

Introduction
Across all human cancers, cutaneous malignant melanoma (MM) genome has one of the highest prevalence of somatic mutations. The type of mutations found is known to result from ultraviolet radiation (UVR) induced DNA damage [1]. The most commonly reported mutations in cutaneous malignant melanomas are located in BRAF and NRAS genes [2–5]. BRAF mutations are found in approximately 50% (22% to 70%) of cutaneous MM [3, 6, 7]. These mutations are more commonly detected in melanomas developing in the skin with intermittent sun exposure (non-chronic sun-damaged) - such as a trunk; than in non-UVR exposed skin surface. The most abundant BRAF mutation is associated with V600E amino acid substitution, and it comprises about 90% of all BRAF mutations found in MM. At the same time, NRAS mutations are detected in approximately 20% of MM and are more commonly reported in melanomas developing in the skin with chronic sun exposure [3, 6, 8]. The most common missense substitutions in NRAS are Q61R and Q61K [3].
Different patterns of genetic alterations, including type and frequency of mutations and chromosomal aberrations, is expected for melanoma subtypes [9]. Genomic profile differs not only between histopathological melanoma subtypes but also between melanomas developing in different anatomical locations [10]. It has been suggested that different melanoma subtypes, in fact, develop as a result of deregulation of type-specific molecular pathways and their spectrum is correlated with the degree of UVR exposure-related mutagenesis [9, 11]. On sun-exposed skin where the rate of cumulative solar damage is high, lentigo maligna and desmoplastic melanoma are observed. Superficial spreading melanoma is connected with low cumulative solar damage, and acral melanomas are described as nonsolar [12].
In the Cancer Genome Atlas study with whole-genome sequencing molecular profile of 333 non-UVR-related melanomas was described. It was shown that structural variants (deletions, duplications, tandem duplications, and foldback inversions) are significantly more common in non-UVR-related melanomas. Occurrence in some studies UVR-related mutational profile in acral sites suggests that nail plate and thick strata corneum do not provide complete UVR protection [11]. An increased number of genetic aberrations may be associated with a poorer prognosis [13].
The current 11. WHO Classification of Skin Tumours recognizes the most common acral melanoma histotype is acral lentiginous melanoma (ALM), followed by nodular cutaneous melanoma (NM) and superficial spreading melanoma (SSM) [12]. The majority of SSM and NM harbor mostly BRAF (50%) or NRAS (17%) mutations. On the contrary, in ALM, BRAF mutation is found in 17% of cases and NRAS - in 15% [6, 14]. On the other hand, ALMs harbor more often KIT mutations than other MMs – in 15% to 40% [6, 14]. ALM subtype was suggested to harbor specific gene amplifications and deletions resulting from unique genomic instability [15]. ALM is not correlated with sunburn or higher UVR exposure as it is developing in the skin unexposed to the sun, but chronic physical stress or pressure to acral locations may be a predisposing factor [8, 16–18].
Cutaneous MM located on the acral part of extremities - hand and foot melanoma (HFM) - comprises a rare group within all melanomas in Caucasians. Whole-genome sequencing study shown that structural changes and mutational signature of acral melanomas were dominated by different than other MMs sites. Acral melanoma presents unknown, non or lower UVR related, etiology [9, 19]. BRAF, NRAS, or NF1 is mutated in HFM only in 42–55% of cases [19]. KIT mutation is detected in 3–40% of acral melanomas [19, 20]. Mutations in the promoter of TERT occur in 9–41% of HFM and are currently recognized as a prognostic factor [19]. TERT inhibitors are potentially available for use in the clinic. Others mutations observed in acral melanomas are PAK1, CDK4, CCND1, CDKN2A, PTEN, P16, MAP2K2, ARID2, MITF, TP53, RAC1, RB1, SPRED1 ALK, ROS1, RET and NTRK1 [19]. Subungual melanoma (SUM) is a subgroup of HFM that arises from structures within the nail apparatus. SUM is exceedingly rare, representing 0.7–3.5% of all MMs in Caucasian populations, with an annual incidence of approximately 0.1 per 100 000 [6, 13, 21–23]. These tumors develop mostly on great toe and thumb. SUM seems to be not related to sun exposure, however, in Australian Melanoma Genome Project UVR signatures on acral melanomas occurred most frequently in subungual parts [11]. Both HFM and SUM seem to have poorer survival outcomes comparing to MMs of the other sites, with 5-year overall survival (OS) in the range 20–60% [22–27], although ‘Lieberherr’s in meta-analysis reports 77% [28]. Poor survival rates are in part as a result of delayed diagnosis since frequent misdiagnosis. In the literature, SUM present the most diverse mutation profile, including oncogenes like NRAS, PIK3CA, EGFR, FGFR3, PTPN11, IDH2, ALK and suppressor genes STK11, TP53, APC and high frequency of copy number aberrations in CCND1 and CDK4 than other MMs [13, 29]. The BRAF mutations occur less frequent in SUM compared to the MMs of other sites.
Due to limited data, the genetic profile of SUM is still under investigation. It is important to know tumor genetic profile as genetic mutations have a fundamental impact on the selection of targeted therapy such as BRAF/MEK inhibitors in BRAF-mutated tumors, tyrosine kinase inhibitors in KIT-mutated tumors and MEK inhibitors in NRAS-mutated tumors [6]. Generally, HFMs are described only by few, heterogeneous studies with various outcomes. SUM is a subgroup of HFM, so there is even fewer data. As next-generation sequencing (NGS, including whole genome/exome sequencing and gene panels) is now more widely used to characterize the genetic profiles of tumors, it was therefore selected as a basic research method for the project. Our study aimed to define the molecular profile of highly selected melanoma population using results from NGS panel of 50 cancer-related genes in homogenous cohort of patients with SUM.
