respectively. Secondary anti-rabbit and anti-mouse HRP-conjugated antibodies were from Cell Signaling Biotechnology.
RP (reversibility potential)
Cells were plated at low densities, treated with senescence inducing drugs for 3-4 days as indicated in figure legends. Then, drugs were washed out and cells were allowed to re-grow in fresh drug-free medium for a few days (as indicated in figure legends). Then cells were either counted or formed colonies were stained with 1% Crystal Violet (Sigma-Aldrich) and counted.
Author Contributions
MVB conceptualised the project, designed the experiments and wrote the manuscript. OL performed the experiments, reviewed the manuscript and gave a feedback.
Acknowledgements and Funding
This work was supported by Roswell Park Cancer Institute, Buffalo, NY 14203
Conflicts of Interests
No conflict to declare.
References
1. Proud CG. The multifaceted role of mTOR in cellular stress responses. DNA Repair. 2004; 3: 927-934.
2. Martin DE, Hall MN. The expanding TOR signaling network. Curr Opin Cell Biol. 2005; 17: 158-166.
3. Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010; 40: 310-322.
4. Duran RV, Hall MN. Glutaminolysis feeds mTORC1. Cell Cycle. 2012; 11: 4107-4108.
5. Blagosklonny MV, Hall MN. Growth and aging: a common molecular mechanism. Aging. (Albany NY) 2009; 1: 357-362.
6. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011; 12: 21-35.
7. Sandri M, Barberi L, Bijlsma AY, Blaauw B, Dyar KA, Milan G, Mammucari C, Meskers CG, Pallafacchina G, Paoli A, Pion D, Roceri M, Romanello V, Serrano AL, Toniolo L, Larsson L et al. Signalling pathways regulating muscle mass in ageing skeletal muscle: the role of the IGF1-Akt-mTOR-FoxO pathway. Biogerontology. 2014; 14: 303-323.
8. Bentzinger CF, Lin S, Romanino K, Castets P, Guridi M, Summermatter S, Handschin C, Tintignac LA, Hall MN, Ruegg MA. Differential response of skeletal muscles to mTORC1 signaling during atrophy and hypertrophy. Skelet Muscle. 2013; 3: 6.
9. Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr Biol. 2009; 19: R1046-1052.
10. Yeh WC, Bierer BE, McKnight SL. Rapamycin inhibits clonal expansion and adipogenic differentiation of 3T3-L1 cells. Proc Natl Acad Sci U S A. 1995; 92: 11086-11090.
11. Sugatani T, Hruska KA. Akt1/Akt2 and mammalian target of rapamycin/Bim play critical roles in osteoclast differentiation and survival, respectively, whereas Akt is dispensable for cell survival in isolated osteoclast precursors. J Biol Chem. 2005; 280: 3583-3589.
12. Cho HJ, Park J, Lee HW, Lee YS, Kim JB. Regulation of adipocyte differentiation and insulin action with rapamycin. Biochem Biophys Res Commun. 2004; 321: 942-948.
13. Kofman AE, McGraw MR, Payne CJ. Rapamycin increases oxidative stress response gene expression in adult stem cells. Aging (Albany NY). 2012; 4: 279-289.
14. Park SJ, Kang SY, Kim NS, Kim HM. Phosphatidylinositol 3-kinase regulates PMA-induced differentiation and superoxide production in HL-60 cells. Immunopharmacol Immunotoxicol. 2002; 24: 211-226.
15. Raslova H, Baccini V, Loussaief L, Comba B, Larghero J, Debili N, Vainchenker W. Mammalian target of rapamycin (mTOR) regulates both proliferation of megakaryocyte progenitors and late stages of megakaryocyte differentiation. Blood. 2006; 107: 2303-2310.
16. Martin-Cano FE, Camello-Almaraz C, Hernandez D, Pozo MJ, Camello PJ. mTOR pathway and Ca(2)(+) stores mobilization in aged smooth muscle cells. Aging (Albany NY). 2013; 5: 339-346.
