
INTRODUCTION
In 2017, there will be approximately 220,000 new cases of lung cancer in the United States with approximately 160,000 deaths [1]. Platinum-based chemotherapy delivers a response rate of 20–35% and a median overall survival (OS) of 8–12 months in non-small cell lung cancer (NSCLC) [2, 3]. Improved understanding of the molecular signatures of NSCLC has led to the development of targeted therapeutics against epidermal growth factor receptor (EGFR), Kirsten rat sarcoma viral oncogene homolog (KRAS), or anaplastic lymphoma kinase (ALK) alterations which can result in significant response and extended OS. However, a significant percentage of NSCLC patients do not have a targetable mutation. Moreover, the 5 year OS for NSCLC, even with targeted therapeutics, is 15% [4].
The blockade of immune checkpoints, through inhibition, such as programmed cell death (PD-1)/programmed cell death ligand (PD-L1) targeting, has shown remarkable response in some patients with NSCLC [5]. Pembrolizumab, a PD-1 inhibitor, is approved for first and second line PD-L1 expressing NSCLC, after demonstrating its association with significantly improved OS compared to docetaxel [6]. Nivolumab, another PD-1 inhibitor, and atezolizumab, a PD-L1 inhibitor, are approved to treat ALK/EGFR WT and mutant metastatic NSCLC [7, 8]. Higher PD-L1 expression is associated with better efficacy of PD-1/PD-L1 therapeutics and improved response in NSCLC, [9, 10] however PD-L1 expression is not the sole determinant of response to PD-1/PD-L1 therapy and some patients who do not express PD-L1 have been shown to respond to PD-1/PD-L1 inhibition [11, 12]. While PD-1/PD-L1 inhibitors can be remarkably effective in some patients producing durable responses, only a subset of patients will benefit (e.g. 10-40% of NSCLC) [7, 13].
Attempts have been made to establish a correlation between PD-1/PD-L1 expression and NSCLC driver dysregulation [4, 14, 15]. About 50–65% of patients with EGFR-mutated NSCLC respond to EGFR tyrosine kinase inhibitors (TKIs), [16] but most patients become resistant to these treatments by acquiring a secondary point mutation in EGFR or activation of additional signaling pathways, including mesenchymal-epithelial transition factor (MET) and KRAS [17, 18]. Dysregulation of MET signaling has been shown to drive tumorigenesis in NSCLC [19]. Inhibition of MET signaling in EGFR TKI-resistant cells may restore sensitivity to EGFR inhibitors [20, 21]. Additionally, anti-tumor activity of agents targeting MET has been observed in MET-amplified EGFR WT patients [22]. The correlation between MET amplification and PD-L1 expression, particularly for EGFR WT NSCLC patients is important for therapy and has not been fully explored.
To improve on the success of checkpoint inhibitors as monotherapy, combination therapy is being considered. Conventional chemotherapy and targeted therapy are currently being considered in combination with checkpoint inhibitors [23]. Theoretically, direct correlation between two molecular abnormalities suggests collaboration and selection between these two abnormalities and targeting both drivers may be more effective as a therapeutic approach. Toward this end, we explored the correlation between PD-L1 expression and KRAS, EGFR, TP53, and MET alterations in this study.
MET is a receptor protein-tyrosine kinase for hepatocyte growth factor, which induces the RAS-ERK/MAPKs cascades through the activation of Grb2-associated binding protein 1 (Gab1) [24]. Similarly, the EGFR is a receptor tyrosine kinase that induces RAS-ERK/MAPK and AKT-PI3K-mTOR [25]. Furthermore, inactivation of the TP53 gene is well documented to cooperate with activated RAS to induce tumorigenesis [26].
RESULTS
PD-L1 expression
Positive PD-L1 expression (≥1%) appeared in 55.5% of males and 56.0% of females. Representative examples of PD-L1 protein expression by IHC are shown in Figure 1A-1D: A-C shows different staining of the same tumor with panel C representing PD-L1 staining and panel D shows intermediate PD-L1 staining of a different tumor. As shown in Figure 2, PD-L1 (SP 142) was expressed in 166/397 (41.8%) of NSCLC patients tested in this study. Twenty seven (7%) had expression between 1-5%, 61 (15%) between 1-20%, 92 (23%) between 1-50%, and 74 (18.6%) had PD-L1expression > 50% in tumor cells (TPS; tumor proportion score).
