
Introduction
Melanoma is one of the most aggressive forms of skin cancer with a high frequency of metastasis and a poor prognosis in the metastatic stage [1]. About 132,000 new cases of melanoma are diagnosed worldwide each year, according to the World Health Organization. Incidence rates are higher in women than in men before the age of 50, but by the age of 65, rates in men are double those in women, and by age 80 they are triple [2]. Despite advances in surgery and multi-agent chemotherapy, nearly 80% of patients still die from metastatic melanoma [3]. BRAF inhibitor and interleukin 2 therapy are often used in clinical set up but these regimens elicit severe toxicities with unsatisfactory efficacy and rapid development of resistance [4–6]. If patients were diagnosed early with primary melanoma, surgical resection is the best choice for most of them to reduce mortality [7]. However, a 5-year survival rate in metastatic melanoma is still under 15–20% of patients [8]. Clinicians prefer chemotherapy as a first line treatment option to avoid surgical consequences. High doses of chemotherapy are prescribed depending on the stage of melanoma but drug tolerance level of system is questionable. Conventional chemotherapy dosing schedule is used to balance the toxicity and efficacy, but the severe side effects in particular hepatotoxicity limits the administration of most anticancer agents [9]. Liver is the vital organ responsible for detoxification of drugs, however exposure of multiple doses of drugs leads to cholestatic hepatitis, progression to fibrosis and cirrhosis, malignant transformation, sinusoidal obstruction and fulminant hepatic failure [10–12]. Melanoma treatment is associated with liver injury. Ipilimumab and nivolumab well known target agents were approved by WHO to treat different stages of melanoma. Tanaka et al. reported that administration of both ipilimumab and nivolumab to 59 year old stage IV melanoma patients elicits severe hepatitis and elevation of liver injury associated serum biomarkers [13, 14]. This evidence alarms the urgent need of a safer alternative therapeutic regimen for melanoma.
Natural products (extracts or pure compounds) from various sources (plants, marine organisms, microorganisms, etc) are screened for their ability to act as anti-tumor agents and have shown to have structural diversity and chemical complexity, as well as modulating several cellular signaling pathways [15]. Reportedly, natural products activate anti-inflammatory, anti-tumor and/or anti-metastatic responses [15, 16] and also evade multidrug resistance [17, 18]. Vinblastine and paclitaxel, are well known anti cancer agents obtained from natural sources. Frankincense is an aromatic resin hardened from exuded gums obtained from trees of the genus Boswellia (Burseraceae family). Boswellia sp. includes Boswellia sacra from Oman and Yemen, Boswellia carteri from Somalia, and Boswellia serrata from India and China. It has been commonly used to reduce swelling and alleviate the pain of inflammatory diseases or tumors [19, 20]. Frankincense extract has been used in China for its accelerating effects on blood circulation. It has been used as an antiarthritic in ayuredic medicine in India for thousands of years [19]. Winking et al. reported that a frankincense extract induces apoptosis and prolong survival in a rat glioma model [21]. A methanol extract of Boswellia serrata inhibits abnormal skin cell proliferation induced by 12-O-tetradecanoylphorbol-13-acetate (TPA) and tumor promotion initiated by 7,12-dimethylbenz[a]anthracene (DMBA) in a mouse model [22]. In a human clinical study, a Boswellia serrata resin extract has been shown to reduce cerebral edema and potential anti-cancer activity in patients irradiated for brain tumors [23]. Boswellia sp. essential oil, an extract prepared by distillation of frankincense gum resins, is one of the most commonly used essential oils in aromatherapy and it is reported as potent anti-cancer agent in breast and pancreatic cancer models [24, 25]. Previously, we reported an anti-cancer activity induced by heavy terpenes extracted from frankincense in breast cancer model [26]. These evidences strongly suggests the anti-tumor potential of frankincense, however, the efficacy of frankincense essential oil on melanoma is not yet reported. In this study we assessed the effect of frankincense essential oil on in vitro and in vivo model of melanoma. Further experiments were also carried out to investigate its potential hepatoprotective efficacy.
Results
Cell viability
B16-F10 and FM94 cells were treated with FEO at 3, 5, 7 and 10 μg/ml for 24 h. FEO induced reduction in the cell viability of B16-F10 and FM94 carcinoma cells as compare to the untreated control (Figure 1A and 1B) and not normal human epithelial melanocyte (HNEM) (Figure 1C). FEO treated FM94 cells completely lost their adherence and morphology whereas HNEM cells retained their morphology (Figure 1D and 1E). The data indicated that pre treatment with FEO induced cytotoxicity in tumor cells by sparing normal cells.
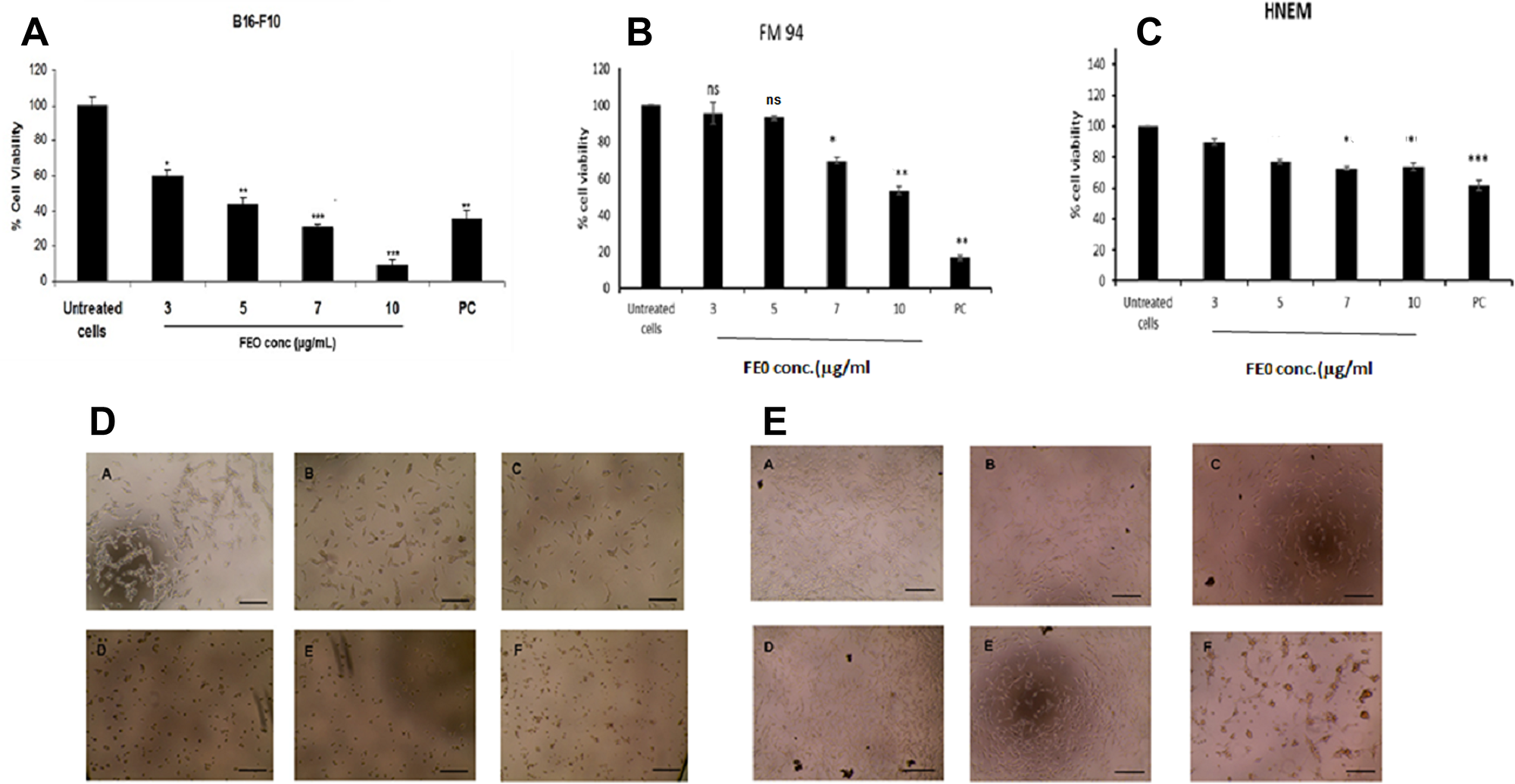
Figure 1: Cytotoxicity of FEO on B16-F10, FM94 and HNEM cells. (A) B16-F10 cells; (B) FM94 cells; (C) HNEM cells; (D) FM94 cells treated with FEO (A-Untreated cells; B-3 μg/ml; C-5 μg/ml; D-7 μg/ml; E-10 μg/ml; F- Dox 5 ug/ml) for 24 h and morphological image was photographed by EVOS image analyser; E: HNEM cells treated with FEO (A-Untreated cells; B-3 μg/ml; C-5 μg/ml; D-7 μg/ml; E-10 μg/ml; F- Dox 5 ug/ml) for 24 h and morphological image was photographed by EVOS image analyser. Data presented as mean ± SD of triplicates of three independent experiments. *Represents significant difference at p < 0.05 compared with control. **Represents significant difference at p < 0.01 compared with control. ***Represents significant difference at p < 0.001 compared with control. NS: Non significant. Scale bar indicates 10 um.
Nuclear fragmentation
24 h of FEO treatment induced apoptosis in B16-F10 cells. Hoechest staining reveals that 5, 7 and 10 μg/ml of FEO induced nuclear fragmentation when compared to untreated control (Figure 2). These data indicate that FEO constituents target the nucleus of melanoma cells and cause DNA damage.
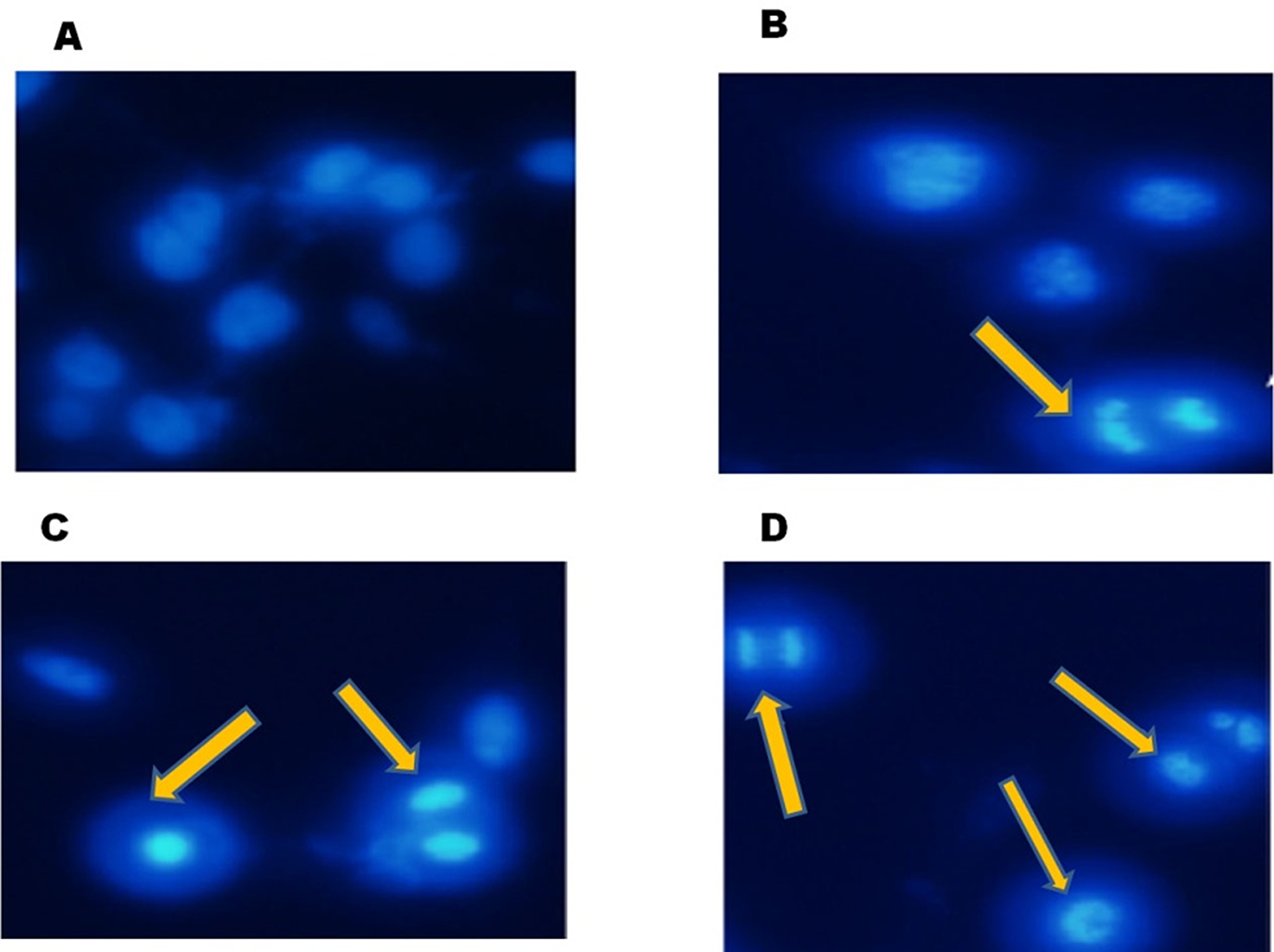
Figure 2: FEO induced nuclear fragmentation in B16-F10 cells: B16-F10 cells were treated with different concentrations of FEO for 24 hours and stained with hoechest stain 33258. (A) Control; (B) 5 ug/ml; (C) 7 ug/ml; (D) 10 ug/ml. The nuclei were observed using fluorescence microscope (20 um).