17. Blagosklonny MV. Big mice die young but large animals live longer. Aging (Albany NY). 2013; 5: 227-233.
18. Blagosklonny MV. M(o)TOR of aging: MTOR as a universal molecular hypothalamus. Aging (Albany NY). 2013; 5: 490-494.
19. Rallis C, Bahler J. Inhibition of TORC1 signaling and increased lifespan: gained in translation? Aging (Albany NY). 2013; 5: 335-336.
20. Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008; 7: 3355-3361.
21. Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging (Albany NY). 2012; 4: 159-165.
22. Blagosklonny MV. Geroconversion: irreversible step to cellular senescence. Cell Cycle. 2014: 0.
23. Sousa-Victor P, Gutarra S, Garcia-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardi M, Ballestar E, Gonzalez S, Serrano AL, Perdiguero E, Munoz-Canoves P. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014; 506: 316-321.
24. Sousa-Victor P, Perdiguero E, Munoz-Canoves P. Geroconversion of aged muscle stem cells under regenerative pressure. Cell Cycle. 2014; 13: 3183-3190.
25. Zhao H, Halicka HD, Li J, Darzynkiewicz Z. Berberine suppresses gero-conversion from cell cycle arrest to senescence. Aging (Albany NY). 2013; 5: 623-636.
26. Leontieva OV, Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging (Albany NY). 2010; 2: 924-935.
27. Adhikari D, Zheng W, Shen Y, Gorre N, Hamalainen T, Cooney AJ, Huhtaniemi I, Lan ZJ, Liu K. Tsc/mTORC1 signaling in oocytes governs the quiescence and activation of primordial follicles. Hum Mol Genet. 2010; 19: 397-410.
28. Lane DP, Verma C, Fang CC. The p53 inducing drug dosage may determine quiescence or senescence. Aging (Albany NY). 2010; 2: 748.
29. Dulic V. Senescence regulation by mTOR. Methods Mol Biol. 2013; 965: 15-35.
30. Gan B, DePinho RA. mTORC1 signaling governs hematopoietic stem cell quiescence. Cell Cycle. 2009; 8: 1003-1006.
31. Gharibi B, Farzadi S, Ghuman M, Hughes FJ. Inhibition of Akt/mTOR attenuates age-related changes in mesenchymal stem cells. Stem Cells. 32: 2256-2266.
32. Adhikari D, Flohr G, Gorre N, Shen Y, Yang H, Lundin E, Lan Z, Gambello MJ, Liu K. Disruption of Tsc2 in oocytes leads to overactivation of the entire pool of primordial follicles. Mol Hum Reprod. 2009; 15: 765-770.
33. Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY). 2010; 2: 344-352.
34. Leontieva OV, Demidenko ZN, Blagosklonny MV. Rapamycin reverses insulin resistance (IR) in high-glucose medium without causing IR in normoglycemic medium. Cell Death Dis. 2014; 5: e1214.
35. Leontieva OV, Demidenko ZN, Blagosklonny MV. S6K in geroconversion. Cell Cycle. 2013; 12: 3249-3252.
36. Leontieva OV, Blagosklonny MV. CDK4/6-inhibiting drug substitutes for p21 and p16 in senescence: duration of cell cycle arrest and MTOR activity determine geroconversion. Cell Cycle. 2013; 12: 3063-3069.
37. Cho S, Hwang ES. Status of mTOR activity may phenotypically differentiate senescence and quiescence. Mol Cells. 2012; 33: 597-604.
38. Gan B, Sahin E, Jiang S, Sanchez-Aguilera A, Scott KL, Chin L, Williams DA, Kwiatkowski DJ, DePinho RA. mTORC1-dependent and -independent regulation of stem cell renewal, differentiation, and mobilization. Proc Natl Acad Sci U S A. 2008; 105: 19384-19389.
39. Leontieva OV, Lenzo F, Demidenko ZN, Blagosklonny MV. Hyper-mitogenic drive coexists with mitotic incompetence in senescent cells. Cell Cycle. 2012; 11: 4642 - 4649.