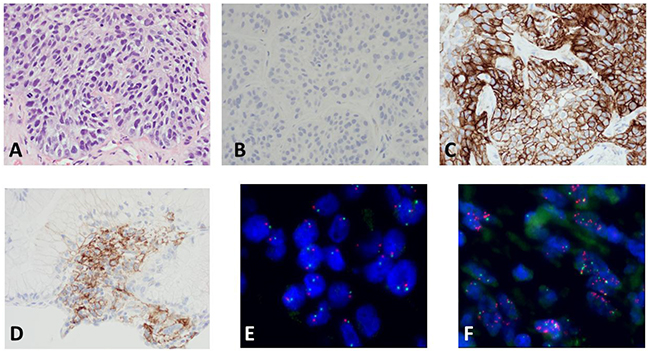
Figure 1: Representative examples of PD-L1 protein expression by IHC and MET amplification by FISH. Top panel shows hematoxylin-eosin stain of the tumor in (A), negative isotopic control in (B), and PD-L1 staining in (C). Intermediate PD-L1 staining in a different tumor is shown in (D). The FISH images show MET amplification (red probe) in one tumor in (E) and no amplification in a different tumor in (F) The green signal represents chromosome 7 centromere.
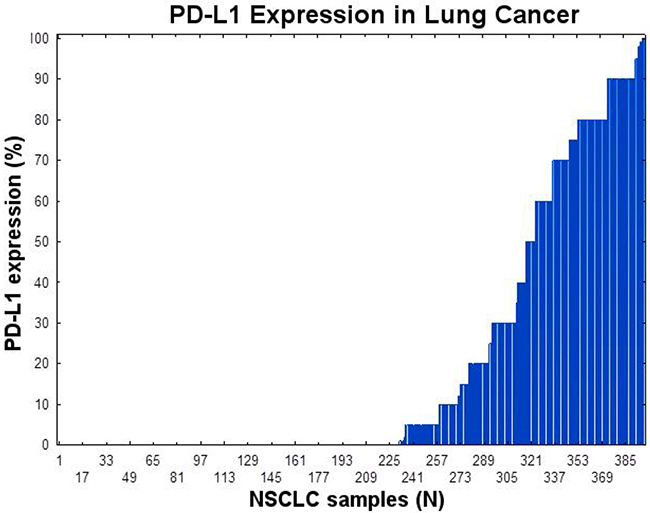
Figure 2: PD-L1 expression in NSCLC using immunohistochemistry using SP147 PD-L1 clone. The tumor proportion score (TPS) is shown.
Inverse correlation between EGFR mutation and PD-L1 expression
Next generation sequencing (NGS) revealed that 17.4% (69/397) patients had EGFR mutations in our study. Of the EGFR WT patients, 251 did not have PD-L1 expression (TPS <50%) and 77 (23.5%) did. Of the patients who did have EGFR mutations, 66 did not have PD-L1 expression (TPS <50%) and 3 did, representing 4.4% (3/69) PD-L1 expressing in EGFR mutated consort. As shown in Figure 3, the distribution of EGFR WT has higher PD-L1 expression than the mutated EGFR. The number of cases with EGFR mutation and PD-L1 expression ≥50% was small, but the data suggests that there is a reverse correlation between PD-L1 expression and EGFR mutational status even when considering PD-L1 expression as a continuous variable (P=0.0025). Additionally, when a cut-off point of 50% is used for PD-L1 expression, there were significantly fewer positive cases in the EGFR mutant as compared to EGFR WT (P=0.0003). This correlation becomes slightly weaker as the PD-L1 expression decreases (P=0.002 @ TPS=20% and P=0.137 @ TPS=5%).