DNA fragmentation
To ensure the DNA targeting ability of FEO, B16-F10 cells were treated with different concentrations (5, 7 and 10 μg/ml) of FEO for 24 hrs and observed for DNA damage. Figure 3 shows that FEO induced DNA ladder formation in dose-dependent manner.
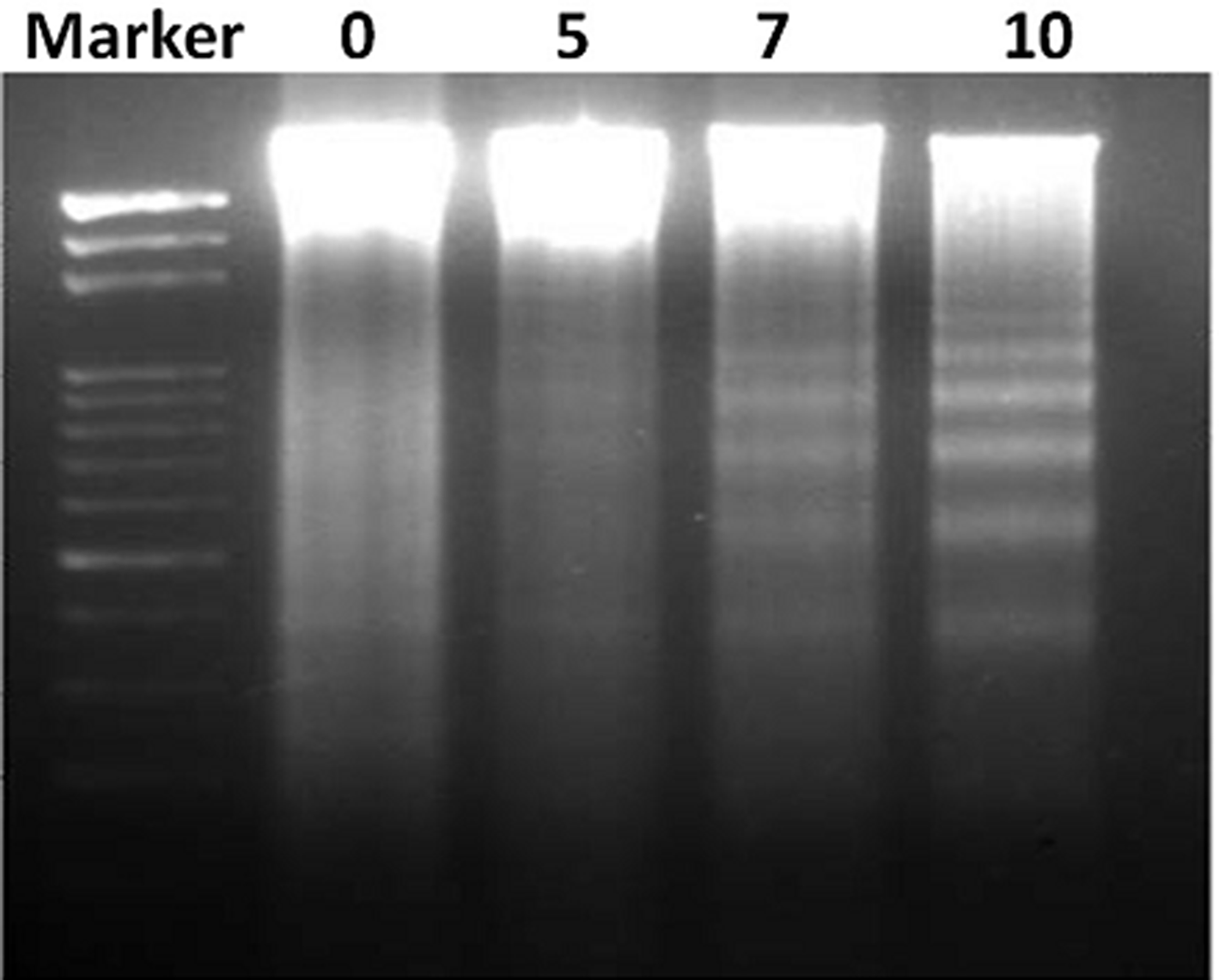
Figure 3: FEO induce DNA fragmentation in B16-F10 cells. B16-F10 cells were treated with 0, 5, 7 and 10 μg/ml for 24 hours. DNA was isolated resolved in agarose gel and examined by ethidium bromide staining.
Apoptosis
B16-F10 cells were treated with FEO at 10 μg/ml for 24 h and analyzed for apoptosis by annexin V staining using flow cytometry. FEO treatment resulted in a significant increase in percentage of cells in early (25%) and late (10%) apoptotic stage whereas only 2–3% of untreated cells reached early and late apoptosis stage (Figure 4).
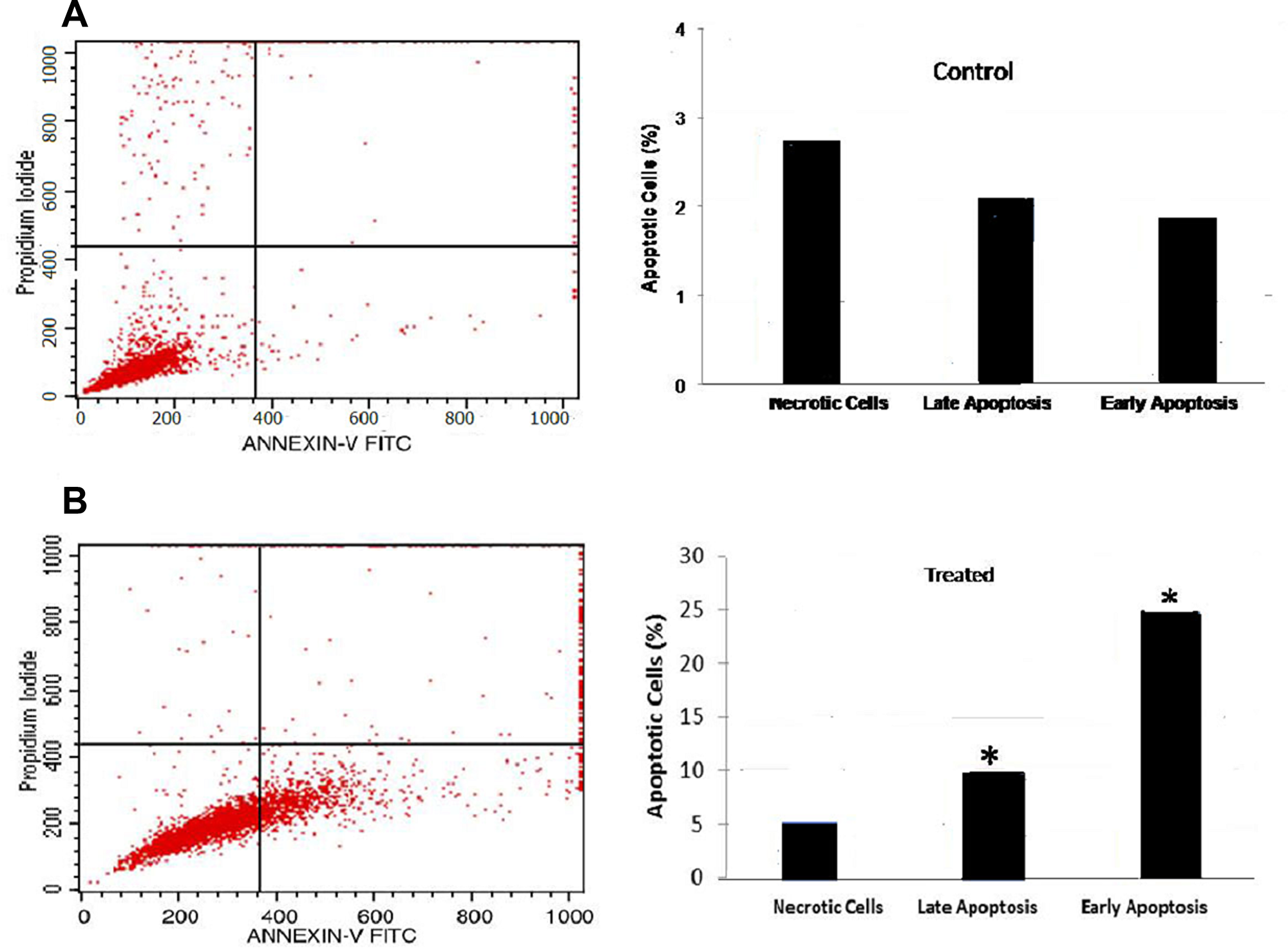
Figure 4: Flow cytometric analysis of apoptosis: B16-F10 cells treated with FEO at 10 μg/ml for 24 h. Annexin-V FITC and propidium iodide staining were used to analyze apoptosis by flow cytometry. (A) Untreated cells showed 2–3% of cells in early or late apoptosis stage. (B) FEO treated cells, significant increase (*P < 0.05) in percentage distribution of cells in early (25%) and late (10%) apoptosis were observed.
Molecular mechanism of inducing apoptosis by FEO
To determine the mechanism of apoptotic induction in B16-F10 cells by FEO treatment, the expression levels of Bcl-2 and Bax by western blot were analyzed. The immunoblot analysis showed gradual decline in the expression levels of Bcl-2 with marked increase in the Bax protein expression levels in a time-dependent manner (Figure 5A). To investigate if the apoptosis was induced by FEO on human melanoma cells, FM94 cells treated with FEO (0, 7, 10 μg/ml) for 24 hours and the level of expression of the apoptotic markers (caspase 3, caspase 9, and PARP) were assessed. Figure 5B illustrates that FEO significantly up regulated the cleaved caspase 3, caspase 9, and PARP expression in dose-dependent manner. Further time-dependent down regulation of MCL-1 a major anti-apoptotic protein in melanoma was observed in FM94 cells by FEO (10 μg/ml) treatment (Figure 5C). This data suggest that FEO induced apoptosis in both mice B16-F10 and human FM94 carcinoma cells.
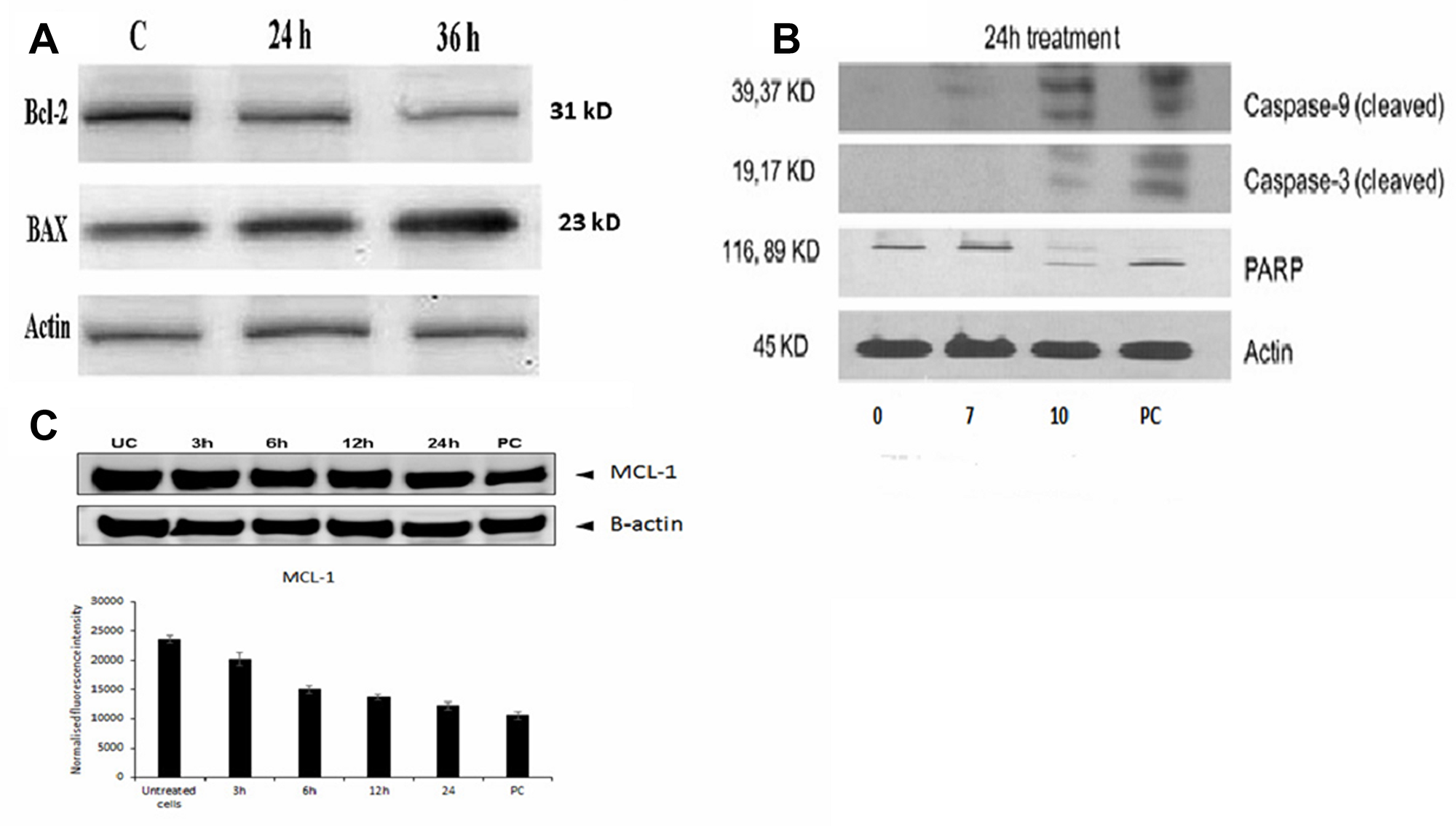
Figure 5: Molecular mechanism of FEO induced apoptosis in B16-F10 and FM94 cells. (A) Bcl-2 and Bax protein expression of B16-F10 cells was determined after 24 and 36 h of treatment with 10 μg/ml of FEO; (B) Cleaved Caspase 9, Caspase 3 and PARP expression of FM94 cells was determined after 24 h treatment with 0, 7, 10 μg/ml of FEO. PC indicates positive control (5 μg/ml of doxorubicin); (C) MCL-1 expression of FM94 cells was determined after 3 h, 6 h, 12 h, and 24 h treatment with 10 μg/ml of FEO. Actin used for normalization of protein expression.