40. Halicka HD, Zhao H, Li J, Lee YS, Hsieh TC, Wu JM, Darzynkiewicz Z. Potential anti-aging agents suppress the level of constitutive mTOR- and DNA damage- signaling. Aging (Albany NY). 2012; 4: 952-965.
41. Mathon NF, Lloyd AC. Cell senescence and cancer. Nature Rev Cancer. 2001; 1: 203-213.
42. Dulic V, Beney GE, Frebourg G, Drullinger LF, Stein GH. Uncoupling between phenotypic senescence and cell cycle arrest in aging p21-deficient fibroblasts. Mol Cell Biol. 2000; 20: 6741-6754.
43. Blagosklonny MV. Cell senescence and hypermitogenic arrest. EMBO Rep. 2003; 4: 358-362.
44. Wang Y, Meng A, Zhou D. Inhibition of phosphatidylinostol 3-kinase uncouples H2O2-induced senescent phenotype and cell cycle arrest in normal human diploid fibroblasts. Exp Cell Res. 2004; 298: 188-196.
45. Campisi J. Suppressing cancer: the importance of being senescent. Science. 2005; 309: 886-887.
46. Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, Iken M, Vucur M, Weiss S, Heikenwalder M, Khan S, Gil J et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011; 479: 547-551.
47. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013; 153: 1194-1217.
48. Mao Z, Ke Z, Gorbunova V, Seluanov A. Replicatively senescent cells are arrested in G1 and G2 phases. Aging (Albany NY). 2012; 4: 431-435.
49. Serrano M. Dissecting the role of mTOR complexes in cellular senescence. Cell Cycle. 2012; 11: 2231-2232.
50. Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. 2014; 28: 99-114.
51. Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013; 123: 966-972.
52. McCubrey JA, Demidenko ZN. Recent discoveries in the cycling, growing and aging of the p53 field. Aging (Albany NY). 2012; 4: 887-893.
53. Busse S, Steiner J, Micheel J, Dobrowolny H, Mawrin C, Krause TJ, Adamaszek M, Bogerts B, Bommhardt U, Hartig R, Busse M. Age-related increase of VGF-expression in T lymphocytes. Aging (Albany NY). 2014; 6: 440-453.
54. van Deursen JM. The role of senescent cells in ageing. Nature. 2014; 509: 439-446.
55. Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014; 15: 482-496.
56. Cenni V, Capanni C, Mattioli E, Columbaro M, Wehnert M, Ortolani M, Fini M, Novelli G, Bertacchini J, Maraldi NM, Marmiroli S, D’Apice MR, Prencipe S, Squarzoni S, Lattanzi G. Rapamycin treatment of Mandibuloacral dysplasia cells rescues localization of chromatin-associated proteins and cell cycle dynamics. Aging (Albany NY). 2014; 6: 755-770.
57. Demaria M, Campisi J. Matters of life and breath: A role for hypoxia in determining cell state. Aging (Albany NY). 2012; 4: 523-524.
58. Driscoll MK, Albanese JL, Xiong ZM, Mailman M, Losert W, Cao K. Automated image analysis of nuclear shape: what can we learn from a prematurely aged cell? Aging (Albany NY). 2012; 4: 119-132.
59. Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010; 107: 9660-9664.
60. Leontieva OV, Natarajan V, Demidenko ZN, Burdelya LG, Gudkov AV, Blagosklonny MV. Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proc Natl Acad Sci U S A. 2012; 109: 13314-13318.
61. Leontieva OV, Demidenko ZN, Blagosklonny MV. Contact inhibition and high cell density deactivate the mammalian target of rapamycin pathway, thus suppressing the senescence program. Proc Natl Acad Sci U S A. 2014; 111: 8832-8837.
62. Leontieva O, Gudkov A, Blagosklonny M. Weak p53 permits senescence during cell cycle arrest. Cell Cycle. 2010; 9: 4323-4327.
63. Sui X, Fang Y, Lou H, Wang K, Zheng Y, Lou F, Jin W, Xu Y, Chen W, Pan H, Wang X, Han W. p53 suppresses stress-induced cellular senescence via regulation of autophagy under the deprivation of serum. Mol Med Rep. 2014.