Results
Thirty one Caucasian patients diagnosed with SUM were enrolled - 18 women and 13 men. The mean age was 62 years. Median Breslow thickness was 5 mm, and ulceration was observed in 74% of cases. In all cases, SLNB was performed with a positive result in 42% of SUM. The most common histological subtype diagnosed was ALM (76% of patients). Lower extremity digits involved 19 of 31 (61%) cases. The clinicopathologic characteristics of enrolled patients are presented in Table 1.
Table 1: Clinicopathologic characteristics of enrolled patients
| Characteristics | Total n = 31 |
|---|---|
| Age at diagnosis, years | |
| Median | 66.24 |
| Mean | 62.05 |
| IQR | 15.12 |
| Sex, n (%) | |
| Male | 13 (42) |
| Female | 18 (58) |
| SUM location, n (%) | |
| Hands | 12 (39) |
| Feet | 19 (61) |
| Primary tumor thickness (mm) | |
| Median | 5 |
| Mean | 5.62 |
| Ulceration, n (%) | |
| Absent | 8 (26) |
| Present | 23 (74) |
| Histopathological subtype, n (%) | |
| NM | 5 (20) |
| SSM | 0 (0) |
| ALM | 19 (76) |
| LMM | 1 (4) |
| NA | 6 |
| SLNB | |
| Positive, n (%) | 13 (42) |
| Negative, n (%) | 18 (58) |
| Median survival, mo (range) | 42 (3,5–173) |
| Alive, n (%) | 11 (35) |
| Dead, n (%) | 20 (65) |
At least one mutation was detected in 12 patients (39%) with the most common mutations in the KIT gene in 5 cases (13%, two mutations in one case) and NRAS - in 4 cases (13%). KRAS mutation was detected in 2 patients, while other mutations in CTNNB1, TP53, ERBB2, SMAD4, BRAF – in single cases. The detailed summary of detected mutations with details is presented in Table 2.
Table 2: Summary of detected mutations
| Patient | Age at diagnosis | Sex | Gene | Nucleotide change | Amino acid change | Location | Histopathologic type | Breslow thickness (mm) | SLNB | Ulceration | Survival status |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 69 | F | KRAS | c.35G>T | p.G12V | foot | NM | 5 | Negative | Yes | Dead |
| 2 | 78 | M | KIT | c.1721_1729delCACAACTTC | p.Gln575_Pro577del | hand | ALM | 1 | Positive | No | Dead |
| 3 | 60 | F | NRAS | c.34G>T | p.G12C | foot | ALM | 13 | Positive | Yes | Dead |
| 4 | 74 | F | NRAS | c.38G>A | p.G13D | foot | ALM | 20 | Negative | Yes | Dead |
| 5 | 49 | F | NRAS | c.182A>G | p.Q61R | hand | NA | 1 | Negative | Yes | Dead |
| 6 | 51 | F | SMAD4 | c.1081C>T | p.R361C | hand | ALM | 4 | Negative | Yes | Alive |
| KRAS | c.35G>A | p.G12D | |||||||||
| 7 | 52 | M | KIT | c.1727T>C | p.L576P | hand | ALM | 7 | Positive | Yes | Dead |
| 8 | 66 | F | KIT | c.1727T>C | p.L576P | hand | ALM | 6 | Negative | Yes | Dead |
| KIT | c.1676T>A | p.V559D | |||||||||
| 9 | 64 | M | ERBB2 | c.2329G>C | p.V777L | hand | NM | 14 | Positive | Yes | Dead |
| 10 | 49 | F | NRAS | c.181C>A | p.Q61K | hand | NA | 1 | Negative | Yes | Dead |
| CTNNB1 | c.134C>T | p.S45F | |||||||||
| TP53 | c.797G>A | p.G266E | |||||||||
| 11 | 64 | M | KIT | c.1733_1735delATG | p.Asp579del | foot | ALM | 9 | Positive | Yes | Dead |
| 12 | 26 | F | BRAF | c.1799T>A | p.V600E | foot | NA | 2 | Negative | No | Alive |
DISCUSSION
The mean patients age in our study was 62 years, which was consistent with 57.8–69 years reported of SUM patients in the literature [13, 21]. Comparing to other studies of acral location where one third of cases had no mutations detected; in our SUM group, no mutation was detected in even more cases (61%) [5]. In our cohort, median Breslow thickness was 5 mm, which is consistent with the literature of SUM [21]. The ulceration in our group was common (74%) which is also consistent with previous studies [21]. In our cohort, 76% of cases were identified as ALM, while in the study of 13 patients with SUM, including seven Caucasians, percentage was higher (85%). Another SUM study of 54 patients reports ALM in 65% of cases. The positive status of SLNB involved 42% of SUM patients, while in the scarce literature, the reported percentage is lower (31%) [21].
KIT
The KIT gene encodes a transmembrane tyrosine kinase receptor, which is associated with activation of mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3 kinase (PI3K/Akt) pathways [6]. MAPK and PI3K/Akt pathways play a role in the melanoma genesis because they regulate different cellular functions (proliferation, differentiation, and survival).
In our study, 5 (13%) SUM harbored KIT mutations. The identified KIT mutations included three single amino acid changes [L576P (n = 2) and p.V559D] and two deletions [p.Gln575_Pro577del, and p.Asp579del]. All of them were previously observed in melanomas [6]. In Reily’s study of 19 SUM cases, KIT mutation occurred in 11% of samples [21]. Although other studies of SUM present a higher percentage of 30–50% [25, 32] or no detected among 13 SUM patients [29]. In HFM/ALM, KIT mutations are frequent - occurring in 8.5–36% of tumors usually as a substitution of a single amino acid in exon 11, 13, or 17 [6, 8–10, 30–34]. Siroy et al. sequenced 54 patients with advanced acral melanoma and reported KIT mutation in 11% of cases [5]. Otherwise, the most common mutations were NRAS (24%), BRAF (19%), and TP53 (6%). In Gong et al. meta-analysis incidence of KIT mutations in melanoma was higher in older patients and was associated with chronic sun-damage, acral and mucosal melanomas [35]. Mutations in KIT are relatively uncommon in MMs overall, about 1–6% [9, 31].