In vivo toxicity experiment
In order to assess the toxicity of FEO, mice were treated with a single dose of FEO (1200 mg/kg body weight) (i.p.) and observed for four weeks. Body weight and histology of major tissues (liver, kidney, brain, and heart) were analyzed. No significant difference in the body weight was observed in FEO treated mice compared to untreated control animals (Figure 6). Histological studies revealed that tissue sections from FEO-treated animals did not indicate any detectable pathologic abnormalities as examined by H&E staining. The liver showed normal hepatic lobular architecture, intact central vein with trapped red blood cells in a liver section from FEO treated animals. Kidney sections showed normal glomeruli, proximal and distal tubules, interstitium, and blood vessels. Brain and heart muscles normal morphology compared to untreated tissue sections (Figure 7).
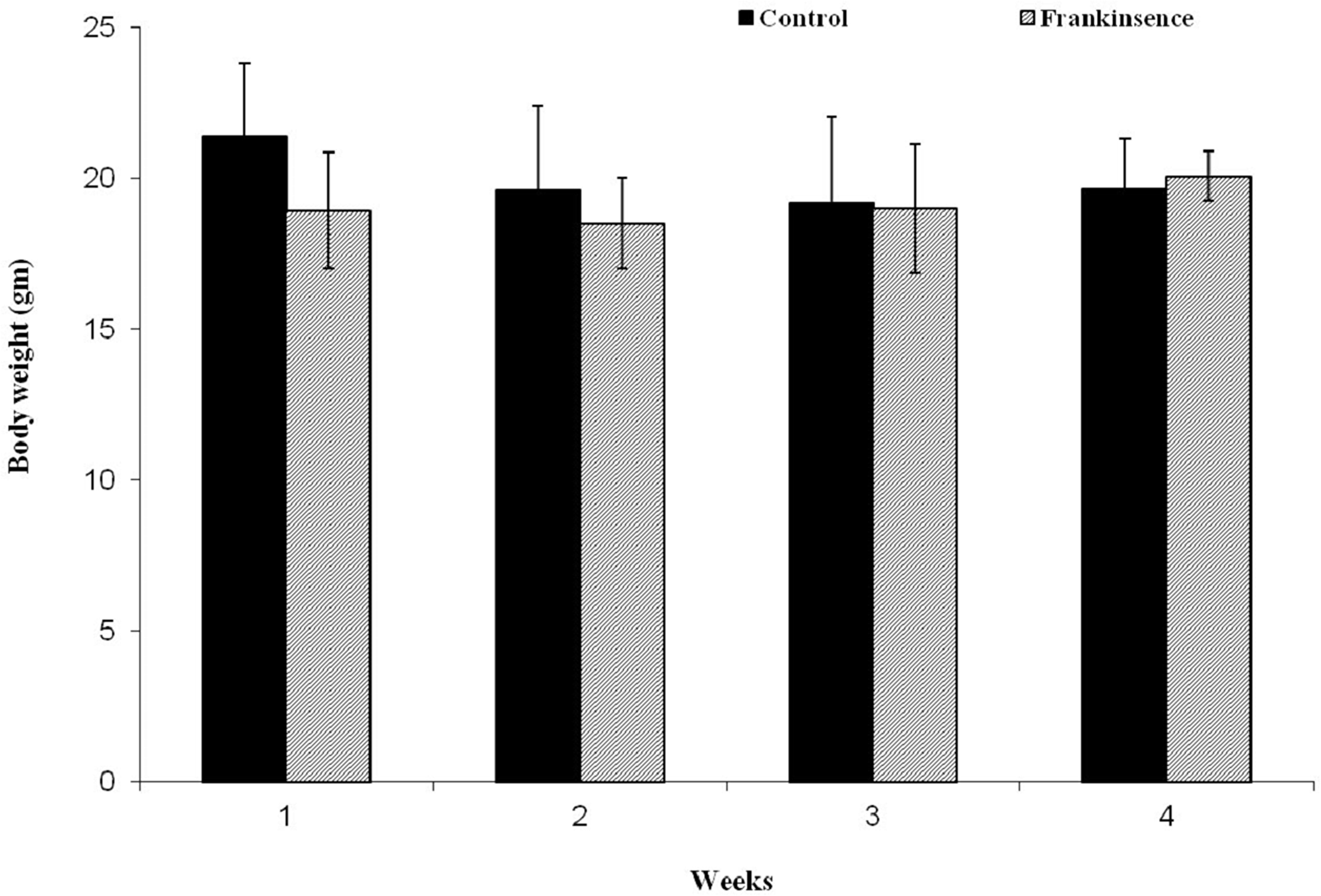
Figure 6: Effect of FEO on animal body weight: Data presented as mean ± SD (n = 8). Mice were treated with FEO (1200 mg/kg body weight) and observed for 30 days at weekly intervals.
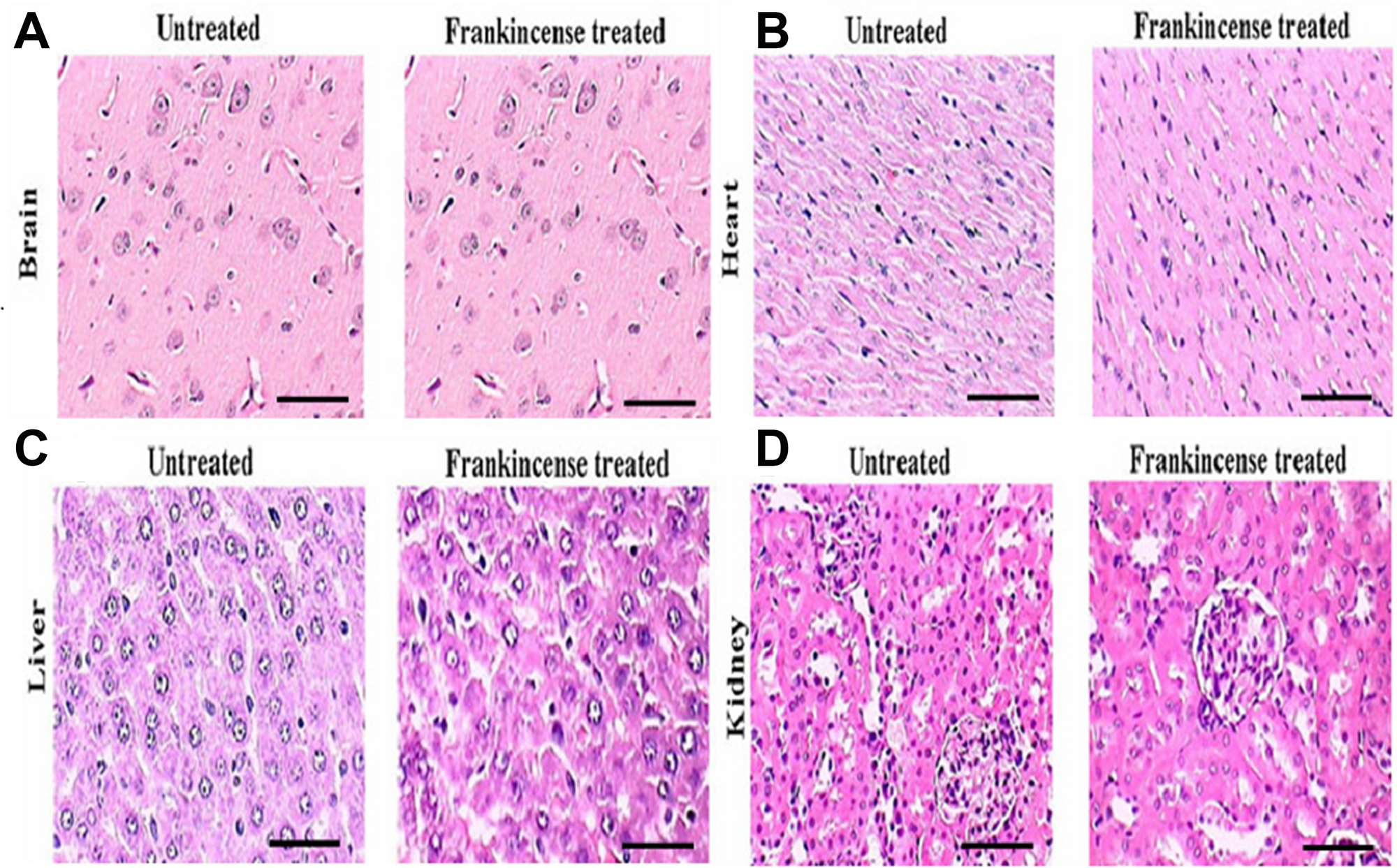
Figure 7: In vivo of toxicity of FEO on major organs: Mice were treated with FEO (1200 mg/kg body weight). At the end of experimental period major organs such as brain, heart, liver and kidney were excised and stained with hemotoxylin and eosin. Scale bar indicates 10 μm.
Tumour remission by FEO
Melanoma tumour remission was observed in control and FEO treated tumour bearing mice. After 14 days the size of tumour was reduced in FEO treated mice compared to untreated control animals (Figure 8). Pattern of tumour growth inhibition revealed that FEO treatment reduced the tumor size dose dependently.
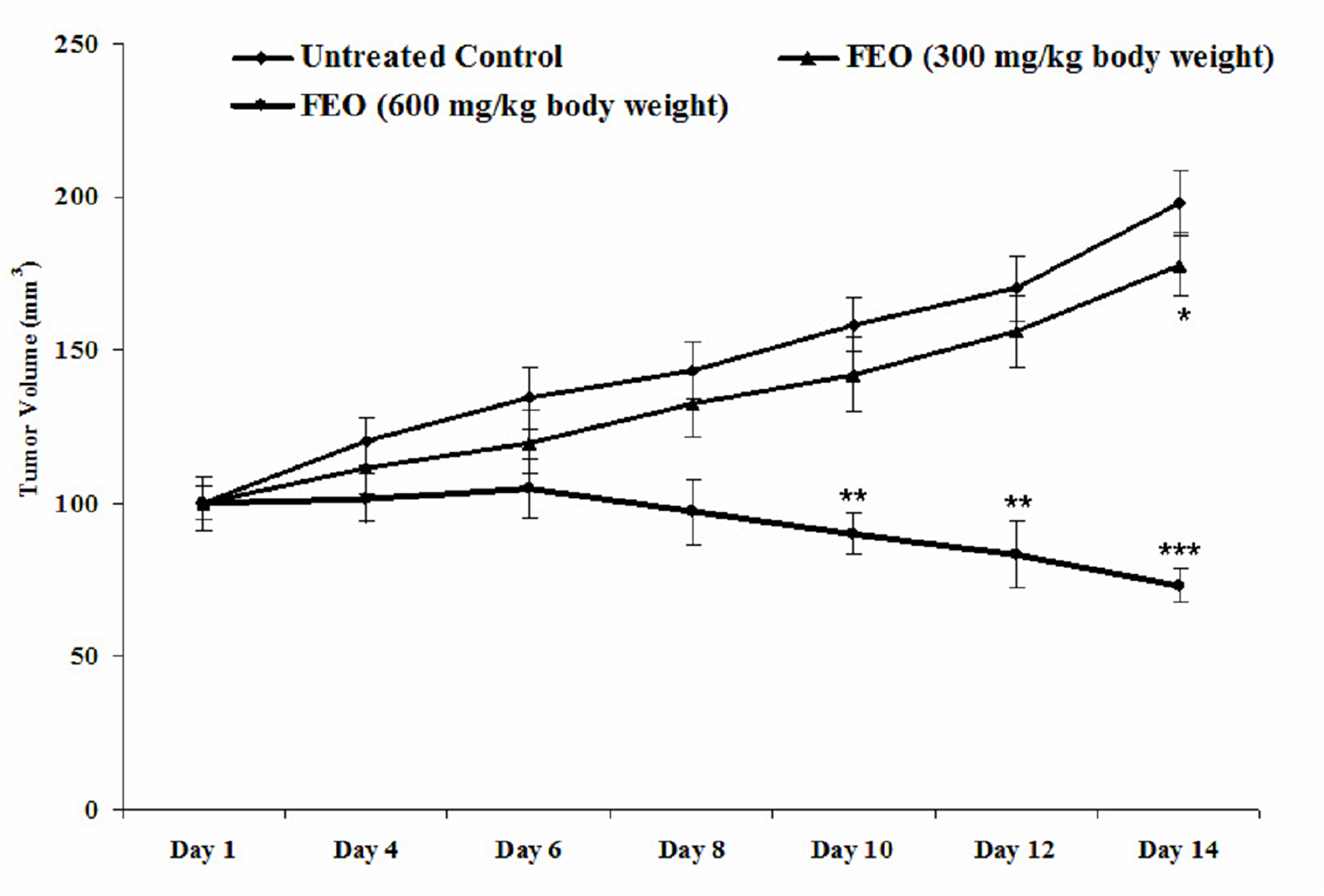
Figure 8: Melanoma tumor remission efficacy of FEO: Data presented as mean ± SD (n = 6). Tumor was induced by transplanting B16F10 cells (5 × 105) in C57BL/6 mice. Tumor bearing mice treated with FEO (300 and 600 mg/kg of body weight) every two days for the total experimental period of 14 days and volume of tumor was measured. FEO 300 mg/kg bwt significantly (*p < 0.05) reduce the tumor volume on day 14 compare to control group. FEO 600 mg/kg bwt significantly (**p < 0.01, ***p < 0.001) reduced tumor burden on day 10, 12, and 14 compared to control group.