64. Blagosklonny MV. Tumor suppression by p53 without apoptosis and senescence: conundrum or rapalog-like gerosuppression? Aging (Albany NY). 2012; 4: 450-455.
65. Cha J, Hirota Y, Dey SK. Sensing senescence in preterm birth. Cell Cycle. 2012; 11: 205-206.
66. Hirota Y, Daikoku T, Tranguch S, Xie H, Bradshaw HB, Dey SK. Uterine-specific p53 deficiency confers premature uterine senescence and promotes preterm birth in mice. J Clin Invest. 120: 803-815.
67. Goldberg AA, Kyryakov P, Bourque SD, Titorenko VI. Xenohormetic, hormetic and cytostatic selective forces driving longevity at the ecosystemic level. Aging (Albany NY). 2010; 2: 461-470.
68. Goldberg AA, Richard VR, Kyryakov P, Bourque SD, Beach A, Burstein MT, Glebov A, Koupaki O, Boukh-Viner T, Gregg C, Juneau M, English AM, Thomas DY, Titorenko VI. Chemical genetic screen identifies lithocholic acid as an anti-aging compound that extends yeast chronological life span in a TOR-independent manner, by modulating housekeeping longevity assurance processes. Aging (Albany NY). 2010; 2: 393-414.
69. Sun P. Contact inhibition against senescence. Oncotarget. 2014; 5: 7212-7213.
70. Kolesnichenko M, Hong L, Liao R, Vogt PK, Sun P. Attenuation of TORC1 signaling delays replicative and oncogenic RAS-induced senescence. Cell Cycle. 2012; 11: 2391-2401.
71. Pospelova TV, Leontieva OV, Bykova TV, Zubova SG, Pospelov VA, Blagosklonny MV. Suppression of replicative senescence by rapamycin in rodent embryonic cells. Cell Cycle. 2012; 11: 2402-2407.
72. Serrano M, Lim AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK1A. Cell. 1997; 88: 593-602.
73. Zhu JY, Woods D, McMahon M, Bishop JM. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 1998; 12: 2997-3007.
74. Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998; 12: 3008-3019.
75. Benanti JA, Galloway DA. The normal response to RAS: senescence or transformation? Cell Cycle. 2004; 3: 715-717.
76. Malumbres M, De Castro IP, Hernandez MI, Jimenez M, Corral T, Pellicer A. Cellular response to oncogenic Ras involves induction of the Cdk4 and Cdk6 inhibitor p15(INK4b). Mol Cell Biol. 2000; 20: 2915-2925.
77. Fonseca BD, Alain T, Finestone LK, Huang BP, Rolfe M, Jiang T, Yao Z, Hernandez G, Bennett CF, Proud CG. Pharmacological and genetic evaluation of proposed roles of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase (MEK), extracellular signal-regulated kinase (ERK), and p90(RSK) in the control of mTORC1 protein signaling by phorbol esters. J Biol Chem. 286: 27111-27122.
78. Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, Sonenberg N, Blenis J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem. 2007; 282: 14056-14064.
79. Martin PM, Aeder SE, Chrestensen CA, Sturgill TW, Hussaini IM. Phorbol 12-myristate 13-acetate and serum synergize to promote rapamycin-insensitive cell proliferation via protein kinase C-eta. Oncogene. 2007; 26: 407-414.
80. Holz MK, Blenis J. Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)-phosphorylating kinase. J Biol Chem. 2005; 280: 26089-26093.
81. Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci U S A. 2004; 101: 13489-13494.
82. Iijima Y, Laser M, Shiraishi H, Willey CD, Sundaravadivel B, Xu L, McDermott PJ, Kuppuswamy D. c-Raf/MEK/ERK pathway controls protein kinase C-mediated p70S6K activation in adult cardiac muscle cells. J Biol Chem. 2002; 277: 23065-23075.