NRAS
The RAS family (N- K- H- RAS) is a family of small proteins that provides to transduce signals triggered by extracellular growth factors. NRAS mutation is associated with activation of both MAPK and the PI3K/Akt pathways and is found in about 12–20% of all MMs [6, 7, 15]. Both MAPK and PI3K/Akt pathways are involved in the proliferation, differentiation, and survival of the cell [8].
In our study in 13% of cases were found NRAS mutation. The identified NRAS mutations included p.G12C, p.G12D, p.G13D, p.Q61K, and p.Q61R amino acid changes. All of them were previously observed in melanomas. Moreover, we have found that BRAF and NRAS were mutually exclusive and occurred only in tumors that were negative for KIT mutations, as reported in the literature [5, 31]. In Dica et al. study of 13 SUM patients, 30.8% presented NRAS mutation [25]. There is no significant difference of NRAS mutation in SUM/HFM/ALM compared to melanomas of the non-acral skin, it is detected in 7.5–25% of acral MM cases [2, 3, 5, 6, 8, 9, 29, 31, 32, 36]. NRAS+ melanomas are correlated with lower tumor-infiltrating lymphocytes (TIL) grade comparing to wild type, the anatomic site different than scalp or neck, and the presence of higher mitotic index. Lower TIL grade suggests a more immunosuppressed microenvironment, which can affect response to immunotherapy. The same study shows that NRAS+ nor BRAF+ was associated with overall melanoma-specific survival, but the risk of death was significantly more reduced for higher-risk NRAS+ or BRAF+ (T2 or higher stage) [2].
KRAS
KRAS (Kristen Rat sarcoma), one of the RAS isoforms, plays an important role in human cancers acting upstream of BRAF [37]. Mutation in KRAS was detected in two (6%) of SUM tumors. The identified KRAS mutations included p.G12V and p.G12D amino acid changes. Both of them were previously observed in melanomas.
KRAS in MMs is infrequently mutated, although it is one of the most frequently mutated proto-oncogenes in human cancers. Siroy et al. identified KRAS mutation in 7.7% advanced MMs [5], and in the Catalogue of Somatic Mutations in Cancer [38] it has been reported in 2% of acral MMs. Also, in Min et al. study of MMs KRAS mutation was reported in 2.3% of cases [7]. There is no information about the present of KRAS mutation in SUM in the literature. KRAS is considered as having a rather weak oncogenic effect in MM [36]. In the study of the genetic profile of melanoma brain metastases, KRAS mutations were associated with reduced overall survival from brain metastasis resection [39].
BRAF
BRAF mutation is associated with activation of the MAPK pathway, which frequently plays a role in human cancers. It is identified as less frequently in MMs on sun-protected skin (acral sites, mucosal). It was found in high frequencies in melanocytic naevi, which suggests that this somatic alternation occurs early in melanoma genesis [34]. In our material, only one patient with SUM was BRAF+ (3%), which is lower than previously reported 6.25–14.4% in SUM cases [21, 25, 29]. The identified most common BRAF mutation is a c. 1799 T>A transversion in exon 15, which causes p.V600E amino acid substitution, the most common mutation observed in melanomas [3]. Siroy et al. (2015) reported 2% of BRAF mutations in acral melanomas. In the literature of ALM, BRAF mutation is presented in 15–20% of cases [3, 6]. Compared to about 50% BRAF mutation in all primary and metastatic MMs, this mutation is under-represented in HFM/ALM [5, 33, 36, 40]. In the Swedish study, which evaluated 88 patients with primary ALM, it has been shown that BRAF, NRAS, and KIT mutations occur in ALM with a similar frequency of about 15%. BRAF mutations were significantly more common in younger patients, in females and in tumors located on the feet [6]. BRAF+ melanomas are correlated with SSM subtype, and the presence of mitoses [2].
CTNNB1
β-catenin (CTNNB1 gene coding protein) is a part of the WNT pathway. It is fortifying cadherin-based adhesion at the plasma membrane and activating transcription in the nucleus [41]. Accumulation of cytoplasmic β-catenin promotes the transcription of proto-oncogenes and other various genes. One of the patients in our study had a mutation in CTNNB1. The identified CTNNB1 mutation was p.S45F amino acid change, which was previously observed in MMs. There is no previously reported information in the literature of CTNNB1 mutation in SUM. In Xu study of acral melanomas the positive expression of β-catenin was observed in 36 (72%) of patients, the expression of β-catenin, was not correlated with gender, age, or diseased body parts, but was significantly positively correlated with the tumor node metastasis stage and metastasis [42]. Shim et al. found a mutation in CTNNB1 in acral melanomas in Korean patients [33]. In all MMs, CTNNB1 mutation occurs in 2–5% of cases [43].
TP53
Mutation in the p53 tumor suppressor gene has been linked to the majority of human cancers. It plays a role in a transcription activating target genes that mediate various functions (including deoxyribonucleic acid repaid, metabolism, apoptosis). Loss of wild-type p53 function, through mutation in p53 or alternation in pathway signaling, may promote cancer cell development, survival, and proliferation [44, 45]. One of our patients had a mutation in TP53. The identified TP53 mutation included p.G266E amino acid change, previously observed in melanoma cell lines. Haugh et al. reported TP53 mutation in 2 of 13 (15.4%) patients with SUM [29]. In the Catalogue of Somatic Mutations in Cancer [38] in acral MMs, the TP53 mutation rate is about 9%, whereas Hayward et al. reported no acral melanomas had a mutation in TP53 [9], and Yeh et al. found 1 case (0.8%) with TP53 mutation [45]. In all sites, MMs mutation in TP53 is presented at a low rate, occurring in 0–24% of MMs [9, 43].