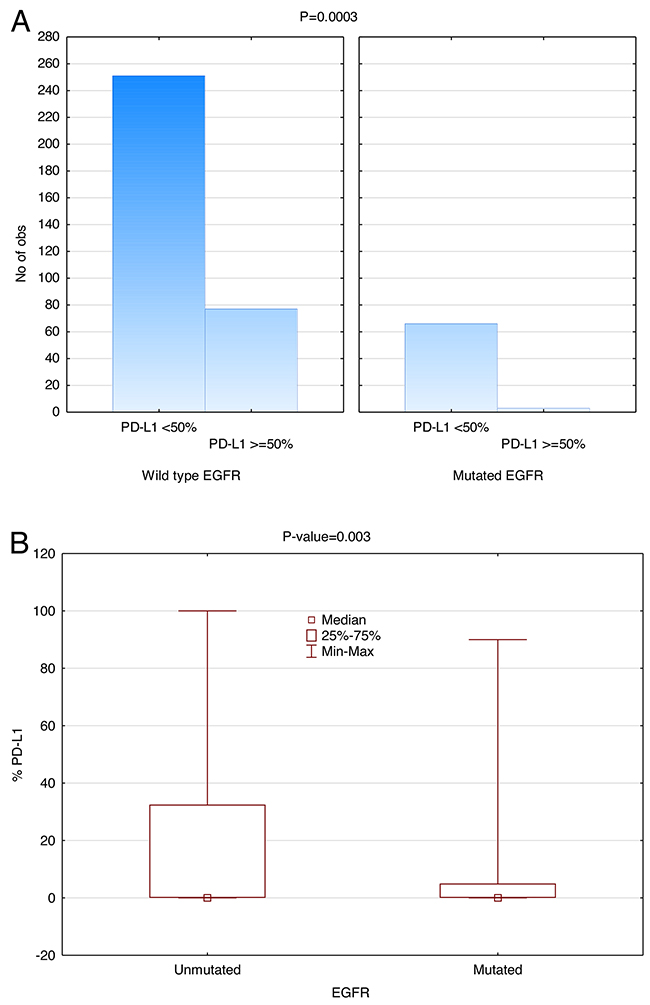
Figure 3: Inverse correlation between EGFR mutation and PD-L1 Expression (SP142). Significantly fewer patients with EGFR mutation had PD-L1 TPS>50% (P=0.0003, Wilcoxon rank-sum test). (A) The height of the histogram represents the number of patients. The two panels compare EGFR WT on the left with mutant EGFR on the right. Within each panel, the first data point represents the number of patients expressing PD-L1 less than 50% and the second data point represents the number of patients expressing PD-L1 more than or at 50%. (B) EGFR versus PD-L1 expression as a continuous variable in box plot form.
Positive correlation between MET amplification and PD-L1 expression
As shown in Figure 1 and Figure 4, based on testing 471 patients with various tumors, including 397 NSCLCs, for MET copies by FISH as compared to chromosome 7 centromere, we established that a cut-off point (CO) of 1.5% is appropriate for determining MET amplification. Of the 397 patients measured for PD-L1 expression, 389 were also measured for MET amplification. MET was amplified (CO > 1.5%) in 4.1% patients with NSCLC. As shown in Figure 5, with MET ratio as the independent variable and PD-L1 as a continuous dependent variable, expression of PD-L1 is correlated with MET amplification. Those patients with MET amplification (≥ 1.5%) 10/16 (62.5%) had PD-L1 expression above the median, whereas for those patients without MET amplification (< 1.5%) 155/373 (41.6%) had PD-L1 expression above the median. Patients with MET amplification (CO > 1.5%) had a significantly higher (P=0.004) percentage of PD-L1 expression as a continuous variable as well as when cut-off points of 5% (P=0.01), 20% (P=0.0006), and 50% (P=0.01) are used.
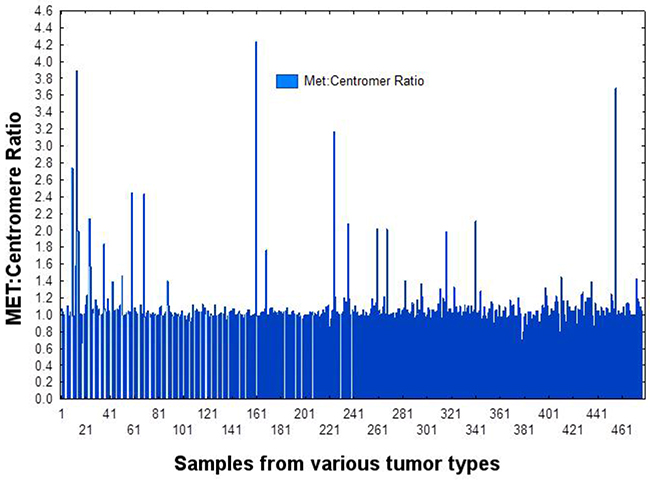
Figure 4: Establishing a cut-off point for MET Amplification by FISH (MET:centromere ratio of 1.5). Based on the signal to noise pattern show in this graph, a ratio ≥1.5 was determined to reflect amplification.
