
INTRODUCTION
Cancer cells exhibit metabolic changes, which enable the malignant cells to sustain cell growth and proliferation [1–3]. Indeed, Otto Warburg was the first to describe that cancer cells preferentially use aerobic glycolysis to produce energy [4–7]. However, in the last 10 years significant effort has been made to the characterization of the metabolic alteration in cancer cells [8, 9]. It is now clear that together with the Warburg effect, the malignant cells show also an increased flux through to the pentose phosphate pathway, high glutamine consumption, maintenance of redox status and increased lipids biosynthesis, which all tighter help is sustaining cell proliferation under metabolic, redox stress or hypoxia [10–12]. Moreover, an increased uptake of glycine and serine has been observed [13–15]. However, serine can be also synthetized within the cells. Indeed, the de novo serine synthesis pathways represent one of the most significant pathways derived from a branching route of glycolysis. Serine can then converted in glycine, which provides the carbon units to fuel the one-carbon metabolism [16, 17]. One-carbon metabolism represents a complex metabolite network that is based on the chemical reactions of folate compounds [18]. This pathway provides the one carbon unit required for the synthesis of proteins, lipids, nucleic acids and other cofactors. The one-carbon unit proceeds in a cyclical pathway from where they are transferred to other metabolic pathways. The importance of this metabolic pathway is underlined by the fact that antimetabolic (anti-folate) chemotherapy is currently widely employed in cancer treatment since its discovery more than 50 years ago [19–21]. It was in fact in 1947 when Sidney Farber at the Children’s Hospital defined the use of antifolate therapy for leukemia, based on the work of the hematologists George Minot, who identified a critical micronutrient later defined as vitamin B12 (1934 Nobel Prize), and Lucy Willis, a clever physician from the London School of Medicine for Women who identified in Bombay a “Willis factor” from the popular yeastly spread “Marmite” which turned out to be folic acid.
In this perspective we will highlight the recent implication of serine and glycine metabolism in cancer biology. Recent reports indicate that the serine biosynthetic pathway is activated in cancer cells and represents an essential process in cancer pathogenesis [22]. Our bioinformatics analysis indicate that selective expression of some metabolic enzymes represents a prognostic factor for cancer, suggesting that activation of this metabolic pathway can be associated to the pathogenesis of different cancer types.
Serine pathway in cancer cells
Glucose and glutamine are two nutrients that cancer cells utilize for supporting energy metabolism and anabolic processes [23, 24]. However, cancer cells also increase de novo synthesis of serine and glycine that provides methyl group required for the biosynthetic pathways and DNA methylation. Indeed, the biosynthesis of serine and glycine was first found increased in lymphomas. In particular, it was shown, by radiolabeling experiments, that serine is formed prior glycine and that the glycolytic intermediate metabolite, 3-phosphoglycerate, is a common precursor [25]. In the last few years this early observation was also observed in breast cancer [26, 27] and melanoma [18]. Within the cells serine is synthetized by 3-phosphoglycerate through a 3-step enzymatic reaction. The first step of this metabolic pathway is the conversion of 3-phosphoglycerate in 3-phosphohydroxypyruvate, reaction catalyzed by phosphoglycerate dehydrogenase (PHGDH). Successively, 3-phosphohydroxypyruvate is converted in phosphoserine by the enzyme phosphoserine phosphatase (PSPH) and then in serine by phosphoserine aminotransferase 1 (PSAT1) (Figure 1) Serine can be also imported from the extracellular compartment by amino acid transporter.
The expression of PHGDH has been found upregulated (amplification of chromosome 1p12) in triple negative breast cancer and in melanoma, suggesting that tumors containing amplification of PHGDH take advantage from serine biosynthesis activity. Indeed, in vitro experiments show that inhibition of PHGDH expression induces a strong decrease in cell proliferation and a reduction in serine biosynthesis. Moreover, when PHDGH were overexpressed in the breast epithelial cells MCF10A (with no upregulated serine biosynthesis), the acinar morphology was disrupt and induces further phenotypic alterations that predispose to malignant transformation.
Oncogenic/oncosuppressor signalling can respond to nutrient stress and thus determine metabolic response in cancer cells. The tumour suppressor p53, beside the canonical response to DNA damage [28, 29] and control of cell cycle arrest [30–35] and apoptosis [36–48], plays a pivotal role in cellular metabolic homeostasis [49–51]. p53 helps cancer cells to face serine starvation, preserving cellular anti-oxidant capacity. Cells lacking p53 failed to respond to serine starvation, due to oxidative stress condition, which leads to reduced viability and severely impaired proliferation. During serine starvation, activation of p53-p21 axis leads to cell cycle arrest, which promotes cell survival by efficiently channeling depleted serine stores to glutathione synthesis [52–55]. The others p53-family members, with all the different expressed isoforms [56–58], determine a complex network which affect cellular metabolism [59]. TAp73 can control the balance of cellular metabolism [60–65], thus exerting its role in development and tumour suppression. TAp73 promotes serine/glycine biosynthetic pathway [66]. Similarly to p63 [67], p73 [68] promotes the expression of the glutaminase-2 (GLS-2), favoring glutaminolysis, which in turn pushes the second step of serine biosynthesis (Fig. 1). Overall, p53-family can influence various metabolic pathways, glycolysis, glutaminolosys, mitochondrial metabolism, fatty acids beta-oxidation etc., enabling cells to respond to metabolic stress [49, 63, 69–72]. However, p53 is mutated in about half of human cancers, resulting not only in a wt p53 loss-of-function but also a mutant p53 (mp53) gain-of-function (GOF) [73–77]. Part of mp53 GOF is due to mp53 interaction and consequent repression of the siblings, p63/p73 [78–81], making this scenario much more complex. Alteration of oncogenic/oncosupressor signaling might determine the expression of specific metabolic enzyme patterns, leading the cancer cells to rely on specific metabolic pathways.