ERBB2
ERBB2 encodes the HER2 receptor tyrosine kinase. Its alternations can lead to uncontrolled cell proliferation, consequently to oncogenesis, in various mechanisms. ERBB2 mutation is uncommon in all types of MMs, a rate of about 1–3% [9]. In one case, we observed ERBB2 mutation. The identified ERBB1 mutation included p.V777L amino acid change. This mutation was not previously observed in melanomas. According to Gottesdiener et al., ERBB2 mutation occurs in 2% in acral MMs, which is consistent with the rate in the Catalogue of Somatic Mutations in Cancer [38, 46]. However, there is no previous information about ERBB2 mutation in SUM patients.
SMAD4
The SMAD4 takes part in transmitting chemical signals from the cell surface to the nucleus. It’s a part of transforming growth factor-beta (TGF-β) signaling pathway by activating SMAD proteins forming a complex with SMAD4 protein. As a central mediator of TGF-β signaling, SMAD4 plays a role in cell differentiation, migration, invasion, and apoptosis [47]. Mouse models suggest that SMAD-mediated signaling in melanoma can play various functions, such as the proliferation of melanoma cells [48]. The loss of SMAD4 can increase DNA instability by disturbing DNA damage response and mechanisms of repair [49]. The identified SMAD4 mutation included p.R361C amino acid change. This mutation was not previously observed in melanomas.
The limitation of this study is a relatively small number of sequenced cases, but this limitation results from disease epidemiology. Another limitation is that used Cancer Hotspot Panel v2 includes only 50 cancer-related genes, so there may be existed loci not examined. For example, this panel does not include recently described human telomerase reverse transcriptase (TERT) promoter mutations, which were observed in approximately 9% of ALMs, and its amplification was correlated with poor outcome [50–52].
This is a first comprehensive analysis that focused on the genetic characterization of SUM in the Caucasian population. There is a lack of information in the literature about genetic profile in this subgroup of melanoma patients. Technological advances in molecular biology, particularly NGS, have increased the opportunities for mutation discovery in different subtypes of MM. We have presented the NGS results of 50 cancer-related genes of exceedingly rare among all MMs SUM cases in a homogenous group of Caucasians patients. Until now, our study is the largest of molecular profile in SUM Caucasian patients. In Reilly’s study of 54 patients with SUM in 19 of the molecular studies were performed with assessing a combination of one or more of BRAF, c-KIT, and NRAS, unfortunately, patients were with unknown racial background [21]. In Haugh’s study of SUM patients, BRAF mutation occurred in 1 in 13 samples [29]. Yeh et al. study of SUM KIT mutation was identified in 3 of 6 cases (more frequently than in acral MMs) [45]. Our findings confirmed previously reported in the literature of SUM amount of KIT and NRAS mutations, and we found a lower rate of BRAF mutations. Moreover, KRAS, CTNNB1, TP53, ERBB2, and SMAD4 mutations not described previously in SUM appeared in our results.
The genetic profile of SUM is different from others sites of MMs, especially those with sun exposure. The most common mutations in our study were KIT (13%) and NRAS (13%) with coherent percentage comparing to previous literature. It stays in concordance with the previous studies that show that the nail plate can block UVB and UVB [9, 11, 53]. It leads to a non-UV-related malignancy pathway.
We have confirmed that SUM arises due to mutations in genes critical for differentiation (KIT, NRAS), cell cycle, and proliferation (KIT, NRAS, KRAS, TP53, ERBB2, SMAD4) and apoptosis (TP53).
Our study offers new insights into the genetics SUM subtype, for understanding pathogenesis and providing potential biomarkers for future studies. Molecular testing is now widely used in patients with advanced melanoma in the process of therapeutic decisions. Mutations reported in melanoma cells provide starting points for the development of the rational design of novel therapies, including immunotherapy agents. They also may provide to find the molecular pathogenesis and natural history of subtypes of this heterogeneous disease. We confirmed that SUM have different than other cutaneous melanomas genetic profile, which due to its rareness and lack of studies should be subjected to further analyzes in multicenter studies.
Materials and Methods
Two thousand five hundred thirty-seven melanoma consecutive cases diagnosed and treated in Maria Sklodowska-Curie National Research Institute of Oncology, Warsaw, Poland, between 01. Jan 1997, and 31. Dec 2014, who underwent sentinel node biopsy, were screened, and 46 SUM patients were selected for the study (Figure 1). For screened MM population inclusion criteria were: diagnosis of primary cutaneous melanoma after excisional biopsy, Breslow thickness ≥ 0.75 mm or presence of ulceration, sentinel lymph node biopsy performed; while exclusion criteria included: a metastatic disease at the time of diagnosis, clinically palpable lymph nodes, incomplete medical records, or lack of primary tumor sample. We have included in this analysis only patients in clinical stage I-II undergoing sentinel node biopsy to constitute the homogenous population, in thirty one cases the high quality pathological specimens were available for molecular analyses for study purposes. Informed consent from all patients was obtained.