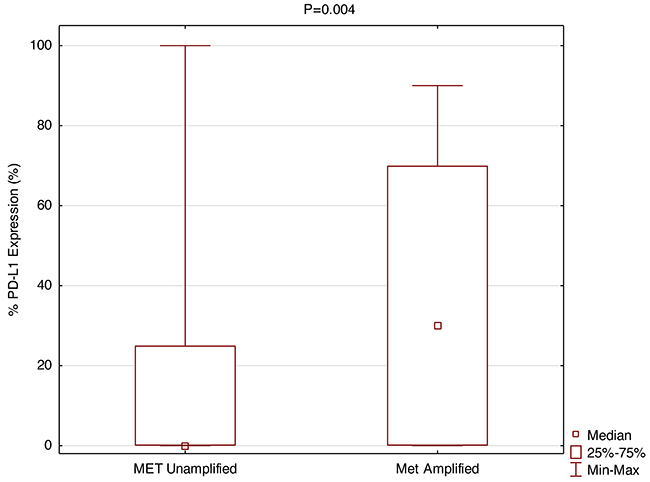
Figure 5: Positive correlation between MET amplification and PD-L1 Expression (SP 142). Patients with MET amplifications had significantly higher PD-L1 TPS (Wilcoxon rank-sum test). MET amplification versus PD-L1 expression as a continuous variable in box plot form.
Positive correlation between TP53 mutation and PD-L1 expression
NGS revealed that 225/397 (56.7%) of NSCLC patients had a TP53 mutation. As shown in Figure 6, there was significant correlation between TP53 mutation and overall PD-L1 expression (P<0.0001) when PD-L1 is used as a continuous variable. This positive correlation is also detected when PD-L1 expression cut-off points of 50%, 20%, or 5% are used (P=0.007, P=0.0003, and P=0.0004, respectively). For TP53 WT, 24/172 (14.0%) were PD-L1 expressing (cut-off ≥ 50%) and 56/225 (24.9%) expressed PD-L1 for TP53 mutated samples. Notably, TP53 mutation was significantly more common in EGFR WT cases (P=0.0002).
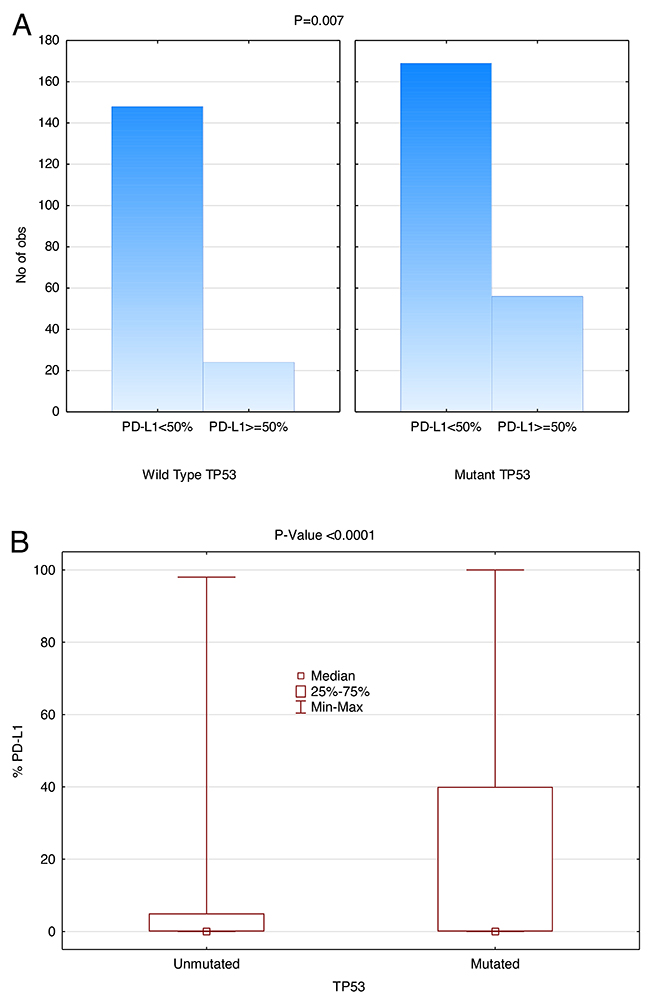
Figure 6: Positive correlation between TP53 mutation and PD-L1 Expression (SP142). Patients with TP53 mutation had significantly higher PD-L1 TPS (Wilcoxon rank-sum test). (A) The height of the histogram represents the number of patients. The two panels compare TP53 WT on the left with mutant TP53 on the right. Within each panel, the first data point represents the number of patients expressing PD-L1 less than 50% and the second data point represents the number of patients expressing PD-L1 more than or at 50%. (B) TP53 versus PD-L1 expression as a continuous variable in box plot form.