In order to evaluate the importance of serine metabolic enzymes expression for cancer cells, we asked to search whether the enzymes of the serine metabolic pathway could function as prognostic marker [82, 83]. We assessed the clinical value of PHGDH in 17 breast and in 7 lung cancers human datasets. In 7 out of 17 breast cancer datasets high PHGDH expression represented a negative prognostic factor, predicting negative patient survival (Table 1, Fig. 2a,b). However, PHGDH did not appear to have any prognostic value in lung cancer datasets, whereas is some datasets PSAT-1, PSPH or SHMT-2 were able to predict patient survival (Table 1). This observation leads to the conclusion that although PHDGH seems to be involved in breast cancer pathogenesis, the alteration of serine biosynthesis might be still involved in other cancer types, employing different mechanisms.
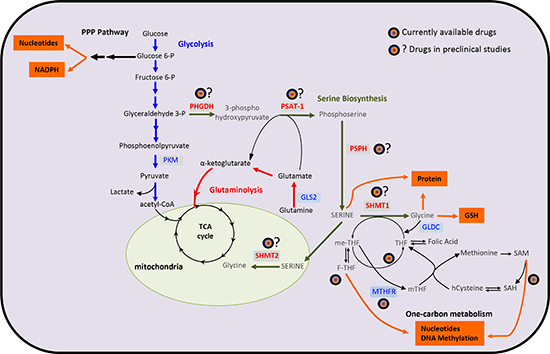
Figure 1: A schematic overview of the metabolic pathways involved in cancer biology. Cancer cells show an increased flux through to the glycolysis, pentose phosphate pathway and high glutamine consumption. Moreover, they also show an increased uptake of glycine and serine. In particular, the serine synthesis pathway utilizes the glycolytic intermediate glycerate-3-phosphate, which is converted by PHGDH, PSAT-1 and PSPH into serine. De novo synthetized serine and glycine fuel on-carbon metabolism. The one-carbon metabolism plays an essential role in the generation of proteins, nucleotides, GSH and substrates for methylation reactions. In red are the cancer-associated genes. PHGDH, phosphoglycerate dehydrogenase; PSAT-1, phosphoserine aminotransferase 1; PSPH, phosphoserine phosphatase; SHMT, Serine hydroxymethyltransferase; GLS-2, glutaminase 2; GLDC, glycine decarboxylase; GSH, glutathione; MTHFR, methylenetetrahydrofolate reductase; SAM, S-adenosylmethionine; SAH, S-adenosylhomomocysteine; THF, tetrahydrofolate; me-THF, 5,10-methylenetetrahydrofolate; F-THF, 10-formyltetrahydrofolate; mTHF, 5-methyltetrahydrofolate; PKM, pyruvate kinase; PPP, pentose phosphate pathway.
Glycine pathway in cancer cells
De novo synthesis of serine plays a crucial role as supplier of precursors for several biosynthetic pathways. Indeed, serine can be converted to glycine by the enzyme serine hydroxymethyltransferase (SHMT) [84, 85]. This reaction represents a major source of methyl groups for the one-carbon pools that are required for the biosynthesis of GSH, proteins, purines and DNA/histones methylation [86]. Therefore, SHMT occupies a critical position at the convergence of two key pathways for chemotherapeutic intervention: serine/glycine metabolism and nucleotide biosynthesis [87]. Within the cell, two isoforms of SHMT are present. SHMT1 is localized in the cytoplasm, whereas SHMT2 is present in the mithondria. Interestingly, c-Myc directly regulates the expression of both shmt1 and shmt2 genes [88–90]. More importantly, the expression and/or activity of the two enzymes are impaired in several tumors [91]. Several experimental evidences indicate that glycine uptake and catabolism can promote tumorigenesis, indicating that glycine metabolism could be a potential target for therapeutic intervention. Indeed, recently has been demonstrated that both glycine consumption and in particular, the expression of the mitochondrial glycine biosynthetic pathway correlate with the rate of proliferation across cancer cells [92–98]. This suggests, that under some circumstances, mitochondria play an essential role in supporting rapid cancer cell proliferation. In fact, inhibiting the expression of mitochondrial SHMT2 gene and deprivation of extracellular glycine were able to reduce HeLa cells proliferation, resulting in a prolonged G1 phase of the cell cycle. On the contrary, the upregulation of the serine/glycine metabolism was correlating with cell proliferation and poor prognosis in several tumors.