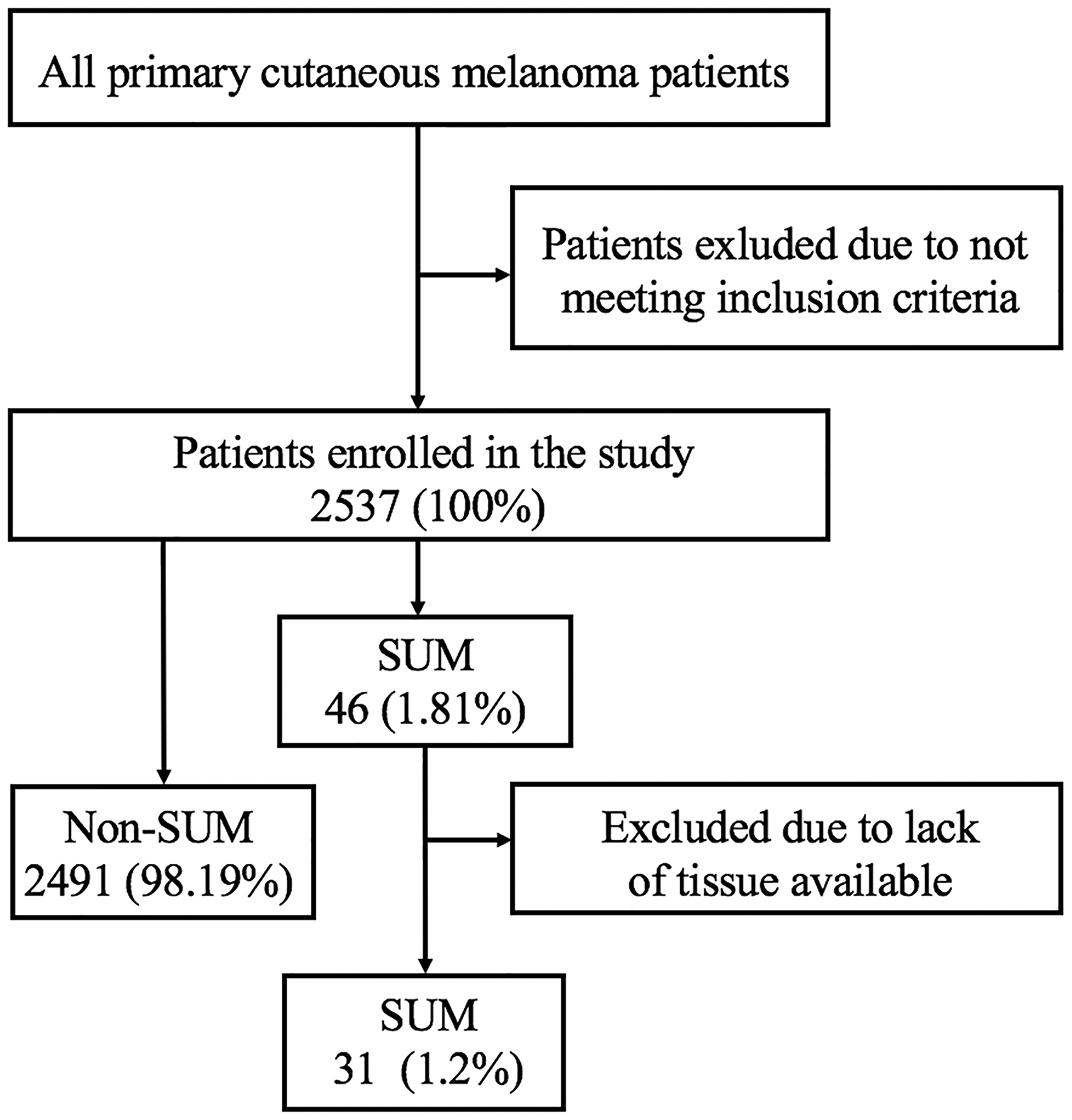
Figure 1: Data extraction. SUM - subungual melanoma.
Next-generation sequencing
SUM DNA was sequenced by the Ion Proton sequencing platform using the Ion AmpliSeq Cancer Hotspot Panel v2 to identify frequently mutated regions in 50 cancer-related genes including ABL1, EZH2, JAK3, PTEN, ACT1, FBXW7, IDH2, PTPN11, ALK, FGFR1, KDR, RB1, APC, FGFR2, KIT, RET, ATM, FGFR3, KRAS, SMAD4, BRAF, FLT3, MET, SMARCB1, CDH1, GNA11, MLH1, SMO, CDKN2A, GNAS, MPL, SRC, CSF1R, GNAQ, NOTCH1, STK11, CTNNB1, HNF1A, NPM1, TP53, EGFR, HRAS, NRAS, VHL, ERBB2, IDH1, PDGFR, ERBB4, JAK2, PIK3CA.
DNA concentration was measured using Qubit fluorimeter and dsDNA HS Assay Kit (Thermo Fisher Scientific). The libraries were prepared using the Ion AmpliSeq™ Library Kit 2.0, Ion AmpliSeq™ Cancer Hotspot Panel v2 (Thermo Fisher Scientific), and the Ion Xpress Barcode Adapters Kit, according to the manufacturer’s instructions (Thermo Fisher Scientific).
The prepared libraries were subjected to double purification using Agencourt AMPure XP (Beckman Coulter Genomics) according to the manufacturer’s instructions (Ion AmpliSeq Library preparation - Thermo Fisher Scientific). Ion Library Quantitation Kit (Thermo Fisher Scientific) was used for real-time PCR analysis. Sequencing was performed on the Ion S5 sequencer (Thermo Fisher Scientific) using the Ion 520™ & Ion 530™ Kit-Chef kit according to the manufacturer’s instructions.
Data analysis
The raw data generated during the sequencing was processed using Torrent Server Suite 5.2 (LT). The obtained sequences were mapped to the reference sequence of the human genome (hg19). Various variants (SNPs, mutations) were detected using Variant Caller v5.2, part of Torrent Server Suite 5.2. The manufacturer’s default parameters for AmpliSeq somatic were used: minimum allele frequency - SNP = 0.018 / INDEL = 0.02, minimum quality - 6, minimum coverage - 100. The called mutations were reviewed using IGV - Integrative Genomics Viewer (Broad Institute). In addition, Torrent Server Suite 5.2 generated FASTQ files that were used for analysis using other software: Biomedic Genomic Workbench 4.0 (QIAGEN) and GALAXY (http://wannovar.wglab.org/). The following default parameters used in the analysis using: Biomedical Genomic Workbench 4.0 (minimum allele frequency - 0.05, minimum quality -10, minimum coverage – 100) and for GALAXY (minimum allele frequency - 0.05, minimum quality -15, minimum coverage – 100). The wANNOVAR software (http://wannovar.wglab.org/) was used to annotate the variants called by TSS and GALAXY.