No correlation between KRAS mutation and PD-L1 expression
NGS revealed that 184/397 (46.3%) of NSCLC patients had KRAS mutations, including activating and nonactivating mutations. There was no correlation between KRAS mutation and overall PD-L1expression (P=0.49) when PD-L1 is considered as a continuous variable or for PD-L1 expression cut-offs 5%, 20%, or 50%. However, when TP53 and KRAS were co-mutated, PD-L1 increased continuously (P=0.0002), and the co-mutation was significantly correlated with PD-L1 expression both at TPS of 5% (P=0.001) and 50% (P=0.0002).
DISCUSSION
In this study we found that PD-L1 expressing EGFR WT NSCLC patients are statistically more likely to also overexpress MET than those who do not express PD-L1. Although measuring MET amplification in NSCLC is valuable for treatment decisions, it is difficult to quantify, with a limited number of putative MET-amplified patients. MET amplification occurs in approximately 2-4% of patients with lung adenocarcinoma, higher in other histologic types of lung cancer, and up to 22% of patients with acquired resistance to EGFR inhibition therapy [27–29]. We were able to establish that a cut-off point (CO) of 1.5% is appropriate for determining MET amplification, which compares favorably with earlier studies [29]. In our study, MET was amplified in 4.1% of NSCLC patients, and patients with MET amplification had significantly (P=0.004) more PD-L1 expression.
Our results indicate that EGFR mutation is inversely correlated with PD-L1 expression in NSCLC. EGFR WT patients included 23.5% that expressed high PD-L1, while EGFR mutated patients included only 4.4% that expressed high PD-L1. Some earlier studies also found this result: PD-L1 expression is higher in EGFR WT [30, 31]. Moreover, activation of the EGFR pathway has been shown to induce PD-L1 expression, [7, 32] while inhibition of EGFR has been shown to down-regulate expression of PD-L1 in EGFR mutated NSCLC cells, but not in the EGFR WT cells [12, 32]. This regulatory role of EGFR on PD-L1 expression, may explain the lack of PD-L1 expression by EGFR-mutant lung tumors and the lack of response rates reported for these patients [31]. Some previous studies reported the opposite findings: PD-L1 expression was higher in EGFR-mutant NSCLC [4, 11, 15, 33, 34]. Most of these studies included an EGFR-mutant patient cohort consisting predominately of patients who have received at least one cycle of EGFR tyrosine kinase inhibitor (TKI). This prior treatment may provide a rationale for the discrepancy, as a significant percentage of these patients may reasonably have acquired MET amplification after treatment, thereby driving PD-L1 expression secondarily. For EGFR WT patients, the relationship between MET amplification and PD-L1 expression may be critical for personalized cancer treatment that downregulates the RAS/MAPK pathway and where PD-L1 inhibitors are likely to be effective given the over-expression of PD-L1 seen in this patient population. A combination of MET targeting and checkpoint inhibitors may be particularly effective for EGFR WT patients, for whom other treatments are lacking.