83. Herbert TP, Tee AR, Proud CG. The extracellular signal-regulated kinase pathway regulates the phosphorylation of 4E-BP1 at multiple sites. J Biol Chem. 2002; 277: 11591-11596.
84. Fonseca BD, Lee VH, Proud CG. The binding of PRAS40 to 14-3-3 proteins is not required for activation of mTORC1 signalling by phorbol esters/ERK. Biochem J. 2008; 411: 141-149.
85. Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005; 123: 569-580.
86. Blagosklonny MV. The mitogen-activated protein kinase pathway mediates growth arrest or E1A-dependent apoptosis in SKBr3 human breast cancer cells. Int J Cancer. 1998; 78: 511-517.
87. Blagosklonny MV. Oncogenic resistance to growth-limiting conditions. Nature Rev Cancer. 2002; 2: 221-225.
88. Leontieva OV, Demidenk ZN, Blagosklonny MV. MEK drives cyclin D1 hyperelevation during geroconversion. Cell Death Diff. 2013; 20: 1241-1249.
89. Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009; 8: 1888-1895.
90. Blagosklonny MV. Molecular damage in cancer: an argument for mTOR-driven aging. Aging (Albany NY). 2011; 3: 1130-1141.
91. Blagosklonny MV. Rapalogs in cancer prevention: Anti-aging or anticancer? Cancer Biol Ther. 2012; 13: 1349-1354.
92. Dazert E, Hall MN. mTOR signaling in disease. Curr Opin Cell Biol. 2011. 23:744-755
93. Kolosova NG, Vitovtov AO, Muraleva NA, Akulov AE, Stefanova NA, Blagosklonny MV. Rapamycin suppresses brain aging in senescence-accelerated OXYS rats. Aging (Albany NY). 2013; 5: 474-484.
94. Zheng XF. Chemoprevention of age-related macular regeneration (AMD) with rapamycin. Aging (Albany NY). 2012; 4: 375-376.
95. Ye L, Widlund AL, Sims CA, Lamming DW, Guan Y, Davis JG, Sabatini DM, Harrison DE, Vang O, Baur JA. Rapamycin doses sufficient to extend lifespan do not compromise muscle mitochondrial content or endurance. Aging (Albany NY). 2013; 5: 539-550.
96. Gems DH, de la Guardia YI. Alternative Perspectives on Aging in C. elegans: Reactive Oxygen Species or Hyperfunction? Antioxid Redox Signal. 2013; 19: 321-329.
97. Blagosklonny MV. Rapamycin extends life- and health span because it slows aging. Aging (Albany NY). 2013; 5: 592-598.
98. Blagosklonny MV. How to save Medicare: the anti-aging remedy. Aging (Albany NY). 2012; 4: 547-552.
99. Blagosklonny MV. Answering the ultimate question “what is the proximal cause of aging?” Aging (Albany NY). 2012; 4: 861-877.
100. Ramos FJ, Chen SC, Garelick MG, Dai DF, Liao CY, Schreiber KH, MacKay VL, An EH, Strong R, Ladiges WC, Rabinovitch PS, Kaeberlein M, Kennedy BK. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci Transl Med. 2012; 4: 144ra103.
101. Anisimov VN. Metformin and rapamycin are master-keys for understanding the relationship between cell senescent, aging and cancer. Aging (Albany NY). 2013; 5: 337-338.
102. Hinojosa CA, Mgbemena V, Van Roekel S, Austad SN, Miller RA, Bose S, Orihuela CJ. Enteric-delivered rapamycin enhances resistance of aged mice to pneumococcal pneumonia through reduced cellular senescence. Exp Gerontol. 2012; 47: 958-965.
103. Comas M, Toshkov I, Kuropatwinski KK, Chernova OB, Polinsky A, Blagosklonny MV, Gudkov AV, Antoch MP. New nanoformulation of rapamycin Rapatar extends lifespan in homozygous p53-/- mice by delaying carcinogenesis. Aging (Albany NY). 2012; 4: 715-722.