Table 1: Survival outcome in human cancer datasets predicted by serine and glycine enzyme expression
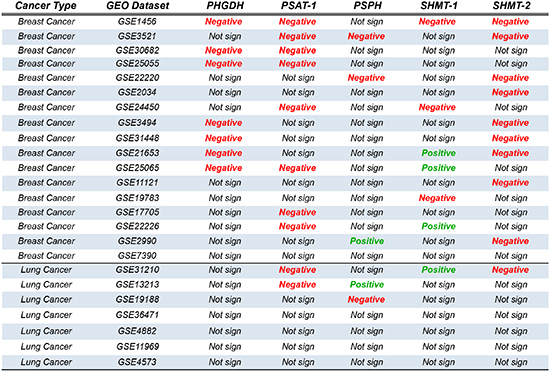
In our survival estimation analysis SHMT-2 resulted the most frequent significant prognostic factor among the serine/glycine biosynthetic enzymes. In 9 out of 17 breast cancer datasets high expression of SHMT-2 predicted negative prognosis (Table 1, Fig. 2c-h). On the other hand prediction ability of SHMT-1 appeared less clear. High expression of SHMT-1 indeed predicted in 3 datasets good prognosis and in 3 datasets negative prognosis, leaving very complex any conclusion (Table 1). From our analysis SHMT-2, even more than PHGDH, appears to be highly clinical relevant for breast cancer. Identification of selective SHMT-1/SHMT-2 selective inhibitors could be key for innovative and successful approaches.
CONCLUSIONS AND PERSPECTIVES
Although the antimetabolites drugs were introduced in cancer therapy more than 50 years ago, they are still the most widely used drugs in cancer chemotherapy. Indeed, the antifolate agents were successfully used in the treatment of children affected by acute lymphoblastic leukemia. In the last decade many scientists have been attracted (or re-attracted) by the metabolic process associated with cancer biology. This boosted part of the scientific community to re-focus their effort in the development of novel antimetabolites drugs and/or in seeking new potential therapeutic targets (druggable metabolic enzymes) [99–105]. In fact, approved inhibitors of thymidylate and purine biosynthesis include methotrexate, pralatrexate, 5-fluorouracil and pemetrexed are currently in clinic. Among these, 5-fluorouracil is a standard agent for several cancer types, including colorectal cancer. The emerging role of serine/glycine/one-carbon metabolism in cancer biology opens the opportunity of alternative chemotherapeutic approaches. Indeed, mimicking uracil, 5-FU inhibits thymidine synthase, resulting in impairment of methylation of dUMP to dTMP and folate cycle disruption [106]. Several of these compounds are currently under pre-clinical evaluation or early-stage clinical trial. In addition, preclinical studies are currently ongoing also for small molecules targeting the catalytic site of metabolic enzymes, such as PHGDH PSAT, PSPH, GLDC [22]. Since the activity of metabolic enzymes can be modulated by the binding of an effector molecule at the allosteric site, it should be also considered to explore the possibility of generate small molecules that target allosteric binding site of the metabolic enzymes. However, our survival analysis (Table 1 and Fig. 2) highlighted that expression of some enzymes instead than others might be associated to the pathogenesis of different cancer types. This observation suggests more cautions. A selective drug targeting design for different cancer types could be critical to achieve therapeutic success. Therefore, it will be of importance to select subsets patients and to find the right combinations of chemical compounds targeting several metabolic enzymes of the serine/glycine pathway.
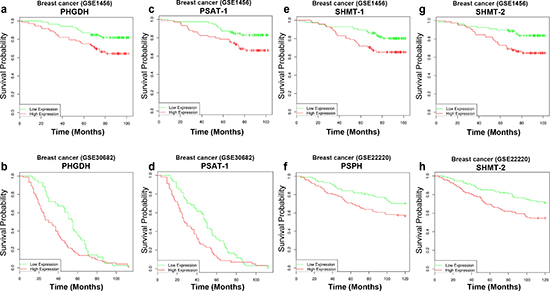
Figure 2: Survival analysis of serine/glycine biosynthetic enzymes. Effect on survival outcome of the (PHGDH, PSAT-1, PSPH, SHMT-2). Clinical follow up data of different breast cancer datasets were censored for survival. Kaplan–Meier analysis with suppressed censoring showed a significant trend toward poor survival when serine biosynthetic enzymes were highly expressed.
Pre-clinical and clinical studies have shown that reducing glucose intake was associated with negative effect on tumor growth [107–111]. Moreover, it has been show that in a tumor in xenograft mouse model, the tumor growth of p53-/- cells was reduced in mice fed with a diet containing no serine and glycine [52]. Overall, these observation indicate that an alternative therapeutic approach could be to associate with pharmacological agents including, a complementary diet or nutrient modification. However, it should be noticed that reduced intake of folate is also associated with breast and colorectal cancer, suggesting the complexity of the relationship between diet and one-carbon metabolism.