In vivo hepatoprotective efficacy of FEO
Efficacy of FEO on hematology of acetaminophen induced hepatic injured mice
Acetaminophen is well known to induce both acute and chronic hepatic failure. Therefore in this study we used acetaminophen to develop hepatic injury mice model and assessed the efficacy of FEO on complete blood count (CBC). The PVC (35.20 ± 1.93), WBC (6100 ± 556.78), neutrophil (78.20 ± 0.58), Hb (6.07 ± 0.18), MCHC (24.1 ± 1.74), and RBC (6.40 ± 0.092), was declined and lymphocytes (20.00 ± 0.50), MCV (133.4 ± 23), MCH (31.1 ± 4.37) counts were significantly (p < 0.05) increased in acetaminophen (750 mg/kg body weight) treated mice. Acetaminophen induced abnormality was significantly (p < 0.05) reversed by FEO (250 and 500 mg/kg body weight) treatments (Table 1).
Table 1: Complete blood count: Values are expressed as mean ± SD (n = 8)
| Parameters | Group I | Group II | Group III | Group IV |
|---|---|---|---|---|
| PVC | 39.33 ± 0.67 | 35.20 ± 1.93 | 37.25 ± 3.04 | 33.75 ± 0.25 |
| White blood cells (10 m/m3) | 6500 ± 288.68 | 6100 ± 556.78 | 6500 ± 540.06 | 5000 ± 204.12 |
| Lymphocytes | 18.23 ± 1.76 | 20.00 ± 0.50* | 20.40 ± 0.58 | 20.00 ± 0.82 |
| Neutrophils | 81.67 ± 1.76 | 78.20 ± 0.58* | 79.50 ± 0.96 | 79.00 ± 0.86 |
| Hb (g/dl) | 8.10 ± 0.29 | 6.07 ± 0.18 | 6.93 ± 0.26 | 7.10 ± 0.70 |
| MCV(μm3) | 72.9 ± 7.97 | 133.4 ± 23 | 105.7 ± 15.6 | 88.0 ± 10.2 |
| MCH (pg) | 20.9 ± 2.21 | 31.1 ± 4.37 | 26.7 ± 4.04 | 22.7 ± 1.75 |
| MCHC (%) | 29.1 ± 1.88 | 24.1 ± 1.74 | 25.9 ± 2.31 | 26.7 ± 1.98 |
| RBC (106 /mm3) | 8.67 ± 0.072 | 6.40 ± 0.092* | 7.47 ± 0.0821** | 8.52 ± 0.079 |
Efficacy of FEO on liver histology of acetaminophen induced hepatic injured mice
Drug induced hepatic injury markedly disturb the morphology and arrangement of cells within liver tissue. Acetaminophen (750 mg/kg body weight) treatment induced mild sinusoidal congestion, hepatocellular necrosis, focal damage around the central vein, feathery degeneration, loss of lobular architecture with damaged cellular outlines, hemorrhage, nuclei have become condensed, and microvesicular fatty change (Figure 9). These detrimental effects of acetaminophen were restored by pre treatment with FEO (250 and 500 mg/kg body weight) and silymarin (25 mg/kg body weight).
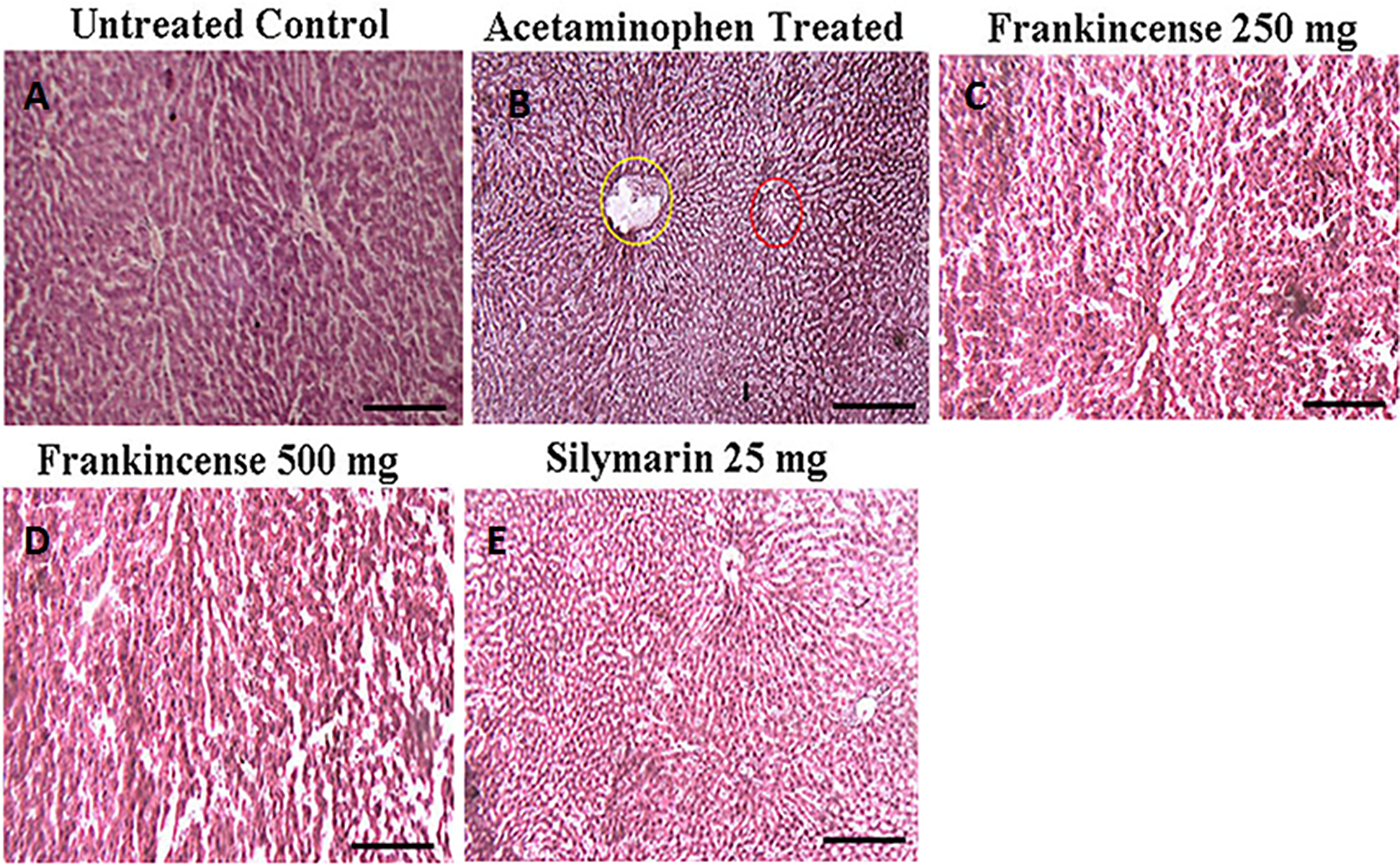
Figure 9: Efficacy of FEO on acetaminophen induced hepatic injury: Mice were divided into five groups (n = 6). (A) Untreated control: Mice received normal saline (0.9% v/w) i.p. for seven days. (B) Acetaminophen treated Acetaminophen (750 mg/kg body weight) alone injected to mice i.p. for seven days to induce hepatic injury. (C) Frankincense 250 mg: Mice treated simultaneously with Acetaminophen (750 mg/kg body weight) and FEO (250 mg/kg body weight) for seven days. (D) Frankincense 500 mg: Mice treated simultaneously with Acetaminophen (750 mg/kg body weight) and FEO (500 mg/kg body weight) for seven days. (E) Silymarin 25 mg: Mice treated simultaneously with Acetaminophen (750 mg/kg body weight) and silymarin (25 mg/kg body weight) (positive drug control). After seven days of treatment period the liver of all the animals was excised and histology was carried out. Scale bar indicates 10 μm.
Efficacy of FEO on biochemical parameters of acetaminophen induced hepatic injured mice
Alteration in biochemical parameters is clinical manifestation of systemic toxicities including hepatotoxicity. In this study, we measured the levels of toxicity associated biomarkers such as AST, ALT, ALK, bilirubin, protein, cholesterol, glucose, albumin/globulin ratio and triglycerides in serum. Acetaminophen (750 mg/kg body weight) treated mice showed elevated levels of AST (118.50 ± 1.72, p < 0.05), ALT (114.00 ±2.67, p < 0.05), ALK (32.24 ± 1.96, p < 0.05), bilirubin (6.20 ± 0.63, p < 0.05), cholesterol (229.67 ± 0.75) and triglyceride (144 ± 14.4) whereas, the levels of protein (5.06 ±0.20, p < 0.05), glucose (90.66 ± 2.45), and albumin/globulin ratio (4.36 ± 0.4) were decreased (Figure 10). Seven days of pre treatment with FEO (250 and 500 mg/kg body weight) reversed the changes caused by acetaminophen and brought back the level of biochemical parameters to near normal control.
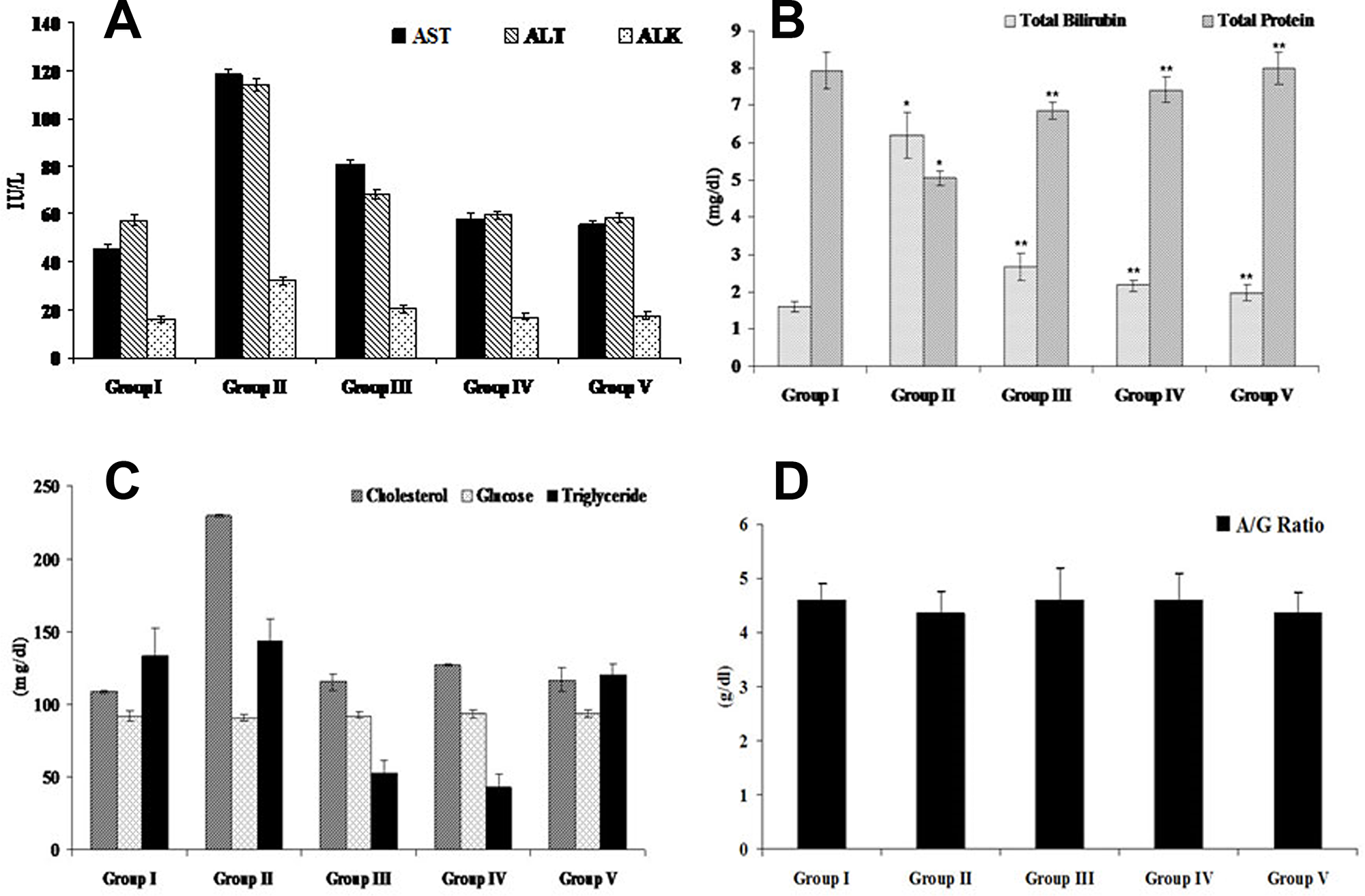
Figure 10: Efficacy of FEO on biochemical parameters of acetaminophen induced hepatic injured mice: Values are expressed as Mean ± SD (n = 6). *p < 0.05 is considered significant when compared with group I; **p < 0.01 is considered significant when compared with group II by Dennett’s multiple comparison test. Group I: Untreated control fed orally with normal saline (0.9% v/w) 5ml/kg body weight daily. Group II as hepatotoxic control treated with acetaminophen (750 mg/kg body weight). Group III and IV received FEO 250 and 500 mg/kg body weight respectively along with acetaminophen (750 mg/kg body weight) for seven days. Group V treated with Silymarin (25 mg/kg body weight) with acetaminophen (750 mg/kg body weight) for seven days. (A) AST, ALT, ALK. (B) Total bilirubin and total protein. (C) Cholesterol, glucose, and triglyceride. (D) Albumin/globulin ratio.