Abbreviations
MM: malignant melanoma; UVR: ultraviolet radiation; ALM: acral lentiginous melanoma; NM: nodular cutaneous melanoma; SSM: superficial spreading melanoma; HFM: hand and foot melanoma; SUM: subungual melanoma; OS: overall survival; NGS: next-generation sequencing; MAPK: mitogen-activated protein kinase; PI3K/Akt: phosphatidylinositol 3 kinase; TIL: tumor-infiltrating lymphocytes; TGF-β: transforming growth factor-beta.
ACKNOWLEDGMENTS AND FUNDING
This work has been supported by Maria-Sklodowska Curie National Research Institute of Oncology statutory funding.
CONFLICTS OF INTEREST
Declared. Rutkowski has received honoraria for lectures and Advisory Baords from MSD, BMS, Novartis, Pierre Fabre, Blueprint Medicines and Pfizer outside the scope of this study.
References
1. Alexandrov L, Nik-Zainal S, Wedge D, Aparicio S, Behjati S, Biankin A, Bignell GR, Stratton MR. Signatures of mutational processes in human cancer. Nature. 2013; 500:415–421. https://doi.org/10.1038/nature12477. [PubMed].
2. Thomas NE, Edmiston SN, Alexander A, Groben PA, Parrish E, Kricker A, Armstrong BK, Anton-Culver H, Gruber SB, From L, Busam KJ, Hao H, Orlow I, et al. Association between NRAS and BRAF mutational status and melanoma-specific survival among patients with higher-risk primary melanoma. JAMA Oncol. 2015; 1:359–368. https://doi.org/10.1001/jamaoncol.2015.0493. [PubMed].
3. Lee JH, Choi JW, Kim YS. Frequencies of BRAF and NRAS mutations are different in histological types and sites of origin of cutaneous melanoma: a meta-analysis. Br J Dermatol. 2011; 164:776–784. https://doi.org/10.1111/j.1365-2133.2010.10185.x. [PubMed].
4. Yaman B, Akalin T, Kandiloğlu G. Clinicopathological Characteristics and Mutation Profiling in Primary Cutaneous Melanoma. Am J Dermatopathol. 2015; 37:389–397. https://doi.org/10.1097/DAD.0000000000000241. [PubMed].
5. Siroy A, Boland G, Milton D, Roszik J, Frankian S, Malke J, Haydu L, Prieto VG, Tetzlaff M, Ivan D, Wang WL, Torres-Cabala C, Curry J, et al. Beyond BRAF(V600): clinical mutation panel testing by next-generation sequencing in advanced melanoma. J Invest Dermatol. 2015; 135:508–515. https://doi.org/10.1038/jid.2014.366. [PubMed].
6. Zebary A, Omholt K, Vassilaki I, Höiom V, Lindén D, Viberg L, Kanter-Lewensohn L, Johansson CH, Hansson J. KIT, NRAS, BRAF and PTEN mutations in a sample of Swedish patients with acral lentiginous melanoma. J Dermatol Sci. 2013; 72:284–289. https://doi.org/10.1016/j.jdermsci.2013.07.013. [PubMed].
7. Min KW, Choe JY, Kwon MJ, Lee HK, Kang HS, Nam ES, Cho SJ, Park HR, Min SK, Seo J, Kim YJ, Kim NY, Kim HY. BRAF and NRAS mutations and antitumor immunity in Korean malignant melanomas and their prognostic relevance: Gene set enrichment analysis and CIBERSORT analysis. Pathol Res Pract. 2019; 215:152671. https://doi.org/10.1016/j.prp.2019.152671. [PubMed].
8. Desai A, Ugorji R, Khachemoune A. Acral melanoma foot lesions. Part 1: epidemiology, aetiology, and molecular pathology. Clin Exp Dermatol. 2017; 42:845–8. https://doi.org/10.1111/ced.13243. [PubMed].
9. Hayward N, Wilmott J, Waddell N, Johansson P, Field M, Nones K, Patch A, Mann G. Whole-genome landscapes of major melanoma subtypes. Nature. 2017; 547:175–180. https://doi.org/10.1038/nature22071. [PubMed].
10. Woodman SE, Davies MA. Targeting KIT in melanoma: A paradigm of molecular medicine and targeted therapeutics. Biochem Pharmacol. 2010; 80:568–574. https://doi.org/10.1016/j.bcp.2010.04.032. [PubMed].
11. Rawson RV, Johansson PA, Hayward NK, Waddell N, Patch AM, Lo S, Pearson JV, Thompson JF, Mann GJ, Scolyer RA, Wilmott JS. Unexpected UVR and non-UVR mutation burden in some acral and cutaneous melanomas. Lab Invest. 2017; 97:130–145. https://doi.org/10.1038/labinvest.2016.143. [PubMed].
12. Edler D, Bastian B, Cree I, Massi D, Scolyer R. The 2018 World Health Organization Classification of Cutaneous, Mucosal, and Uveal Melanoma: Detailed Analysis of 9 Distinct Subtypes Defined by Their Evolutionary Pathway. Arch Pathol Lab Med. 2020; 144:500–522. https://doi.org/10.5858/arpa.2019-0561-RA. [PubMed].
13. Romano R, Shon W, Sukov W. Malignant Melanoma of the Nail Apparatus: A Fluorescence In Situ Hybridization Analysis of 7 Cases. Int J Surg Pathol. 2016; 24:512–518. https://doi.org/10.1177/1066896916648379. [PubMed].
14. Goydos J, Shoen S. Acral lentiginous melanoma. Cancer Treat Res. 2016; 167:321–329. https://doi.org/10.1007/978-3-319-22539-5_14. [PubMed].
15. Whiteman DC, Pavan WJ, Bastian BC. The melanomas: a synthesis of epidemiological, clinical, histopathological, genetic, and biological aspects, supporting distinct subtypes, causal pathways, and cells of origin. Pigment Cell Melanoma Res. 2011; 24:879–897. https://doi.org/10.1111/j.1755-148X.2011.00880.x. [PubMed].