In conclusion, more work is needed in order to define the complexity of the metabolic pathways involved in cancer biology and the relationship between them. Moreover, we also need to understand not only the differences between normal and tumor cells, but also why some cancer cells are more dependent on specific metabolic pathways than others. This it could potentially improve the selectivity and the outcome of the therapy.
ACKNOWLEDGEMENTS
This work has been supported by the Medical Research Council, UK; grants from AIRC (2011-IG11955), AIRC 5xmille (MCO #9979), Min. Salute (Ric oncol 26/07) and IDI-IRCCS (RF08 c.15, RF07 c.57) to GM.
Conflict of interest
All the authors declare no conflict of interest.
REFERENCES
1. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nature reviews Cancer. 2011; 11:85–95.
2. Wallace DC. Mitochondria and cancer. Nature reviews Cancer. 2012; 12:685–698.
3. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell metabolism. 2008; 7:11–20.
4. Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nature reviews Cancer. 2011; 11:325–337.
5. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324:1029–1033.
6. Sotgia F, Martinez-Outschoorn UE, Lisanti MP. Genetic induction of the Warburg effect inhibits tumor growth. Oncotarget. 2012; 3:1266–1267.
7. Galluzzi L, Kepp O, Kroemer G. Reverse Warburg: straight to cancer. Cell cycle. 2012; 11:1059.
8. Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012; 491:364–373.
9. Munoz-Pinedo C, El Mjiyad N, Ricci JE. Cancer metabolism: current perspectives and future directions. Cell death & disease. 2012; 3:e248.
10. Lyssiotis CA, Vander-Heiden MG, Munoz-Pinedo C, Emerling BM. Emerging concepts: linking hypoxic signaling and cancer metabolism. Cell death & disease. 2012; 3:e303.
11. Rotblat B, Grunewald TG, Leprivier G, Melino G, Knight RA. Anti-oxidative stress response genes: bioinformatic analysis of their expression and relevance in multiple cancers. Oncotarget. 2013; 4:2577–2590.
12. Ackerman D, Simon MC. Hypoxia, lipids, and cancer: surviving the harsh tumor microenvironment. Trends in cell biology. 2014; 24:472–478.
13. Amelio I, Cutruzzola F, Antonov A, Agostini M, Melino G. Serine and glycine metabolism in cancer. Trends in biochemical sciences. 2014; 39:191–198.
14. Kim SK, Jung WH, Koo JS. Differential expression of enzymes associated with serine/glycine metabolism in different breast cancer subtypes. PloS one. 2014; 9:e101004.
15. Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nature reviews Cancer. 2013; 13:572–583.
16. Kalhan SC, Hanson RW. Resurgence of serine: an often neglected but indispensable amino Acid. The Journal of biological chemistry. 2012; 287:19786–19791.
17. Tedeschi PM, Markert EK, Gounder M, Lin H, Dvorzhinski D, Dolfi SC, Chan LL, Qiu J, DiPaola RS, Hirshfield KM, Boros LG, Bertino JR, Oltvai ZN, Vazquez A. Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell death & disease. 2013; 4:e877.
18. Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, Sasaki AT, Anastasiou D, Mullarky E, Vokes NI, Sasaki M, Beroukhim R, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nature genetics. 2011; 43:869–874.
19. Farber S, Diamond LK. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. The New England journal of medicine. 1948; 238:787–793.
20. Chabner BA, Roberts TG, Jr. Timeline: Chemotherapy and the war on cancer. Nature reviews Cancer. 2005; 5:65–72.
21. Wood LD. Folate receptor alpha: a new tool in the diagnosis and treatment of lung cancer. Oncotarget. 2012; 3:668–669.
22. DeBerardinis RJ. Serine metabolism: some tumors take the road less traveled. Cell metabolism. 2011; 14:285–286.
23. Rajagopalan KN, DeBerardinis RJ. Role of glutamine in cancer: therapeutic and imaging implications. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2011; 52:1005–1008.
24. Ruiz-Perez MV, Sanchez-Jimenez F, Alonso FJ, Segura JA, Marquez J, Medina MA. Glutamine, glucose and other fuels for cancer. Current pharmaceutical design. 2014; 20:2557–2579.
25. Kit S. The biosynthesis of free glycine and serine by tumors. Cancer research. 1955; 15:715–718.
26. Pollari S, Kakonen SM, Edgren H, Wolf M, Kohonen P, Sara H, Guise T, Nees M, Kallioniemi O. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast cancer research and treatment. 2011; 125:421–430.
27. Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, Chen WW, Barrett FG, Stransky N, Tsun ZY, Cowley GS, Barretina J, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011; 476:346–350.
28. Carrera S, Cuadrado-Castano S, Samuel J, Jones GD, Villar E, Lee SW, Macip S. Stra6, a retinoic acid-responsive gene, participates in p53-induced apoptosis after DNA damage. Cell death and differentiation. 2013; 20:910–919.