Efficacy of FEO on Phase I and II drug metabolizing enzymes
Drug metabolizing enzymes such as cytochrome p450 (Cytp450), cytochrome b5 (Cytb5) and glutathione-s-transferase (GST) play pivotal role in detoxification of drugs. To develop FEO based drug formulation for melanoma treatment it is imperative to study efficacy of FEO on above mentioned enzymes. Administration of 250 and 500 mg/kg body weight of FEO for seven days changed the levels of Cytp450 (2.7 ± 0.89, p < 0.001; 3.9 ± 0.74, p < 0.001 respectively), Cytb5 (0.31 ± 0.19 p < 0.01; 0.36 ± 0.14, p < 0.01 respectively), and GST (3.46 ± 1.08, p < 0.001; 3.89 ± 1.45, p < 0.001) significantly (Figure 11). Interestingly, treatment with FEO enhanced levels of these enzymes and this data clearly stated that FEO can retain hepatic physiological functions in particular detoxification. BHA was used positive diet control.
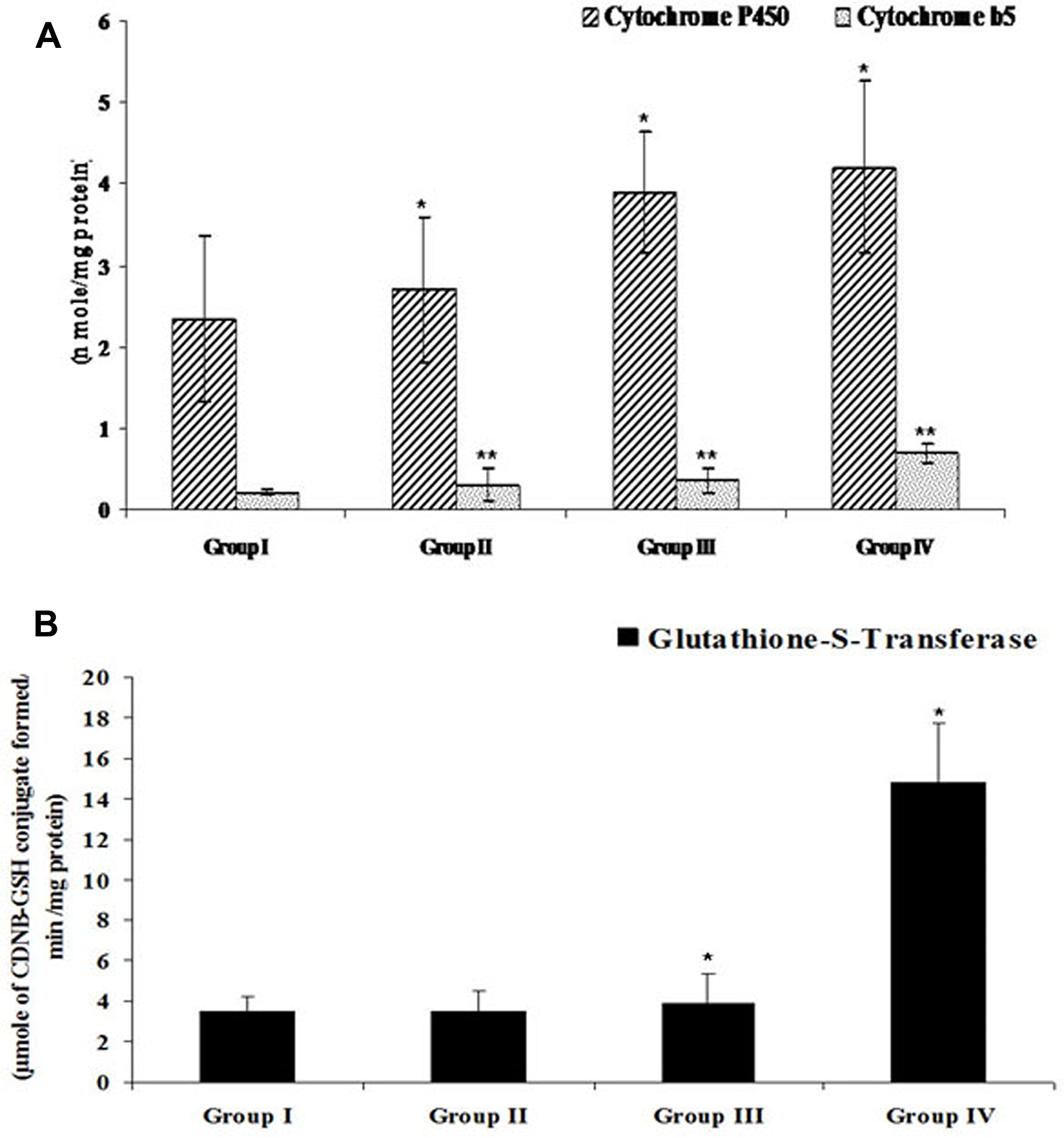
Figure 11: Efficacy of FEO on drug metabolizing enzymes of acetaminophen induced hepatic injured mice: Values are expressed as mean ± SD (n = 8) animals. *p < 0.05, **p < 0.01 represent significant changes against control. Group I: Untreated control and fed orally with distilled water daily for seven days. Group II: Mice treated with FEO 250 mg/kg body weight. Group III: Mice treated with FEO 500 mg/kg body. Group IV: Mice treated with 0.75% BHA along with diet (positive control). (A) CytP450 and Cytb5, (B) GST. BHA – butylated hydroxyanisole; Cyt P450 – Cytochrome P450; Cyt b5 – Cytochrome b5; GST – Glutathione S-transferase.
DISCUSSION
Surgical resection for early stage and drug (Ipilimumab and nivolumab) therapies for advanced stage of melanoma (metastasis) are often chosen by the oncologist but poor prognosis status and a decline in the patient’s survival accompanied with post treatment toxicities are major concerns. Diagnosis at early stage and developing tolerable agents for therapy are major challenges. Thus natural products have attracted attention in recent years for developing effective and safe anti-cancer drugs. About 36% of the low molecular weight compounds approved by the Food and Drug Administration (FDA) are natural products or their derivatives [27]. Epidemiological studies states that natural products including resveratrol, lycopene, dioscin and polyunsaturated omega-3 fatty acids (PUFA), play an indispensable role in preventing cancers with lower toxicities [28–32]. Frankincense is well known plant in Middle Eastern regions (Yemen and Oman), India and China. Since ancient times it is a common practice that inhaling of frankincense resin smoke, drinking of aqueous extract of frankincense resin and chewing fresh resin for healing various ailments. Different varieties of frankincense are available and some of them are edible too. In this study, we used hojari variety of frankincense resin (Boswellia sacra) which is edible and it is consumed by local people. Frankincense extract has been studied for various biological activities in particular as an anti-cancer agent and different forms of boswellic acids have been isolated as it is principal components [33]. The use of frankincense essential oil (FEO) in aromatherapy is well known and it is also reported its potential as anti-cancer agent against breast and pancreatic cancer in vitro [24, 25]. Despite the potential of FEO no concrete scientific evidence is available to translate this in to clinic. To explore further herein we report for the first time anti melanoma (in vitro and in vivo) efficacy and hepatoprotective capacity of FEO.
Cytotoxicity of FEO was tested in B16-F10, FM94 and HNEM cells. 24 h treatment with FEO significantly reduced the cell viability of B16-F10 and FM94 cells by sparing HNEM cells (Figure 1A–1C). This clearly indicates that FEO selectivity inducing cytotoxicity in carcinoma cells and not in normal cells. To unravel the mechanism FEO mediated cell death, FEO treated cells were subjected to apoptotic analysis. Biochemical signatures accompanied with apoptosis include chromosomal DNA cleavage into inter-nucleosomal fragments, phosphatidylserine externalization and a number of intracellular substrate cleavages by specific proteolysis [34]. Hoechest 33258 staining reveals that FEO treatment induced dose dependent nuclear damage in B16-F10 cells (Figure 2). Further experiment, indicated that FEO induces DNA damage in a dose dependent manner (Figure 3). It is well established that the principal component of FEO is α-pinene with other terpene mixtures [35]. These complex terpenes mixtures could intercalate with DNA to form adducts. Similarly α-pinene isolated from Schinus terebinthifolius Raddi induced DNA fragmentation in B16-F10 cells [36]. Previous works demonstrated that essential oils and other plant derived compounds can induce genomic DNA fragmentation in apoptotic cells [37, 38]. Though FEO treatment targeted the DNA in B16-F10 cells, it is imperative to understand apoptotic state of B16-F10 cells upon FEO treatment. Flow cytometric cell cycle analysis showed that FEO facilitates early apoptosis in B16-F10 cells than late apoptosis compared to untreated cells (Figure 4). Nuclear condensation and endonuclease G also translocates to the nucleus where it cleaves nuclear chromatin to produce oligonucleosomal DNA fragments which is referred to as early form of apoptosis [39, 40]. This is in agreement with our results where we observed FEO elicits extensive DNA damage with nuclear fragmentation. Induction of apoptosis is considered as a predominant therapeutic approach for various bioactive agents and natural products. Previous studies have indicated that Bcl-2 family proteins and pro-apoptotic proteins (Bax and Bak) play an important role in the intrinsic apoptotic pathway by regulation of mitochondria integrity [41]. While reduced Bcl-2 expression accompanied with high expression of Bax may promote apoptotic response to anticancer drugs, increased expression of Bcl-2 leads to resistance to chemotherapeutic drugs [42]. In our study, we found that FEO down regulates the expression of Bcl-2 with simultaneous up regulation of pro apoptotic Bax in B16-F10 cells (Figure 5A). FEO elicits apoptosis by down regulating the Bcl-2 which leads to formation of apoptosome in the cytosol and initiation of caspase signaling cascade.
In order to evaluate the molecular mechanism of FEO induced apoptosis, we treated human melanoma cells (FM94) with FEO and assessed caspase 3, caspase 9 and PARP expression. The hallmark of apoptosis is upon receiving death signal in the form of ligands (Fas, Death receptor 3, 4, 5, and TRAIL) or drugs, proapoptotic proteins such as caspase 3, 8 and 9 undergo post translational modifications that include dephosphorylation and cleavage leading to their activation and formation of death inducing signaling complex and translocated in to mitochondria where apoptosis initiated [43]. Moreover caspase-3 activation is responsible for cleaving most apoptotic substrates such as poly-(ADP-ribose) polymerase (PARP) [44, 45]. In this study we observed profound expression of cleaved caspase 3, caspase 9 and PARP in FM94 cells upon FEO treatment (Figure 5B). Our data indicates that induction of apoptosis by FEO was caspase-dependent and appeared to be mediated by the mitochondrial pathway. Similarly centipedegrass extract and docetaxel induced apoptosis in melanoma cells via caspase signaling [46, 47]. Studies found that MCL-1 major anti-apoptotic protein was frequently overexpressed and identified as an important target in majority of human cancers [48]. MCL-1 is known to be critical for survival of melanoma cells under various stress conditions [49, 50]. The expression of MCL-1 is also known to increase in melanoma with disease progression [51]. Reduction of MCL-1 expression weakens the link between pro- and anti-apoptotic Bcl-2 family proteins, such as Noxa/Mcl-1 and Bim/Mcl-1, resulting in the release of pro-apoptotic Bcl-2 family proteins. Thus Mcl-1 plays a unique role in regulating apoptosis, as elimination of Mcl-1 is required at an early stage for induction of apoptosis [49, 50] Therefore we studied the efficacy of FEO on MCL-1 expression in FM94 cells. We found that FEO down regulates MCL-1 time dependently (Figure 5C). This suggest a key role for MCL-1 in regulating apoptosis in FM94 cells induced by FEO. Similarly demethylzeylasteral and 9, 11-dehydroergosterol peroxide induce apoptosis in melanoma cells by down regulating MCL-1 [52, 53].
Toxicity is a limitation of chemotherapy therefore the search for safer and system tolerated agents is always encouraged by the WHO. To determine whether 1200 mg/kg body weight of FEO treatment elicits changes in body weight and toxicities to normal major organs, we examined brain, heart, liver and kidney of treated and untreated groups of Swiss albino mice. Histological sections of these tissues from FEO treated mice did not show any detectable pathologic abnormalities as examined by H&E staining (Figure 7). The mouse melanoma cell B16F10, derived from C57BL/6 mice, is one of the most malignant cancers in mice and can serve as a good model of human melanoma to evaluate various treatments including vaccines [54–56]. Therefore in this study we used melanoma xenograft model by injecting B16F-10 cells in C57BL/6 mice. FEO (300 and 600 mg/kg body weight) treatment reduced tumor size significantly compared to untreated control animals (Figure 8). This is in agreement with our in vitro data. Generally essential oils are usually devoid of long term genotoxic risks and many of them show antimutagenic capacity that can be linked to anticarcinogenic activity. Furthermore, studies have demonstrated that essential oil constituents are very efficient in reducing the melanoma tumour volume or tumour cell proliferation by exerting apoptotic effects [57, 58].