16. Costa Svedman F, Pillas D, Kaur M, Linder R, Hansson J, Aliki T. Stage-specific survival and recurrence in patients with cutaneous malignant melanoma in Europe – a systematic review of the literature. Clin Epidemiol. 2016; 8:109–122. https://doi.org/10.2147/CLEP.S99021. [PubMed].
17. Piliang M. Acral lentiginous melanoma. Clin Lab Med. 2011; 31:281–288. https://doi.org/10.1016/j.cll.2011.03.005. [PubMed].
18. Jung HJ, Kweon SS, Lee JB, Lee SC, Yun SJ. A Clinicopathologic Analysis of 177 Acral Melanomas in Koreans. JAMA Dermatol. 2013; 149:1281–8. https://doi.org/10.1001/jamadermatol.2013.5853. [PubMed].
19. Rabbie R, Ferguson P, Molina-Aguilar C, Adams DJ, Robles-Espinoza CD. Melanoma subtypes: genomic profiles, prognostic molecular markers and therapeutic possibilities. J Pathol. 2019; 247:539–551. https://doi.org/10.1002/path.5213. [PubMed].
20. Kim K, Alrwas A. Treatment of KIT-mutated metastatic mucosal melanoma. Chin Clin Oncol. 2014; 3:35. https://doi.org/10.3978/j.issn.2304-3865.2014.08.02. [PubMed].
21. Reilly DJ, Aksakal G, Gilmour RF, Gyorki DE, Chauhan A, Webb A, Henderson MA. Subungual melanoma: Management in the modern era. J Plast Reconstr Aesthet Surg. 2017; 70:1746–1752. https://doi.org/10.1016/j.bjps.2017.08.001. [PubMed].
22. Spencer JM. Nail-apparatus melanoma. Lancet. 1999; 353:84–85. https://doi.org/10.1016/S0140-6736(05)76149-3. [PubMed].
23. Mole R, MacKenzie D. Cancer, Melanoma, Subungual. StatPearls Publishing; 2018. [PubMed].
24. Banfield CC, Redburn JC, Dawber RP. The incidence and prognosis of nail apparatus melanoma. A retrospective study of 105 patients in four English regions. Br J Dermatol. 1998; 139:276–279. https://doi.org/10.1046/j.1365-2133.1998.02365.x. [PubMed].
25. Dika E, Patrizi A, Fanti PA, Chessa MA, Reggiani C, Barisani A, Piraccini BM. The Prognosis of Nail Apparatus Melanoma: 20 Years of Experience from a Single Institute. Dermatology. 2016; 232:177–184. https://doi.org/10.1159/000441293. [PubMed].
26. Kottschade LA, Grotz TE, Dronca RS, Salomao DR, Pulido JS, Wasif N, Jakub JW, Bagaria SP, Kumar R, Kaur JS, Morita SY, Moran SL, Nguyen JT, et al. Rare Presentations of Primary Melanoma and Special Populations. Am J Clin Oncol. 2014; 37:635–641. https://doi.org/10.1097/COC.0b013e3182868e82. [PubMed].
27. Moehrle M, Metzger S, Schippert W, Garbe C, Rassner G, Breuninger H. “Functional” Surgery in Subungual Melanoma. Dermatol Surg. 2003; 29:366–374. https://doi.org/10.1097/00042728-200304000-00009. [PubMed].
28. Lieberherr S, Cazzaniga S, Haneke E, Hunger RE, Seyed Jafari SM. Melanoma of the nail apparatus: a systematic review and meta-analysis of current challenges and prognosis. J Eur Acad Dermatol Venereol. 2020; 34:967–976. https://doi.org/10.1111/jdv.16121. [PubMed].
29. Haugh AM, Zhang B, Quan VL, Garfield EM, Bubley JA, Kudalkar E, Verzi AE, Walton K, VandenBoom T, Merkel EA, Lee CY, Tan T, Isales MC, et al. Distinct Patterns of Acral Melanoma Based on Site and Relative Sun Exposure. J Invest Dermatol. 2018; 138:384–393. https://doi.org/10.1016/j.jid.2017.08.022. [PubMed].
30. Durbec F, Martin L, Derancourt C, Grange F. Melanoma of the hand and foot: epidemiological, prognostic and genetic features. A systematic review. Br J Dermatol. 2012; 166:727–739. https://doi.org/10.1111/j.1365-2133.2011.10772.x. [PubMed].
31. Beadling C, Jacobson-Dunlop E, Hodi FS, Le C, Warrick A, Patterson J, Town A, Harlow A, Cruz F, Azar S, Rubin BP, Muller S, West R, et al. KIT Gene Mutations and Copy Number in Melanoma Subtypes. Clin Cancer Res. 2008; 14:6821–6828. https://doi.org/10.1158/1078-0432.CCR-08-0575. [PubMed].
32. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic Activation of KIT in Distinct Subtypes of Melanoma. J Clin Oncol. 2006; 24:4340–4346. https://doi.org/10.1200/JCO.2006.06.2984. [PubMed].
33. Shim JH, Shin HT, Park J, Park JH, Lee JH, Yang JM, Kim DH, Jang KT, Lee DY. Mutational profiling of acral melanomas in Korean populations. Exp Dermatol. 2017; 26:883–888. https://doi.org/10.1111/exd.13321. [PubMed].
34. Tsao H, Chin L, Garraway LA, Fisher DE. Melanoma: from mutations to medicine. Genes Dev. 2012; 26:1131–1155. https://doi.org/10.1101/gad.191999.112. [PubMed].
35. Gong HZ, Zheng HY, Li J. The clinical significance of KIT mutations in melanoma. Melanoma Res. 2018; 28:259–270. https://doi.org/10.1097/cmr.0000000000000454. [PubMed].