29. Chen YC, Chan JY, Chiu YL, Liu ST, Lozano G, Wang SL, Ho CL, Huang SM. Grail as a molecular determinant for the functions of the tumor suppressor p53 in tumorigenesis. Cell death and differentiation. 2013; 20:732–743.
30. Amundson SA, Myers TG, Fornace AJ, Jr. Roles for p53 in growth arrest and apoptosis: putting on the brakes after genotoxic stress. Oncogene. 1998; 17:3287–3299.
31. Dixit D, Sharma V, Ghosh S, Mehta VS, Sen E. Inhibition of Casein kinase-2 induces p53-dependent cell cycle arrest and sensitizes glioblastoma cells to tumor necrosis factor (TNFalpha)-induced apoptosis through SIRT1 inhibition. Cell death & disease. 2012; 3:e271.
32. Kracikova M, Akiri G, George A, Sachidanandam R, Aaronson SA. A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell death and differentiation. 2013; 20:576–588.
33. Brosh R, Assia-Alroy Y, Molchadsky A, Bornstein C, Dekel E, Madar S, Shetzer Y, Rivlin N, Goldfinger N, Sarig R, Rotter V. p53 counteracts reprogramming by inhibiting mesenchymal-to-epithelial transition. Cell death and differentiation. 2013; 20:312–320.
34. Mazza D, Infante P, Colicchia V, Greco A, Alfonsi R, Siler M, Antonucci L, Po A, De Smaele E, Ferretti E, Capalbo C, Bellavia D, Canettieri G, Giannini G, Screpanti I, Gulino A, et al. PCAF ubiquitin ligase activity inhibits Hedgehog/Gli1 signaling in p53-dependent response to genotoxic stress. Cell death and differentiation. 2013; 20:1688–1697.
35. Lee MS, Seo J, Choi DY, Lee EW, Ko A, Ha NC, Yoon JB, Lee HW, Kim KP, Song J. Stabilization of p21 (Cip1/WAF1) following Tip60-dependent acetylation is required for p21-mediated DNA damage response. Cell death and differentiation. 2013; 20:620–629.
36. Nayak G, Cooper GM. p53 is a major component of the transcriptional and apoptotic program regulated by PI 3-kinase/Akt/GSK3 signaling. Cell death & disease. 2012; 3:e400.
37. Malek M, Guillaumot P, Huber AL, Lebeau J, Petrilli V, Kfoury A, Mikaelian I, Renno T, Manie SN. LAMTOR1 depletion induces p53-dependent apoptosis via aberrant lysosomal activation. Cell death & disease. 2012; 3:e300.
38. Sorrentino G, Mioni M, Giorgi C, Ruggeri N, Pinton P, Moll U, Mantovani F, Del Sal G. The prolyl-isomerase Pin1 activates the mitochondrial death program of p53. Cell death and differentiation. 2013; 20:198–208.
39. Wiman KG. p53 talks to PARP: the increasing complexity of p53-induced cell death. Cell death and differentiation. 2013; 20:1438–1439.
40. Bouvard V, Zaitchouk T, Vacher M, Duthu A, Canivet M, Choisy-Rossi C, Nieruchalski M, May E. Tissue and cell-specific expression of the p53-target genes: bax, fas, mdm2 and waf1/p21, before and following ionising irradiation in mice. Oncogene. 2000; 19:649–660.
41. Haupt S, Gamell C, Wolyniec K, Haupt Y. Interplay between p53 and VEGF: how to prevent the guardian from becoming a villain. Cell death and differentiation. 2013; 20:852–854.
42. Kim SY, Cordeiro MH, Serna VA, Ebbert K, Butler LM, Sinha S, Mills AA, Woodruff TK, Kurita T. Rescue of platinum-damaged oocytes from programmed cell death through inactivation of the p53 family signaling network. Cell death and differentiation. 2013; 20:987–997.
43. Craxton A, Butterworth M, Harper N, Fairall L, Schwabe J, Ciechanover A, Cohen GM. NOXA, a sensor of proteasome integrity, is degraded by 26S proteasomes by an ubiquitin-independent pathway that is blocked by MCL-1. Cell death and differentiation. 2012; 19:1424–1434.
44. Huang BH, Zhuo JL, Leung CH, Lu GD, Liu JJ, Yap CT, Hooi SC. PRAP1 is a novel executor of p53-dependent mechanisms in cell survival after DNA damage. Cell death & disease. 2012; 3:e442.
45. Esposito F, Tornincasa M, Federico A, Chiappetta G, Pierantoni GM, Fusco A. High-mobility group A1 protein inhibits p53-mediated intrinsic apoptosis by interacting with Bcl-2 at mitochondria. Cell death & disease. 2012; 3:e383.
46. Ehrhardt H, Hofig I, Wachter F, Obexer P, Fulda S, Terziyska N, Jeremias I. NOXA as critical mediator for drug combinations in polychemotherapy. Cell death & disease. 2012; 3:e327.
47. Carpinelli MR, Wise AK, Arhatari BD, Bouillet P, Manji SS, Manning MG, Cooray AA, Burt RA. Anti-apoptotic gene Bcl2 is required for stapes development and hearing. Cell death & disease. 2012; 3:e362.