Chemotherapy induced liver injury can cause necrosis, steatosis, fibrosis, cholestasis, and vascular injury [59]. Stage IV melanoma patient treated with ipilimumab and nivolumab exhibit elevation of hepatic injury biomarkers [13, 14]. Drug hepatotoxicity, is the leading cause of acute liver failure in the clinic worldwide [60]. Boswellia sacra extract shown to be hepatoprotective agent [61] however, FEO has not yet been reported for hepatoprotection effect. Acetaminophen is a classical hepatotoxin that is responsible for almost 50% of all acute liver failure cases in the USA, the UK and many Western countries [60]. Hepatotoxicity is a direct liver injury caused by the toxic metabolite of acetaminophen [62]. In this study, acetaminophen (750 mg/kg body weight) was used to induce hepatic injury. Hepatoprotective efficacy of FEO (250 and 500 mg/kg body weight) was studied on hepatic injured mice by assessing hematology (complete blood count), liver tissue histology, serum biochemical parameters, and phase I and II drug metabolizing enzymes. The analysis of blood cell count and Hb concentration are preliminary parameters to indicate abnormality. Pre treatment with acetaminophen altered the PVC, WBC, Lymphocytes, Neutrophils, Hb, MCV, MCH, MCHC%, and RBC. The administration of FEO reversed the altered counts in to normal level (Table 1). Our results evidenced that FEO can protect hepatic injury and associated blood cells abnormalities. Heavy dose of cancer drugs weakens the immune cells such as WBC, lymphocytes, and neutrophils which lead to their malfunction. A retrospective analysis of several ipilimumab Phase I–III trials in patients with advanced melanoma showed that immune related adverse events occurred in about 64.2% of treated patients and resulted in death [63]. Further Hb concentration along with RBC count determines the exchange of gases (O2 and CO2) to maintain homeostasis. FEO treatment restored blood cell counts and Hb level to normal values however; detail mechanistic study is required to delineate the role of FEO on blood components.
Levels of transaminases (AST and ALT) and liver histopathological and morphometrical changes were used as indicators of hepatotoxicity [64]. In this study we analyzed AST and ALT levels in acetaminophen along with FEO treated mice. Treatment with FEO (250 and 500 mg/kg body weight) reduced elevated levels of AST and ALT induced by acetaminophen administration (Figure 10). FEO could retain the liver structural integrity which was disturbed by acetaminophen because transaminases present in cytoplasmic location is released into the circulating blood only after structural damage [65, 66]. ALK is a marker of intra or extra hepatic cholestasis, its synthesis increases and the enzyme is thus released into plasma under such conditions. Drug-induced liver injury may present with a cholestatic pattern conjugated with hyperbilirubinemia accompanied by an increase in ALK levels [67]. In our study we found that treatment with acetaminophen enhanced the levels of serum ALK and bilirubin (Figure 10). Administration of FEO significantly reduced the ALK and bilirubin levels. Hepatotoxic agents induced biliary obstruction which is responsible for hyperbilirubinemia and enhanced ALK levels [68]. Our data indicates that treatment with FEO could protect the billiary obstruction caused by acetaminophen. Liver is the primary site of the synthesis of plasma proteins [69], disturbances of protein synthesis therefore occur as a consequence of impaired hepatic function which will lead to a decrease in the plasma concentrations. In our study we observed that acetaminophen treatment significantly reduced serum protein concentration. Seven days of treatment with FEO restored the total protein levels. This data shows that impairment of hepatic function caused by acetaminophen has been rescued by FEO. Albumin and globulin (A/G) accounts for the largest proportion of serum protein and they are generated by the liver and A/G ratio acting as one of the biomarkers for evaluating liver function [70]. In this study, we found that treatment with acetaminophen diminished the level of A/G ratio. However, treatment with FEO brought back the A/G ratio to normal levels (Figure 10). It was reported that clinical observation of decrease of serum albumin levels in hepatic diseases are associated with destruction or loss of parenchymal elements [71]. In hepatic diseases gamma globulin may be produced in increasing amounts by the hepatic reticuloendothelial cells [72] which eventually alters A/G ratio. Failure to return to normal should raise a suspicion of delayed recovery or chronic active hepatitis. Our data reveals that FEO might protect parenchymal and reticuloendothelial cells to balance the A/G ratio. High triglyceride and cholesterol level in hepatic injury is the clinical manifestation of cholestasis or hepatitis. Generally, the level of plasma lipids tends to decrease with the severity of liver disease [73, 74]. This is in agreement with our results, where we found acetaminophen (750 mg/kg body weight) increased the level of cholesterol and triglycerides (Figure 10). The results evidenced that acetaminophen might induce reduction in lipase activity, which could lead to decrease in triglyceride hydrolysis and damage of hepatic parenchymal cells that subsequently leads to disturbance of lipid metabolism in the liver. Administration of FEO (250 and 500 mg/kg body weight) diminished levels of cholesterol and triglyceride. These finding showed that mechanism of lipid lowering effects of FEO might be attributed to an inhibitory activity on microsomal acyl coenzyme A: cholesterol acyltransferase enzyme. This enzyme is responsible for acylation of cholesterol to cholesterol esters in the liver. Furthermore, level of glucose was measured in acetaminophen-induced hepatic injured mice since maintenance of blood glucose requires the relatively intact liver cells to release glucose and hepatic insufficiency. The latter might leads to production of several abnormalities directly or indirectly associated with carbohydrate metabolism [75]. In the present study administration of FEO maintained the level of glucose similar to control level (Figure 10). It is suggested that FEO might protect glucose releasing hepatocytes to maintain the constant/normal glucose level.
Histopathological study of liver tissue revealed that normal control group showed normal hepatic cells with intact cytoplasm and well defined nucleus with restricted number of inflammatory cells (Figure 9A); whereas acetaminophen treated group, showed a central vein dilated along with hemorrhage, nuclei condensation, cytoplasmic membrane destruction. In addition, scattered lymphocyte and plasma cells were also seen around portal triad (Figure 9B). The FEO and silymarin treated groups showed less damage with intact cytoplasm and nucleus and appeared to have normal hepatic cells (Figure 9C, 9D, and 9E). Phase I and Phase II drug metabolizing enzymes play an important role in converting toxic agents into non toxic and soluble substances. The rate limiting step in the activation and detoxification of chemical carcinogens and xenobiotics have been postulated to be dependent on the rate of reduction of cytochrome 450 (CytP450) substrate complex which in turn is dependent on the content of cytochrome b5 (Cytb5) and on the activity of NADPH cytochrome-c reductase [76]. Glutathione S-transferase (GST) is one of the detoxification systems in most human and animal tissues [77]. Organs with low levels GST are more susceptible to the toxic action of a variety of alkylating and therapeutic agents. In this study FEO (250 and 500 mg/kg body weight) treated mice showed increased levels of Cyt P450, Cyt b5, and GST (Figure 11). Therefore, it is suggested that FEO treatment mediated enhancement of Cyt P450, Cyt b5, and GST might act as a defense mechanism to protect the liver and probably other organs against the toxicity.
Materials and Methods
Boswellia sacra essential oil preparation
Certified Hojari grade Boswellia sacra gum resins were obtained from the local market. Essential oil was extracted by hydrodistillation method as described earlier [24]. Briefly, Boswellia sacra resins were loaded into 55° C water with a ratio of 1:2.5 (w/v), and mixed with an electromechanical agitator for 30–45 min or until a thick homogenous mucilage was formed. Temperatures of the hydrodistiller were monitored by an infrared thermometer; and pressures were recorded at the condenser terminal. To remove any residual water, collected Boswellia sacra essential oil (FEO) was immediately transferred into a –20° C freezer; and ice crystals were separated from the essential oil.
Cell lines and culture method
Mouse melanoma (B16-F10) cells were purchased from American Type Culture Collection, USA. Human melanoma cells (FM94) is kind gift from Dr. Sobia Kauser, University of Bradford, UK. Human normal epithelial melanocytes (HNEM) purchased from Promo cells, UK. B16-F10 and FM94 cells were cultured in Roswell Park Memorial Institute (RPMI 1640) medium with 10% fetal bovine serum and 1% antibiotics (penicillin/streptomycin) and HNEM cells were cultured in Promo cell melanocytes growth medium. Cells were maintained in humidified cell incubator at 37° C and 5% CO2.
Drug preparation
Stock solution of FEO was prepared in dimethyl sulfoxide. Different concentrations of (3, 5, 7 and 10 μg/ml) were added in the cell culture medium.
MTT assay
B16-F10, FM94, HNEM cells (1 × 105/well) were seeded in 96 well plates (100 μL/well) and allowed to adhere firmly overnight in respective medium with 10% fetal bovine serum. Then cells were treated with different concentrations (3, 5, 7 and 10 μg/ml) of FEO for 24 h. After 24h elapsed medium was removed and cells were incubated with MTT reagent (5 mg/mL) for 4 h and violet formazan crystals were dissolved in dimethylsulfoxide and absorbance was read at 540/690 nm. Absorbance of control (without treatment) was considered as 100% cell viability. Doxorubicin was used as positive drug control.
Fluorescence microscopic analysis of apoptosis
B16-F10, 1 × 105 cells/ml were seeded in 96 well plates (100 μ/well) and allowed to adhere firmly over night. The cells were then treated with different concentrations of FEO (5, 7 and 10 μg/ml). After 24 h treatment, 20 μl of trypsin was added into each well. Trypsinized cell suspension washed with phosphate buffered saline (PBS) and 25 μl cell suspension was transferred to glass slides. Hoechest 33258 (Sigma, St. Louis, MO, USA) staining solution was added to each suspension and then covered with a coverslip. The morphology of apoptotic cells was examined and visualized under a fluorescence microscope (Nikon Eclipse, Inc, Japan) at 400× magnification.
DNA fragmentation by agarose gel electrophoresis
Inter-nucleosomal cleavage of DNA was analyzed as described previously [78]. B16-F10 cells were seeded in 6-well plates at a concentration of 1 × 106 cells per ml of medium. Cells were then treated with FEO (5, 7 and 10 μg/ml). Cells were harvested after 24 hours of treatment and DNA fragmentation was assessed by gel electrophoresis.
Annexin-V assay
B16-F10 (2 × 10 5cells/10 cm dish) cells were treated with 10 μg/ml of FEO for 24 h. Then cells were harvested by centrifugation, washed with ice-cold PBS, and then resuspended with ice-cold 70% ethanol overnight. The following day, cells were treated with 10 μg/ml of RNase at 37° C, then spun down and stained with 40 μg/ml of propidium iodide (PI) and Annexin-V-FITC for 30 min. The apoptotic state of cells was measured by flow cytometry (FACS, BD Bioscience).
Western blot analysis
The whole cell lysate was prepared from B16-F10 cells treated with FEO (10 μg/ml) for 24 and 36 hours, and FEO (0, 7, and 10 μg/ml) treated FM94 cells for 24 hours as described earlier [79]. Then whole cell lysate were resolved in a 10% SDS polyacrylamide gel electrophoretically and electro transferred onto a nitrocellulose membrane. The immunoblots were probed with Bcl-2, Bax, caspase 3 (cleaved), Caspase 9 (cleaved), and PARP antibodies and were visualized with the NBT/BCIP chromogenic substrate. Further MCL-1 expression was determined in FM94 cells treated with FEO at 10 μg/ml for 3, 6, 12, 24 hours as described above. Antibodies purchased from Cell signaling technology, UK.
In vivo experiments
Animals
The swiss albino and C57BL/6 mice were selected from a random breed colony maintained in the animal house of Jawaharlal Nehru Cancer Hospital and Research Center, Bhopal, India. The mice were housed in polypropylene cages containing sterile paddy husk (procured locally) as bedding and maintained under controlled conditions of temperature (23 ± 20° C), humidity (50 ± 5%) and light and dark (10–14 h) respectively. The animals are fed with standard mice feed (formula obtained from Cancer Research institute, Mumbai, India) and filtered acidified water ad libitum. Mice of either sex, 6–8 weeks old and weighing 22 ± 2 g, were selected from the above colony for the experiments. The study was carried out as per OECD guidelines for testing for chemicals under IAEC number JNCHRC/13/IAEC/PN-185/B. All the experiments were conducted under the guidelines of Institutional Animal Ethical Committee (Project No.500/01/08/2016 /project 5 /28/07/2019) and confirms to the guidelines set by WHO (World health organizations, Geneva, Switzerland and INSA, New Delhi, India.
In vivo toxicity assessment
Swiss albino mice were divided in to two groups (n = 16). Group I treated with vehicle (saline) and group II treated with FEO (1200 mg/ kg of body weight) intraperitoneally (i.p.) every week of total four weeks. Body weight was measured for 30 days on weekly basis.
Histopathological analysis
At the end of the experimental period, animals were euthanized by cervical dislocation and liver, kidney, brain, and heart tissues were dissected and examined by gross and microscopic anatomy. Then tissue sections were stained with hematoxylin and eosin staining as described earlier [80]. Microscopic evaluations were done by the pathologist.
Induction of tumour (melanoma) in mice
Cell suspension of B16F10 cells (5 × 105) was implanted in C57BL/6 mice subcutaneously at the shaved part. The mice bearing the tumour were randomly divided into 3 groups with 6 mice in each group. The dosing was started when tumour has reached a mean diameter of 100 mm3 and this day was designated as day 0.
Experimental design
Mice (n = 18) were randomized into following three groups: Group I (Control (vehicle) treated with normal saline (0.9% v/w) i.p., Group II treated with FEO 300 mg/kg of body weight (i.p), and Group III treated with FEO 600 mg/kg of body weight (i.p) every two days for two weeks of experimental period. The tumour size was measured every alternate day using vernier calipers, and the tumour volume was calculated.