104. Komarova EA, Antoch MP, Novototskaya LR, Chernova OB, Paszkiewicz G, Leontieva OV, Blagosklonny MV, Gudkov AV. Rapamycin extends lifespan and delays tumorigenesis in heterozygous p53+/- mice. Aging (Albany NY). 2012; 4: 709-714.
105. Donehower LA. Rapamycin as longevity enhancer and cancer preventative agent in the context of p53 deficiency. Aging (Albany NY). 2012; 4: 660-661.
106. Kaeberlein M. mTOR Inhibition: From Aging to Autism and Beyond. Scientifica (Cairo). 2013; 2013: 849186.
107. Kennedy BK, Pennypacker JK. Drugs that modulate aging: the promising yet difficult path ahead. Transl Res. 2013; 163:456-465
108. Luo Y, Li L, Zou P, Wang J, Shao L, Zhou D, Liu L. Rapamycin enhances long-term hematopoietic reconstitution of ex vivo expanded mouse hematopoietic stem cells by inhibiting senescence. Transplantation. 2014; 97: 20-29.
109. Halloran J, Hussong SA, Burbank R, Podlutskaya N, Fischer KE, Sloane LB, Austad SN, Strong R, Richardson A, Hart MJ, Galvan V. Chronic inhibition of mammalian target of rapamycin by rapamycin modulates cognitive and non-cognitive components of behavior throughout lifespan in mice. Neuroscience. 2012; 223: 102-113.
110. Liu Y, Huang Y, Wang Z, Li X, Louie A, Wei G, Mao JH. Temporal mTOR inhibition protects Fbxw7-deficient mice from radiation-induced tumor development. Aging (Albany NY). 2013; 5: 111-119.
111. Livi CB, Hardman RL, Christy BA, Dodds SG, Jones D, Williams C, Strong R, Bokov A, Javors MA, Ikeno Y, Hubbard G, Hasty P, Sharp ZD. Rapamycin extends life span of Rb1+/- mice by inhibiting neuroendocrine tumors. Aging (Albany NY). 2013; 5: 100-110.
112. Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A, Oh K, Wasko BM, Ramos FJ, Palmiter RD, Rabinovitch PS, Morgan PG et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science. 2013; 342: 1524-1528.
113. Blagosklonny MV. Once again on rapamycin-induced insulin resistance and longevity: despite of or owing to. Aging (Albany NY). 2012; 4: 350-358.
114. Blagosklonny MV. TOR-centric view on insulin resistance and diabetic complications: perspective for endocrinologists and gerontologists. Cell Death Dis. 2013; 4: e964.
115. Blagosklonny MV. Prospective treatment of age-related diseases by slowing down aging. Am J Pathol. 2012; 181: 1142-1146.
116. Leontieva OV, Novototskaya LR, Paszkiewicz GM, Komarova EA, Gudkov AV, Blagosklonny MV. Dysregulation of the mTOR pathway in p53-deficient mice. Cancer Biol Ther. 2013; 14:1182-1188.
117. Checkley LA, Rho O, Angel JM, Cho J, Blando J, Beltran L, Hursting SD, DiGiovanni J. Metformin inhibits skin tumor promotion in overweight and obese mice. Cancer Prev Res (Phila). 2014; 7: 54-64.
118. Checkley LA, Rho O, Moore T, Hursting S, DiGiovanni J. Rapamycin is a potent inhibitor of skin tumor promotion by 12-O-tetradecanoylphorbol-13-acetate. Cancer Prev Res (Phila). 2011: 4: 1011-1020.
119. Athar M, Kopelovich L. Rapamycin and mTORC1 inhibition in the mouse: skin cancer prevention. Cancer Prev Res (Phila). 2011; 4: 957-961.
120. Chang BD, Xuan Y, Broude EV, Zhu H, Schott B, Fang J, Roninson IB. Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene. 1999; 18: 4808-4818.
121. Broude EV, Swift ME, Vivo C, Chang BD, Davis BM, Kalurupalle S, Blagosklonny MV, Roninson IB. p21(Waf1/Cip1/Sdi1) mediates retinoblastoma protein degradation. Oncogene. 2007; 26:6954-6958.