48. Valentino T, Palmieri D, Vitiello M, Pierantoni GM, Fusco A, Fedele M. PATZ1 interacts with p53 and regulates expression of p53-target genes enhancing apoptosis or cell survival based on the cellular context. Cell death & disease. 2013; 4:e963.
49. Berkers CR, Maddocks OD, Cheung EC, Mor I, Vousden KH. Metabolic regulation by p53 family members. Cell metabolism. 2013; 18:617–633.
50. Kang MY, Kim HB, Piao C, Lee KH, Hyun JW, Chang IY, You HJ. The critical role of catalase in prooxidant and antioxidant function of p53. Cell death and differentiation. 2013; 20:117–129.
51. Montero J, Dutta C, van Bodegom D, Weinstock D, Letai A. p53 regulates a non-apoptotic death induced by ROS. Cell death and differentiation. 2013; 20:1465–1474.
52. Maddocks OD, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, Vousden KH. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature. 2013; 493:542–546.
53. Sachweh MC, Drummond CJ, Higgins M, Campbell J, Lain S. Incompatible effects of p53 and HDAC inhibition on p21 expression and cell cycle progression. Cell death & disease. 2013; 4:e533.
54. Sanchez-Macedo N, Feng J, Faubert B, Chang N, Elia A, Rushing EJ, Tsuchihara K, Bungard D, Berger SL, Jones RG, Mak TW, Zaugg K. Depletion of the novel p53-target gene carnitine palmitoyltransferase 1C delays tumor growth in the neurofibromatosis type I tumor model. Cell death and differentiation. 2013; 20:659–668.
55. Dalleau S, Baradat M, Gueraud F, Huc L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell death and differentiation. 2013; 20:1615–1630.
56. Grespi F, Amelio I, Tucci P, Annicchiarico-Petruzzelli M, Melino G. Tissue-specific expression of p73 C-terminal isoforms in mice. Cell cycle. 2012; 11:4474–4483.
57. Wei J, Zaika E, Zaika A. p53 Family: Role of Protein Isoforms in Human Cancer. Journal of nucleic acids. 2012; 2012:687359.
58. Li C, Chang DL, Yang Z, Qi J, Liu R, He H, Li D, Xiao ZX. Pin1 modulates p63alpha protein stability in regulation of cell survival, proliferation and tumor formation. Cell death & disease. 2013; 4:e943.
59. Budanov AV. The Role of Tumor Suppressor p53 in the Antioxidant Defense and Metabolism. Sub-cellular biochemistry. 2014; 85:337–358.
60. Du W, Jiang P, Mancuso A, Stonestrom A, Brewer MD, Minn AJ, Mak TW, Wu M, Yang X. TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nature cell biology. 2013; 15:991–1000.
61. Fets L, Anastasiou D. p73 keeps metabolic control in the family. Nature cell biology. 2013; 15:891–893.
62. Cutruzzola F, Avigliano L, Ci E. p73 keeps metabolic control in balance. Cell cycle. 2014; 13:179–180.
63. He Z, Liu H, Agostini M, Yousefi S, Perren A, Tschan MP, Mak TW, Melino G, Simon HU. p73 regulates autophagy and hepatocellular lipid metabolism through a transcriptional activation of the ATG5 gene. Cell death and differentiation. 2013; 20:1415–1424.
64. He Z, Simon HU. Autophagy protects from liver injury. Cell death and differentiation. 2013; 20:850–851.
65. Chang CP, Su YC, Hu CW, Lei HY. TLR2-dependent selective autophagy regulates NF-kappaB lysosomal degradation in hepatoma-derived M2 macrophage differentiation. Cell death and differentiation. 2013; 20:515–523.
66. Amelio I, Markert EK, Rufini A, Antonov AV, Sayan BS, Tucci P, Agostini M, Mineo TC, Levine AJ, Melino G. p73 regulates serine biosynthesis in cancer. Oncogene. 2013.
67. Giacobbe A, Bongiorno-Borbone L, Bernassola F, Terrinoni A, Markert EK, Levine AJ, Feng Z, Agostini M, Zolla L, Agro AF, Notterman DA, Melino G, Peschiaroli A. p63 regulates glutaminase 2 expression. Cell cycle. 2013; 12:1395–1405.
68. Velletri T, Romeo F, Tucci P, Peschiaroli A, Annicchiarico-Petruzzelli M, Niklison-Chirou MV, Amelio I, Knight RA, Mak TW, Melino G, Agostini M. GLS2 is transcriptionally regulated by p73 and contributes to neuronal differentiation. Cell cycle. 2013; 12:3564–3573.
69. Vousden KH, Ryan KM. p53 and metabolism. Nature reviews Cancer. 2009; 9:691–700.
70. Kim DY, Kim W, Lee KH, Kim SH, Lee HR, Kim HJ, Jung Y, Choi JH, Kim KT. hnRNP Q regulates translation of p53 in normal and stress conditions. Cell death and differentiation. 2013; 20:226–234.