Hepatoprotective activity
Hematological analysis
Animals were randomized and divided into four groups (n = 8). Group I served as untreated control (treated with normal saline (0.9% v/w) i.p. daily for seven days). Group II as hepatotoxic control treated with acetaminophen (750 mg/kg body weight) i.p. for seven days. Group III received acetaminophen (750 mg/kg body weight) and FEO (250 mg/kg body weight) concurrently i.p. for seven days. Group IV received acetaminophen (750 mg/kg body weight) and FEO (500 mg/kg body weight) concurrently i.p. for seven days. At the end of experimental period blood samples were collected via vein puncture into sterile sample tubes containing the anticoagulant, EDTA. Packed cell volume (PVC), White blood cells (WBC) count, Lymphocytes count, Neutrophils count, hemoglobin concentration (Hb), Mean corpuscular volume (MCV), Mean Corpuscular Hemoglobin (MCH), Percent of Mean Corpuscular Hemoglobin Concentration (MCHC%), red blood cell (RBC) were analysed using an automated analyser, Cell Dyne, model 331430, Abbott laboratories, IL, USA.
Determination of biochemical parameters and histology of liver
Animals were randomized and divided into five groups (n = 8). Group I served as untreated control (treated with normal saline (0.9% v/w) i.p. daily for seven days). Group II as hepatotoxic control treated with acetaminophen (750 mg/kg body weight) i.p., for seven days. Group III received aacetaminophen (750 mg/kg body weight) and FEO (250 mg/kg body weight) concurrently i.p. for seven days. Group IV received aacetaminophen (750 mg/kg body weight) and FEO (500 mg/kg body weight) concurrently i.p. for seven days. Group V treated with acetaminophen (750 mg/kg body weight) and positive drug control Silymarin (25 mg/kg body weight) i.p., concurrently for seven days. At the end of experimental period blood samples were collected by puncturing retro-orbital plexus. The blood samples were allowed to clot for 45 min at room temperature. Serum was separated by centrifugation at 2500 rpm for 15 min at room temperature. Serum biochemical parameters such as serum glutamic oxaloacetic transaminase (AST), serum glutamic pyruvic transaminase, (ALT), serum alkaline phosphatase (ALK), bilirubin, protein, cholesterol, glucose, albumin/globulin ratio, triglyceride were measured as described earlier. Furthermore, liver was excised from animals and subjected to histological analysis by hematoxylin and eosin staining method.
Determination of phase I and phase II drug metabolizing enzymes
Animals were randomized and divided into four groups (n = 8). Group I served as untreated control and fed orally with distilled water daily for seven days. Group II and III received FEO 250 and 500 mg/kg body weight (i.p), respectively for seven days. Group IV served as positive control and treated with 0.75% butylated hydroxyanisole (BHA) along with diet. At the end of experimental period livers were excised immediately and washed in ice cold normal saline, followed by 0.15 M Tris-Hcl (pH 7.4) blotted dry and weighed. A 10% (w/v) of homogenate was prepared in 0.15 M Tris-Hcl buffer. A part of homogenate after precipitating proteins with trichloroacetic acid (TCA) was used for analysis.
Cytochrome P450 and Cytochrome b5
Cytochrome P450 was determined using the carbon monoxide difference spectra. Both cytochrome P450 and cytochrome b5 content were assayed in microsomal suspension by the method of Omura and Sato [81], using an absorption coefficient of 91 and 185 cm2 M-1 m-1, respectively.
Glutathione S-transferase
Glutathione S-transferase (GST) activity was determined spectrophotometrically at 37° C as described earlier [82]. Briefly, the reaction mixture (1ml) contained 0.334 ml of 100 mM phosphate buffer (pH 6.5), 0.033 ml of 30 mM CDNB and 0.033 ml of 30 mM of reduced glutathione. After pre incubating the reaction mixture for 2 minutes, the reaction was started by adding 0.01 ml of diluted homogenate and the absorbance was followed for 3 minutes at 340 nm. The specific activity of GST is expressed as μ moles of GSH-CDNB conjugate formed/min/mg protein using extinction co-efficient of 9.6 mM–1 cm–1.
Statistical analysis
The data represents mean ± STD. The Dunnett’s multiple comparison test was used for the analysis. P value < 0.001, <0.01, <0.05 was considered significant.
Conclusions
On the basis of above finding it is concluded that FEO exhibited in vitro anti-melanoma activity by reducing the viability in B16-F10 cell, FM94 cells and not in HNEM cells by the induction of apoptosis via caspase signaling and MCL-1 dependent pathway. Furthermore, FEO can reduce tumour size in C57BL/6 mice melanoma tumour model. FEO can be most effective drug for prevention of hepatic injury along with improved hematological biochemical parameters, liver histology, phase I and phase II drug metabolizing enzymes.
ACKNOWLEDGMENTS
We would like to thank Dr. Shobia Kauser of University of Bradford for donating human melanoma cells.
CONFLICTS OF INTEREST
Authors declare that there is no conflicts of interest.
References
1. Vaid M, Prasad R, Sun Q, Katiyar SK. Silymarin targets β-catenin signaling in blocking migration/invasion of human melanoma cells. PLoS One. 2011; 6:e23000. https://doi.org/10.1371/journal.pone.0023000. [PubMed].
2. American Cancer Society. Cancer Facts and Figures 2018. Atlanta: American Cancer Society. 2019.
3. Orgaz JL, Sanz-Moreno V. Emerging molecular targets in melanoma invasion and metastasis. Pigment Cell Melanoma Res. 2013; 26:39–57. https://doi.org/10.1111/pcmr.12041. [PubMed].
4. Callahan MK, Postow MA, Wolchok JD. Immunomodulatory therapy for melanoma: ipilimumab and beyond. Clin Dermatol. 2013; 31:191–99. https://doi.org/10.1016/j.clindermatol.2012.08.006. [PubMed].
5. Poust JC, Woolery JE, Green MR. Management of toxicities associated with high-dose interleukin-2 and biochemotherapy. Anticancer Drugs. 2013; 24:1–13. https://doi.org/10.1097/CAD.0b013e32835a5ca3. [PubMed].
6. Sullivan RJ, Flaherty KT. Resistance to BRAF-targeted therapy in melanoma. Eur J Cancer. 2013; 49:1297–1304. https://doi.org/10.1016/j.ejca.2012.11.019. [PubMed].
7. Zeng G, Liu J, Chen H, Liu B, Zhang Q, Li M, Zhu R. Dihydromyricetin induces cell cycle arrest and apoptosis in melanoma SK-MEL-28 cells. Oncol Rep. 2014; 31:2713–19. https://doi.org/10.3892/or.2014.3160. [PubMed].
8. Farias CF, Massaoka MH, Girola N, Azevedo RA, Ferreira AK, Jorge SD, Tavares LC, Figueiredo CR, Travassos LR. Benzofuroxan derivatives N-Br and N-I induce intrinsic apoptosis in melanoma cells by regulating AKT/BIM signaling and display anti metastatic activity in vivo. BMC Cancer. 2015; 15:807. https://doi.org/10.1186/s12885-015-1835-3. [PubMed].
9. Bai RZ, Wu Y, Liu Q, Xie K, Wei YQ, Wang YS, Liu K, Luo Y, Su JM, Hu B, Liu JY, Li Q, Niu T, et al. Suppression of lung cancer in murine model: treated by combination of recombinant human endostsatin adenovirus with low-dose cisplatin. J Exp Clin Cancer Res. 2009; 28:31. https://doi.org/10.1186/1756-9966-28-31. [PubMed].
10. Zafrani ES, Leclercq B, Vernant JP, Pinaudeau Y, Chomette G, Dhumeaux D. Massive blastic infiltration of the liver: a cause of fulminant hepatic failure. Hepatology. 1983; 3:428–32. https://doi.org/10.1002/hep.1840030324. [PubMed].
11. World Health Organization. Common terminology criteria for adverse events v4.0 (CTCAE). May 28, 2009 (v4.03: June 14, 2010).
12. National Cancer Institute. Cancer therapy evaluation program. National Cancer Institute. 2005.
13. Della Vittoria Scarpati G, Fusciello C, Perri F, Sabbatino F, Ferrone S, Carlomagno C, Pepe S. Ipilimumab in the treatment of metastatic melanoma: management of adverse events. Onco Targets Ther. 2014; 7:203–09. https://doi.org/10.2147/OTT.S57335. [PubMed].
14. Tanaka R, Fujisawa Y, Sae I, Maruyama H, Ito S, Hasegawa N, Sekine I, Fujimoto M. Severe hepatitis arising from ipilimumab administration, following melanoma treatment with nivolumab. Jpn J Clin Oncol. 2017; 47:175–78. https://doi.org/10.1093/jjco/hyw167. [PubMed].
15. Argyropoulou A, Aligiannis N, Trougakos IP, Skaltsounis AL. Natural compounds with anti-ageing activity. Nat Prod Rep. 2013; 30:1412–37. https://doi.org/10.1039/c3np70031c. [PubMed].
16. Aravindaram K, Yang NS. Anti-inflammatory plant natural products for cancer therapy. Planta Med. 2010; 76:1103–17. https://doi.org/10.1055/s-0030-1249859. [PubMed].
17. Long S, Sousa E, Kijjoa A, Pinto MM. Marine natural products as models to circumvent multidrug resistance. Molecules. 2016; 21:E892. https://doi.org/10.3390/molecules21070892. [PubMed].
18. Sklirou A, Papanagnou ED, Fokialakis N, Trougakos IP. Cancer chemoprevention via activation of proteostatic modules. Cancer Lett. 2018; 413:110–21. https://doi.org/10.1016/j.canlet.2017.10.034. [PubMed].
19. Safayhi H, Mack T, Sabieraj J, Anazodo MI, Subramanian LR, Ammon HP. Boswellic acids: novel, specific, nonredox inhibitors of 5-lipoxygenase. J Pharmacol Exp Ther. 1992; 261:1143–46. [PubMed].
20. Fan AY, Lao L, Zhang RX, Zhou AN, Wang LB, Moudgil KD, Lee DY, Ma ZZ, Zhang WY, Berman BM. Effects of an acetone extract of Boswellia carterii Birdw. (Burseraceae) gum resin on adjuvant-induced arthritis in lewis rats. J Ethnopharmacol. 2005; 101:104–09. https://doi.org/10.1016/j.jep.2005.03.033. [PubMed].
21. Winking M, Sarikaya S, Rahmanian A, Jödicke A, Böker DK. Boswellic acids inhibit glioma growth: a new treatment option? J Neurooncol. 2000; 46:97–103. https://doi.org/10.1023/A:1006387010528. [PubMed].
22. Huang MT, Badmaev V, Ding Y, Liu Y, Xie JG, Ho CT. Anti-tumor and anti-carcinogenic activities of triterpenoid, beta-boswellic acid. Biofactors. 2000; 13:225–30. https://doi.org/10.1002/biof.5520130135. [PubMed].
23. Kirste S, Treier M, Wehrle SJ, Becker G, Abdel-Tawab M, Gerbeth K, Hug MJ, Lubrich B, Grosu AL, Momm F. Boswellia serrata acts on cerebral edema in patients irradiated for brain tumors: a prospective, randomized, placebo-controlled, double-blind pilot trial. Cancer. 2011; 117:3788–95. https://doi.org/10.1002/cncr.25945. [PubMed].
24. Suhail MM, Wu W, Cao A, Mondalek FG, Fung KM, Shih PT, Fang YT, Woolley C, Young G, Lin HK. Boswellia sacra essential oil induces tumor cell-specific apoptosis and suppresses tumor aggressiveness in cultured human breast cancer cells. BMC Complement Altern Med. 2011; 11:129. https://doi.org/10.1186/1472-6882-11-129. [PubMed].
25. Ni X, Suhail MM, Yang Q, Cao A, Fung KM, Postier RG, Woolley C, Young G, Zhang J, Lin HK. Frankincense essential oil prepared from hydrodistillation of Boswellia sacra gum resins induces human pancreatic cancer cell death in cultures and in a xenograft murine model. BMC Complement Altern Med. 2012; 12:253. https://doi.org/10.1186/1472-6882-12-253. [PubMed].
26. Hakkim FL, Al-Buloshi M, Al-Sabahi J, Mohammed Al-Buloshi, Jamal Al-Sabahi. Frankincense derived heavy terpene cocktail boosting breast cancer cell (MDA-MB-231) death in vitro. Asian Pac J Trop Biomed. 2015; 5:824–28. https://doi.org/10.1016/j.apjtb.2015.06.008.
27. Swinney DC, Anthony J. How were new medicines discovered? Nat Rev Drug Discov. 2011; 10:507–19. https://doi.org/10.1038/nrd3480. [PubMed].
28. Hosseini A, Ghorbani A. Cancer therapy with phytochemicals: evidence from clinical studies. Avicenna J Phytomed. 2015; 5:84–97. [PubMed].
29. Scarpa ES, Ninfali P. Phytochemicals as innovative therapeutic tools against cancer stem cells. Int J Mol Sci. 2015; 16:15727–42. https://doi.org/10.3390/ijms160715727. [PubMed].
30. Tetè S, Nicoletti M, Saggini A, Maccauro G, Rosati M, Conti F, Cianchetti E, Tripodi D, Toniato E, Fulcheri M, Salini V, Caraffa A, Antinolfi P, et al. Nutrition and cancer prevention. Int J Immunopathol Pharmacol. 2012; 25:573–81. https://doi.org/10.1177/039463201202500303. [PubMed].
31. Martin-Cordero C, Leon-Gonzalez AJ, Calderon-Montano JM, Burgos-Moron E, Lopez-Lazaro M. Pro-oxidant natural products as anticancer agents. Curr Drug Targets. 2012; 13:1006–28. https://doi.org/10.2174/138945012802009044. [PubMed].
32. Shanmugam MK, Kannaiyan R, Sethi G. Targeting cell signaling and apoptotic pathways by dietary agents: role in the prevention and treatment of cancer. Nutr Cancer. 2011; 63:161–73. https://doi.org/10.1080/01635581.2011.523502. [PubMed].