The PD-1/PD-L1 pathway is a critical therapeutic target for advanced NSCLC, particularly in earlier disease settings. Although different anti-PD-L1 antibody clones are currently used in evaluating PD-L1 expression by immunohistochemistry (IHC), there is a concordance between antibodies for tumor cell scoring, but significant differences in the expression of PD-L1 and staining among 28-8, 22c3, SP142, and E1L3N, with SP142 producing the least stain [35, 36]. Various levels of PD-L1 expression have been noted in NSCLC studies depending on cut-off and clone [10]. Several investigators have suggested reporting both PD-L1 expression at any level in addition to expression ≥ 50% TPS by IHC staining [9]. However, the PD-L1 expression observed in this study using the SP142 clone was 41.8% (cut-off ≥ 1%) and 18.6% (cut-off ≥ 50%), and compares favorably with other studies that show similar results (e.g. 42.5% at any level and 18.4% with a PD-L1 expression level cut-off of ≥ 50%) [37, 38].
In our study, 46.7% of NSCLCs had KRAS mutations. In other studies, KRAS mutation was seen in 6-39%, varying with ethnicity, histology, and smoking status [39–41]. There was no correlation between KRAS mutation and overall PD-L1 expression (P=0.4). Also there was no correlation between KRAS mutation and PD-L1 when 5%, 20%, or 50% cut-off points were used. Other studies also failed to find a correlation between KRAS mutation and PD-L1 expression, [30, 34] though a few studies have found a significant positive correlation [11, 42]. Although it has not been clear why there have been discrepant results, concurrent dysregulation of downstream pathways, such as TP53, in contributing to the regulation of PD-L1 against the background of KRAS mutation, may help to explain the findings [42, 43]. In order to test this hypothesis, we calculated the expression of PD-L1 against combinations of TP53 and KRAS mutant and WT. Indeed we found that although there was no correlation between KRAS mutant status and PD-L1 expression overall or in the presence of WT TP53, the co-mutation of KRAS with TP53 was highly correlated with PD-L1 expression. Multiple studies have shown that mutations in TP53 induce PD-L1 expression. This induction is reported be mediated by miR-34. On the other hand, PD-L1 expression has been reported to be suppressed by EGFR kinase inhibitors. Furthermore, MET overexpression is reported to be associated resistance to EGFR inhibitors. This may explain our findings.
MET amplification drives constitutive activation of the RAF/MAPK pathway, as do KRAS and EGFR mutations. Not surprisingly, except for two patients in our study, all samples with KRAS mutation were EGFR WT. TP53 mutation, on the other hand, was correlated to both EGFR WT patient status and PD-L1 expression. Moreover, TP53 is more common in EGFR WT cases, and together these data point to TP53 being a driver of PD-L1 expression. In this study, EGFR mutant NSCLCs have significantly lower PD-L1 expression, but those without EGFR mutation have higher levels of PD-L1, TP53 mutation, and MET activation; patients with EGFR WT NSCLC who overexpress PD-L1 are statistically more likely to also overexpress MET and have mutated TP53.
It has been shown previously that patients with TP53 mutations are significantly more common in EGFR WT cases (P=0.0002) [44]. In our study, NGS revealed that 56.7% (225/397) of NSCLC patients had a TP53 mutation, a bit higher than described in earlier reports (45-46%) [45]. Patients with TP53 mutation had strikingly higher PD-L1 expression using the SP142 clone. In our study patients with TP53 mutations had a higher copy number of the MET gene (CO ≥ 1.5%; P = 0.01), in keeping with other studies [44]. TP53 mutations are seen more frequently in earlier stages of NSCLC [46, 47]. TP53 has also been previously suggested to modulate the expression of immuno-regulatory genes, such as the TLR family and PD-L1 [48, 49]. Gain of function TP53 mutation may play a role in metastasis early in the disease, suggesting patients harboring TP53 mutation may benefit from personalized combination therapy. Because TP53 mutations allow for the accumulation of a wide variety of mutated proteins, neoantigens, checkpoint inhibition and immune stimulation is an obvious choice.
Our data suggests that in NSCLC, both MET and TP53 genes play a direct role in up-regulating PD-L1 expression. TP53 mutation may play a role in metastasis early in the disease, suggesting patients without EGFR, ALK, or ROS1 mutations, but harboring a TP53 mutation, belong to a unique population subset that may benefit from combination therapy. Using PD-L1 or other immuno-oncology approaches in early stage NSCLCs, particularly those with TP53 mutations, may delay metastasis, and allow conventional treatment to be more effective. Additionally, combination therapy targeting MET with checkpoint inhibition or TP53 with checkpoint inhibition should be considered in treating EGFR WT, TP53 and/or MET amplified NSCLC.