36. Platz A, Egyhazi S, Ringborg U, Hansson J. Human cutaneous melanoma; a review of NRAS and BRAF mutation frequencies in relation to histogenetic subclass and body site. Mol Oncol. 2007; 1:395–405. https://doi.org/10.1016/j.molonc.2007.12.003. [PubMed].
37. Dietrich P, Kuphal S, Spruss T, Hellerbrand C, Bosserhoff AK. Wild-type KRAS is a novel therapeutic target for melanoma contributing to primary and acquired resistance to BRAF inhibition. Oncogene. 2018; 37:897–911. https://doi.org/10.1038/onc.2017.391. [PubMed].
38. Cosmic. COSMIC - Catalogue of Somatic Mutations in Cancer. cancer.sanger.ac.uk. Available 2020 Apr 13, from http://www.sanger.ac.uk/genetics/CGP/cosmic.
39. Rabbie R, Ferguson P, Wong K, Moran U, Turner C, Emanuel P, Haas K, Saunus JM, Davidson MR, Lakhani SR, Shivalingam B, Long GV, Parkinson C, et al. Hotspot KRAS mutations in brain metastases at the first metastatic recurrence of cutaneous melanoma. bioRxiv. 2020 Feb 18. [Epub ahead of print]. https://doi.org/10.1101/2020.02.17.952630.
40. Shtivelman E, Davies MA, Hwu P, Yang J, Lotem M, Oren M, Flaherty KT, Fisher DE. Pathways and therapeutic targets in melanoma. Oncotarget. 2014; 5:1701–52. https://doi.org/10.18632/oncotarget.1892. [PubMed].
41. Grossmann AH, Yoo JH, Clancy J, Sorensen LK, Sedgwick A, Tong Z, Ostanin K, Rogers A, Grossmann KF, Tripp SR, Thomas KR, D’Souza-Schorey C, Odelberg SJ, et al. The Small GTPase ARF6 Stimulates -Catenin Transcriptional Activity During WNT5A-Mediated Melanoma Invasion and Metastasis. Sci Signal. 2013; 6:ra14. https://doi.org/10.1126/scisignal.2003398. [PubMed].
42. Xu S, Yang Z, Zhang J, Jiang Y, Chen Y, Li H, Liu X, Xu D, Chen Y, Yang Y, Zhang Y, Li D, Xia J. Increased Levels of β-catenin, LEF-1, and HPA-1 Correlate with Poor Prognosis for Acral Melanoma with Negative BRAF and NRAS Mutation in BRAF Exons 11 and 15 and NRAS Exons 1 and 2. DNA Cell Biol. 2015; 34:69–77. https://doi.org/10.1089/dna.2014.2590. [PubMed].
43. Atlas of Genetics and Cytogenetics in Oncology and Haematology. Available 2020 Apr 13, from http://www.atlasgeneticsoncology.org/.
44. Muller P, Vousden K. p53 mutations in cancer. Nat Cell Biol. 2013; 15:2–8. https://doi.org/10.1038/ncb2641. [PubMed].
45. Yeh I, Jorgenson E, Shen L, Xu M, North J, Shain A, Reuss D, Asgari M. Targeted genomic profiling of acral melanoma. J Natl Cancer Inst. 2019; 111:1068–1077. https://doi.org/10.1093/jnci/djz005. [PubMed].
46. Gottesdiener L, O’Connor S, Busam K, Won H, Solit D, Hyman D, Shoushtari A. Rates of ERBB2 Alterations across Melanoma Subtypes and a Complete Response to Trastuzumab Emtansine in an ERBB2-Amplified Acral Melanoma. Clin Cancer Res. 2018; 24:5815–5819. https://doi.org/10.1158/1078-0432.CCR-18-1397. [PubMed].
47. Pu W, Shang Y, Shao Q, Yuan X. miR-146a promotes cell migration and invasion in melanoma by directly targeting SMAD4. Oncol Lett. 2018; 15:7111–7117. https://doi.org/10.3892/ol.2018.8172. [PubMed].
48. Tuncer E, Calçada RR, Zingg D, Varum S, Cheng P, Freiberger SN, Deng CX, Kleiter I, Levesque MP, Dummer R, Sommer L. SMAD signaling promotes melanoma metastasis independently of phenotype switching. J Clin Invest. 2019; 129:2702–2716. https://doi.org/10.1172/JCI94295. [PubMed].
49. Zhao M, Mishra L, Deng CX. The role of TGF-β/SMAD4 signaling in cancer. Int J Biol Sci. 2018; 14:111–123. https://doi.org/10.7150/ijbs.23230. [PubMed].
50. de Lima Vazquez V, Vicente AL, Carloni A, Berardinelli G, Soares P, Scapulatempo C, Martinho O, Reis RM. Molecular profiling, including TERT promoter mutations, of acral lentiginous melanomas. Melanoma Res. 2016; 26:93–99. https://doi.org/10.1097/CMR.0000000000000222. [PubMed].
51. Liang WS, Hendricks W, Kiefer J, Schmidt J, Sekar S, Carpten J, Craig DW, Adkins J, Cuyugan L, Manojlovic Z, Halperin RF, Helland A, Nasser S, et al. Integrated genomic analyses reveal frequent TERT aberrations in acral melanoma. Genome Res. 2017; 27:524–532. https://doi.org/10.1101/gr.213348.116. [PubMed].
52. O’Leary JA, Berend KR, Johnson JL, Levin LS, Seigler HF. Subungual Melanoma. A Review of 93 Cases With Identification of Prognostic Variables. Clin Orthop Relat Res. 2000; 378:206–212. https://doi.org/10.1097/00003086-200009000-00031. [PubMed].
53. Stern DK, Creasey AA, Quijije J, Lebwohl MG. UV-A and UV-B Penetration of Normal Human Cadaveric Fingernail Plate. Arch Dermatol. 2011; 147:439–41. https://doi.org/10.1001/archdermatol.2010.375. [PubMed].