33. Roy NK, Deka A, Bordoloi D, Mishra S, Kumar AP, Sethi G, Kunnumakkara AB. The potential role of boswellic acids in cancer prevention and treatment. Cancer Lett. 2016; 377:74–86. https://doi.org/10.1016/j.canlet.2016.04.017. [PubMed].
34. Cohen GM, Sun XM, Fearnhead H, MacFarlane M, Brown DG, Snowden RT, Dinsdale D. Formation of large molecular weight fragments of DNA is a key committed step of apoptosis in thymocytes. J Immunol. 1994; 153:507–16. [PubMed].
35. Han X, Rodriguez D, Parker TL. Biological activities of frankincense essential oil in human dermal fibroblasts. Biochim Open. 2017; 4:31–35. https://doi.org/10.1016/j.biopen.2017.01.003. [PubMed].
36. Matsuo AL, Figueiredo CR, Arruda DC, Pereira FV, Scutti JA, Massaoka MH, Travassos LR, Sartorelli P, Lago JH. α-Pinene isolated from Schinus terebinthifolius Raddi (Anacardiaceae) induces apoptosis and confers antimetastatic protection in a melanoma model. Biochem Biophys Res Commun. 2011; 411:449–54. https://doi.org/10.1016/j.bbrc.2011.06.176. [PubMed].
37. Yang Y, Yue Y, Runwei Y, Guolin Z. Cytotoxic, apoptotic and antioxidant activity of the essential oil of Amomum tsao-ko. Bioresour Technol. 2010; 101:4205–11. https://doi.org/10.1016/j.biortech.2009.12.131. [PubMed].
38. Kilani S, Ben Sghaier M, Limem I, Bouhlel I, Boubaker J, Bhouri W, Skandrani I, Neffatti A, Ben Ammar R, Dijoux-Franca MG, Ghedira K, Chekir-Ghedira L. In vitro evaluation of antibacterial, antioxidant, cytotoxic and apoptotic activities of the tubers infusion and extracts of Cyperus rotundus. Bioresour Technol. 2008; 99:9004–08. https://doi.org/10.1016/j.biortech.2008.04.066. [PubMed].
39. Susin SA, Daugas E, Ravagnan L, Samejima K, Zamzami N, Loeffler M, Costantini P, Ferri KF, Irinopoulou T, Prévost MC, Brothers G, Mak TW, Penninger J, et al. Two distinct pathways leading to nuclear apoptosis. J Exp Med. 2000; 192:571–80. https://doi.org/10.1084/jem.192.4.571. [PubMed].
40. Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001; 412:95–99. https://doi.org/10.1038/35083620. [PubMed].
41. Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010; 37:299–310. https://doi.org/10.1016/j.molcel.2010.01.025. [PubMed].
42. Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B, Bao JK. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012; 45:487–98. https://doi.org/10.1111/j.1365-2184.2012.00845.x. [PubMed].
43. Shamas-Din A, Brahmbhatt H, Leber B, Andrews DW. BH3-only proteins: orchestrators of apoptosis. Biochim Biophys Acta. 2011; 1813:508–20. https://doi.org/10.1016/j.bbamcr.2010.11.024. [PubMed].
44. Chang HY, Yang X. Proteases for cell suicide: functions and regulation of caspases. Microbiol Mol Biol Rev. 2000; 64:821–46. https://doi.org/10.1128/MMBR.64.4.821-846.2000. [PubMed].
45. Oliver L, Vallette FM. The role of caspases in cell death and differentiation. Drug Resist Updat. 2005; 8:163–70. https://doi.org/10.1016/j.drup.2005.05.001. [PubMed].
46. Mhaidat NM, Wang Y, Kiejda KA, Zhang XD, Hersey P. Docetaxel-induced apoptosis in melanoma cells is dependent on activation of caspase-2. Mol Cancer Ther. 2007; 6:752–61. https://doi.org/10.1158/1535-7163.MCT-06-0564. [PubMed].
47. Badaboina S, Bai HW, Park CH, Jang DM, Choi BY, Chung BY. Molecular mechanism of apoptosis induction in skin cancer cells by the centipedegrass extract. BMC Complement Altern Med. 2013; 13:350. https://doi.org/10.1186/1472-6882-13-350. [PubMed].
48. Mukherjee N, Lu Y, Almeida A, Lambert K, Shiau CW, Su JC, Luo Y, Fujita M, Robinson WA, Robinson SE, Norris DA, Shellman YG. Use of a MCL-1 inhibitor alone to de-bulk melanoma and in combination to kill melanoma initiating cells. Oncotarget. 2017; 8:46801–17. https://doi.org/10.18632/oncotarget.8695. [PubMed].
49. Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, Wang X. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003; 17:1475–86. https://doi.org/10.1101/gad.1093903. [PubMed].
50. Wang Y, Lv J, Cheng Y, Du J, Chen D, Li C, Zhang J. Apoptosis induced by Ginkgo biloba (EGb761) in melanoma cells is Mcl-1-dependent. PLoS One. 2015; 10:e0124812. https://doi.org/10.1371/journal.pone.0124812. [PubMed].
51. Zhuang L, Lee CS, Scolyer RA, McCarthy SW, Zhang XD, Thompson JF, Hersey P. Mcl-1, Bcl-XL and Stat3 expression are associated with progression of melanoma whereas Bcl-2, AP-2 and MITF levels decrease during progression of melanoma. Mod Pathol. 2007; 20:416–26. https://doi.org/10.1038/modpathol.3800750. [PubMed].
52. Zhao Y, He J, Li J, Peng X, Wang X, Dong Z, Zhao E, Liu Y, Wu Z, Cui H. Demethylzeylasteral inhibits cell proliferation and induces apoptosis through suppressing MCL1 in melanoma cells. Cell Death Dis. 2017; 8:e3133. https://doi.org/10.1038/cddis.2017.529. [PubMed].
53. Zheng L, Wong YS, Shao M, Huang S, Wang F, Chen J. Apoptosis induced by 9,11dehydroergosterol peroxide from Ganoderma Lucidum mycelium in human malignant melanoma cells is Mcl1 dependent. Mol Med Rep. 2018; 18:938–44. https://doi.org/10.3892/mmr.2018.9035. [PubMed].
54. Zhao F, Dou J, He XF, Wang J, Chu L, Hu W, Yu F, Hu K, Wu Y, Gu N. Enhancing therapy of B16F10 melanoma efficacy through tumor vaccine expressing GPI-anchored IL-21 and secreting GM-CSF in mouse model. Vaccine. 2010; 28:2846–52. https://doi.org/10.1016/j.vaccine.2010.01.057. [PubMed].
55. van den Boorn JG, Konijnenberg D, Tjin EP, Picavet DI, Meeuwenoord NJ, Filippov DV, van der Veen JP, Bos JD, Melief CJ, Luiten RM. Effective melanoma immunotherapy in mice by the skin-depigmenting agent monobenzone and the adjuvants imiquimod and CpG. PLoS One. 2010; 5:e10626. https://doi.org/10.1371/journal.pone.0010626. [PubMed].
56. Tanaka A, Jensen JD, Prado R, Riemann H, Shellman YG, Norris DA, Chin L, Yee C, Fujita M. Whole recombinant yeast vaccine induces antitumor immunity and improves survival in a genetically engineered mouse model of melanoma. Gene Ther. 2011; 18:827–34. https://doi.org/10.1038/gt.2011.28. [PubMed].
57. da Silva JK, da Trindade R, Alves NS, Figueiredo PL, Maia JG, Setzer WN. Essential oils from neotropical piper species and their biological activities. Int J Mol Sci. 2017; 18:e2571. https://doi.org/10.3390/ijms18122571. [PubMed].
58. Kim DY, Won KJ, Hwang DI, Park SM, Kim B, Lee HM. Chemical composition, antioxidant and anti-melanogenic activities of essential oils from Chrysanthemum boreale Makino at different harvesting stages. Chem Biodivers. 2018; 15:e1700506. https://doi.org/10.1002/cbdv.201700506. [PubMed].
59. King PD, Perry MC. Hepatotoxicity of chemotherapy. Oncologist. 2001; 6:162–76. https://doi.org/10.1634/theoncologist.6-2-162. [PubMed].
60. Lee WM. Acute liver failure. Semin Respir Crit Care Med. 2012; 33:36–45. https://doi.org/10.1055/s-0032-1301733. [PubMed].
61. Asad M, Alhumoud M. Hepatoprotective effect and GC-MS analysis of traditionally used Boswellia sacra oleo gum resin (frankincense) extract in rats. Afr J Tradit Complement Altern Med. 2015; 12:1–5. https://doi.org/10.4314/ajtcam.v12i2.1.
62. Black M. Acetaminophen hepatotoxicity. Gastroenterology. 1980; 78:382–92. https://doi.org/10.1016/0016-5085(80)90593-4. [PubMed].
63. Ibrahim RA, Berman DM, DePril V, Humphrey RW, Chen T, Messina KM, Chin M, Liu HY, Bielefield M, Hoos A. Ipilimumab safety profile: summary of findings from completed trials in advanced melanoma. J Clin Oncol. 2011; 29:29. https://doi.org/10.1200/jco.2011.29.15_suppl.8583.
64. Ozcanli T, Erdogan A, Ozdemir S, Onen B, Ozmen M, Doksat K, Sonsuz A. Severe liver enzyme elevations after three years of olanzapine treatment: a case report and review of olanzapine associated hepatotoxicity. Prog Neuropsychopharmacol Biol Psychiatry. 2006; 30:1163–66. https://doi.org/10.1016/j.pnpbp.2006.03.014. [PubMed].
65. Janbaz KH, Gilani AH. Studies on preventive and curative effects of berberine on chemical-induced hepatotoxicity in rodents. Fitoterapia. 2000; 71:25–33. https://doi.org/10.1016/S0367-326X(99)00098-2. [PubMed].
66. Hagar HH. The protective effect of taurine against cyclosporine A-induced oxidative stress and hepatotoxicity in rats. Toxicol Lett. 2004; 151:335–43. https://doi.org/10.1016/j.toxlet.2004.03.002. [PubMed].
67. Velayudham LS, Farrell GC. Drug-induced cholestasis. Expert Opin Drug Saf. 2003; 2:287–304. https://doi.org/10.1517/14740338.2.3.287. [PubMed].
68. Giannini EG, Testa R, Savarino V. Liver enzyme alteration: a guide for clinicians. CMAJ. 2005; 172:367–79. https://doi.org/10.1503/cmaj.1040752. [PubMed].
69. Keith G, Tolman MD, Robert R. Liver function. In: Burtis CA, Ashwood ER, editors. Tietz Textbook of Clinical Chemistry. 3rd ed. Philadelphia: WB Saunders Company; 1999. pp. 1125–77.
70. Cholongitas E, Papatheodoridis GV, Vangeli M, Terreni N, Patch D, Burroughs AK. Systematic review: the model for end-stage liver disease—should it replace Child-Pugh’s classification for assessing prognosis in cirrhosis? Aliment Pharmacol Ther. 2005; 22:1079–89. https://doi.org/10.1111/j.1365-2036.2005.02691.x. [PubMed].
71. Sherlock S. Diseases of the Liver and Biliary System. Oxford; 1975. pp. 30–5.
72. Teloh HA. Serum proteins in hepatic disease. Ann Clin Lab Sci. 1978; 8:127–29. [PubMed].
73. Selimoglu MA, Aydogdu S, Yagci RV. Lipid parameters in childhood cirrhosis and chronic liver disease. Pediatr Int. 2002; 44:400–03. https://doi.org/10.1046/j.1442-200X.2002.01590.x. [PubMed].
74. Ooi K, Shiraki K, Sakurai Y, Morishita Y, Nobori T. Clinical significance of abnormal lipoprotein patterns in liver diseases. Int J Mol Med. 2005; 15:655–60. https://doi.org/10.3892/ijmm.15.4.655. [PubMed].
75. Davidson CS. Protein metabolism in severe liver disease, especially alcoholics with cirrhosis. Am J Med. 1956; 21:129–30. https://doi.org/10.1016/0002-9343(56)90014-6.
76. Zhang H, Hamdane D, Im SC, Waskell L. Cytochrome b5 inhibits electron transfer from NADPH-cytochrome P450 reductase to ferric cytochrome P450 2B4. J Biol Chem. 2008; 283:5217–25. https://doi.org/10.1074/jbc.M709094200. [PubMed].
77. Baillie TA, Slatter JG. Glutathione: A vehicle for transport of chemically reactive metaboloities in vivo. Acc Chem Res. 1991; 24:264–70. https://doi.org/10.1021/ar00009a003.
78. Kim T, Jung U, Cho DY, Chung AS. Se-methylselenocysteine induces apoptosis through caspase activation in HL-60 cells. Carcinogenesis. 2001; 22:559–65. https://doi.org/10.1093/carcin/22.4.559. [PubMed].
79. Zhu P, Zhang J, Zhu J, Shi J, Zhu Q, Gao Y. MiR-429 induces gastric carcinoma cell apoptosis through Bcl-2. Cell Physiol Biochem. 2015; 37:1572–80. https://doi.org/10.1159/000438524. [PubMed].
80. Landen JW, Lang R, McMahon SJ, Rusan NM, Yvon AM, Adams AW, Sorcinelli MD, Campbell R, Bonaccorsi P, Ansel JC, Archer DR, Wadsworth P, Armstrong CA, et al. Noscapine alters microtubule dynamics in living cells and inhibits the progression of melanoma. Cancer Res. 2002; 62:4109–14. [PubMed].
81. Omura T, Sato R. A new cytochrome in liver microsomes. J Biol Chem. 1962; 237:1375–76. [PubMed].
82. Habig WH, Pabst MJ, Jakoby WB. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J Biol Chem. 1974; 249:7130–39. [PubMed].