71. Bartoletti-Stella A, Mariani E, Kurelac I, Maresca A, Caratozzolo MF, Iommarini L, Carelli V, Eusebi LH, Guido A, Cenacchi G, Fuccio L, Rugolo M, Tullo A, Porcelli AM, Gasparre G. Gamma rays induce a p53-independent mitochondrial biogenesis that is counter-regulated by HIF1alpha. Cell death & disease. 2013; 4:e663.
72. Darzynkiewicz Z. Perturbation of nucleotide metabolism—the driving force of oncogene-induced senescence. Oncotarget. 2013; 4:649–650.
73. Strano S, Dell'Orso S, Di Agostino S, Fontemaggi G, Sacchi A, Blino G. Mutant p53: an oncogenic transcription factor. Oncogene. 2007; 26:2212–2219.
74. Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, Bissell MJ, Osborne TF, Tian B, Lowe SW, Silva JM, Borresen-Dale AL. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012; 148:244–258.
75. Mello SS, Attardi LD. Not all p53 gain-of-function mutants are created equal. Cell death and differentiation. 2013; 20:855–857.
76. Hanel W, Marchenko N, Xu S, Yu SX, Weng W, Moll U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell death and differentiation. 2013; 20:898–909.
77. Manzl C, Fava LL, Krumschnabel G, Peintner L, Tanzer MC, Soratroi C, Bock FJ, Schuler F, Luef B, Geley S, Villunger A. Death of p53-defective cells triggered by forced mitotic entry in the presence of DNA damage is not uniquely dependent on Caspase-2 or the PIDDosome. Cell death & disease. 2013; 4:e942.
78. Tucci P, Agostini M, Grespi F, Markert EK, Terrinoni A, Vousden KH, Muller PA, Dotsch V, Kehrloesser S, Sayan BS, Giaccone G, Lowe SW, Takahashi N, Vandenabeele P, Knight RA, Levine AJ. Loss of p63 and its microRNA-205 target results in enhanced cell migration and metastasis in prostate cancer. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109:15312–15317.
79. Weissmueller S, Manchado E, Saborowski M, Morris JPt, Wagenblast E, Davis CA, Moon SH, Pfister NT, Tschaharganeh DF, Kitzing T, Aust D, Markert EK, Wu J, Grimmond SM, Pilarsky C, Prives C. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell. 2014; 157:382–394.
80. Alexandrova EM, Petrenko O, Nemajerova A, Romano RA, Sinha S, Moll UM. DeltaNp63 regulates select routes of reprogramming via multiple mechanisms. Cell death and differentiation. 2013; 20:1698–1708.
81. Alexandrova EM, Talos F, Moll UM. p73 is dispensable for commitment to neural stem cell fate, but is essential for neural stem cell maintenance and for blocking premature differentiation. Cell death and differentiation. 2013; 20:368.
82. Antonov AV, Krestyaninova M, Knight RA, Rodchenkov I, Melino G, Barlev NA. PPISURV: a novel bioinformatics tool for uncovering the hidden role of specific genes in cancer survival outcome. Oncogene. 2014; 33:1621–1628.
83. Amelio I, Gostev M, Knight RA, Willis AE, Melino G, Antonov AV. DRUGSURV: a resource for repositioning of approved and experimental drugs in oncology based on patient survival information. Cell death & disease. 2014; 5:e1051.
84. Garrow TA, Brenner AA, Whitehead VM, Chen XN, Duncan RG, Korenberg JR, Shane B. Cloning of human cDNAs encoding mitochondrial and cytosolic serine hydroxymethyltransferases and chromosomal localization. The Journal of biological chemistry. 1993; 268:11910–11916.
85. Anderson DD, Stover PJ. SHMT1 and SHMT2 are functionally redundant in nuclear de novo thymidylate biosynthesis. PloS one. 2009; 4:e5839.
86. Sassone-Corsi P. Physiology. When metabolism and epigenetics converge. Science. 2013; 339:148–150.
87. Renwick SB, Snell K, Baumann U. The crystal structure of human cytosolic serine hydroxymethyltransferase: a target for cancer chemotherapy. Structure. 1998; 6:1105–1116.
88. Nikiforov MA, Chandriani S, O'Connell B, Petrenko O, Kotenko I, Beavis A, Sedivy JM, Cole MD. A functional screen for Myc-responsive genes reveals serine hydroxymethyltransferase, a major source of the one-carbon unit for cell metabolism. Molecular and cellular biology. 2002; 22:5793–5800.
89. Piskac-Collier AL, Monroy C, Lopez MS, Cortes A, Etzel CJ, Greisinger AJ, Spitz MR, El-Zein RA. Variants in folate pathway genes as modulators of genetic instability and lung cancer risk. Genes, chromosomes & cancer. 2011; 50:1–12.
90. Sarin M, Wang Y, Zhang F, Rothermund K, Zhang Y, Lu J, Sims-Lucas S, Beer-Stolz D, Van Houten BE, Vockley J, Goetzman ES, Graves JA, Prochownik EV. Alterations in c-Myc phenotypes resulting from dynamin-related protein 1 (Drp1) -mediated mitochondrial fission. Cell death & disease. 2013; 4:e670.
91. Zhang WC, Shyh-Chang N, Yang H, Rai A, Umashankar S, Ma S, Soh BS, Sun LL, Tai BC, Nga ME, Bhakoo KK, Jayapal SR, Nichane M, Yu Q, Ahmed DA, Tan C, et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell. 2012; 148:259–272.
92. di Salvo ML, Contestabile R, Paiardini A, Maras B. Glycine consumption and mitochondrial serine hydroxymethyltransferase in cancer cells: the heme connection. Medical hypotheses. 2013; 80:633–636.
93. Markert EK, Levine AJ, Vazquez A. Proliferation and tissue remodeling in cancer: the hallmarks revisited. Cell death & disease. 2012; 3:e397.
94. Barbieri E, Scorrano L. When numbers matters: mitochondrial DNA and gliomagenesis. Cell death and differentiation. 2013; 20:1601–1602.
95. Cook CC, Kim A, Terao S, Gotoh A, Higuchi M. Consumption of oxygen: a mitochondrial-generated progression signal of advanced cancer. Cell death & disease. 2012; 3:e258.
96. Wei L, Zhou Y, Dai Q, Qiao C, Zhao L, Hui H, Lu N, Guo QL. Oroxylin A induces dissociation of hexokinase II from the mitochondria and inhibits glycolysis by SIRT3-mediated deacetylation of cyclophilin D in breast carcinoma. Cell death & disease. 2013; 4:e601.
97. Maillet A, Yadav S, Loo YL, Sachaphibulkij K, Pervaiz S. A novel Osmium-based compound targets the mitochondria and triggers ROS-dependent apoptosis in colon carcinoma. Cell death & disease. 2013; 4:e653.
98. Wang Y, Yang F, Zhang HX, Zi XY, Pan XH, Chen F, Luo WD, Li JX, Zhu HY, Hu YP. Cuprous oxide nanoparticles inhibit the growth and metastasis of melanoma by targeting mitochondria. Cell death & disease. 2013; 4:e783.
99. Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell death & disease. 2013; 4:e532.
100. Favaro E, Harris AL. Targeting glycogen metabolism: a novel strategy to inhibit cancer cell growth?. Oncotarget. 2013; 4:3–4.
101. Sotgia F, Martinez-Outschoorn UE, Lisanti MP. Cancer metabolism: new validated targets for drug discovery. Oncotarget. 2013; 4:1309–1316.
102. Harris AL. Metabolic compartments in tumor tissue: implications for therapy. Cell cycle. 2012; 11:13–14.
103. Li W, Tai Y, Zhou J, Gu W, Bai Z, Zhou T, Zhong Z, McCue PA, Sang N, Ji JY, Kong B, Jiang J, Wang C. Repression of endometrial tumor growth by targeting SREBP1 and lipogenesis. Cell cycle. 2012; 11:2348–2358.
104. Jitschin R, Hofmann AD, Bruns H, Giessl A, Bricks J, Berger J, Saul D, Eckart MJ, Mackensen A, Mougiakakos D. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood. 2014; 123:2663–2672.
105. Ross SJ, Critchlow SE. Emerging approaches to target tumor metabolism. Current opinion in pharmacology. 2014; 17C:22–29.
106. Spears CP, Shahinian AH, Moran RG, Heidelberger C, Corbett TH. In vivo kinetics of thymidylate synthetase inhibition of 5-fluorouracil-sensitive and -resistant murine colon adenocarcinomas. Cancer research. 1982; 42:450–456.
107. Myers AP, Cantley LC. Sugar free, cancer free?. Nutrition. 2012; 28:1036.
108. MacFarlane M, Robinson GL, Cain K. Glucose—a sweet way to die: metabolic switching modulates tumor cell death. Cell cycle. 2012; 11:3919–3925.
109. Rodriguez OC, Choudhury S, Kolukula V, Vietsch EE, Catania J, Preet A, Reynoso K, Bargonetti J, Wellstein A, Albanese C, Avantaggiati ML. Dietary downregulation of mutant p53 levels via glucose restriction: mechanisms and implications for tumor therapy. Cell cycle. 2012; 11:4436–4446.
110. Garufi A, Ricci A, Trisciuoglio D, Iorio E, Carpinelli G, Pistritto G, Cirone M, D'Orazi G. Glucose restriction induces cell death in parental but not in homeodomain-interacting protein kinase 2-depleted RKO colon cancer cells: molecular mechanisms and implications for tumor therapy. Cell death & disease. 2013; 4:e639.
111. Yang HC, Chen TL, Wu YH, Cheng KP, Lin YH, Cheng ML, Ho HY, Lo SJ, Chiu DT. Glucose 6-phosphate dehydrogenase deficiency enhances germ cell apoptosis and causes defective embryogenesis in Caenorhabditis elegans. Cell death & disease. 2013; 4:e616.