MATERIALS AND METHODS
Patients, samples, and study approval
In this retrospective study, tissue samples were collected from 471 patients with various tumors, including 397 lung cancers. The median age of the females was 66 and 68 for males (P=0.04). There were 389 patients with available data on both MET amplification and PD-L1 expression. NSCLC patients in this retrospective study had TP53 mutations (57%), KRAS mutations (46.7%), PD-L1 overexpression (41.8%), EGFR mutations (17.4%), and MET amplifications (4%). A summary of correlations uncovered in this study are summarized in Table 1. The present studies were reviewed and approved by the institutional review board and all were performed in accordance with relevant guidelines and regulations. The histological subtype of tumors was confirmed by a board-certified pathologist with expertise in thoracic malignancies.
Table 1: Correlations among NSCLC genomic markers and PD-L1. Wilcoxon rank-sum test was used for generating the P-values
P-value |
q-Value* |
PD-L1(TPS+) |
|
|---|---|---|---|
EGFR/PD-L1 |
0.0025 |
0.01 |
continuous |
0.0137 |
0.05 |
5% |
|
0.002 |
0.009 |
20% |
|
0.0003 |
0.001 |
50% |
|
MET/PD-L1 |
0.004 |
0.01 |
continuous |
0.01 |
0.05 |
5% |
|
0.0006 |
0.001 |
20% |
|
0.01 |
0.05 |
50% |
|
TP53/PD-L1 |
<0.0001 |
<0.0001 |
continuous |
0.0004 |
0.001 |
5% |
|
0.0003 |
0.001 |
20% |
|
0.007 |
0.01 |
50% |
|
KRAS+TP53/PD-L1 |
0.0002 |
0.0008 |
continuous |
0.001 |
0.01 |
5% |
|
0.0002 |
0.001 |
50% |
|
EGFR/TP53 |
0.0002 |
0.001 |
N/A |
KRAS/PD-L1 |
No Correlation |
||
*P-value after adjusting for multiple testing.
+Tumor proportion score.
Formalin 7μm fixed paraffin embedded sections were examined by a US-certified pathologist for tumor content, and circled for each patient. One slide was used for FISH testing. Four to six consecutive sections were scrapped by a trained licensed clinical technologist for DNA extraction. Only samples with > 20% tumor were used for testing.
PD1/PD-L1 IHC
PD-L1 expression on 397 lung cancer samples was evaluated with IHC using the SP142 clone and standard immunohistochemistry procedure. The tumor proportion score (TPS) was determined by calculating the percentage of tumor cells showing partial or complete membrane staining at any intensity. PD-L1 expression in tumor cells was considered positive if ≥1% of tumor cells had membranous staining of any intensity and high if ≥50%. Biopsies were reviewed and scored by trained/certified pathologists at NeoGenomics Laboratories. Membranous PD-L1 expression on tumor cells was defined by PD-L1 expression cut-offs of ≥50% (high) and 1%–49% (positive) [6].
Next generation sequencing analysis
Lung cancer samples from 397 patients were also sequenced using next generation sequencing (NGS) for mutations in TP53, KRAS, and EGFR. DNA was sequenced using Illumina NGS protocols, including Illumina TruSeq library preparation, Illumina sample indexing, and Illumina synthesis by sequencing (SBS) protocols as recommended by the Illumina (San Diego, CA, USA). In brief, tumor DNA was amplified using either TruSeq kit or custom primers, and amplification products were confirmed with gel electrophoresis using a 2% agarose E-gel (Thermofisher, Carlsbad, CA, USA). Samples were indexed and pooled. Libraries were then loaded on to an Illumina MiSeq (Illumina, San Diego, CA, USA) or Nextseq Instrument for SBS using 150 bp Illumina sequencing kit with Illumina midoutput flow cells. An experiment sheet was generated using Illumina Experiment Manager for each sequencing run. MiSeq Reporter was used for alignment and variant calling using the proper panel bed/manifest file. Exons 2, 3, and 4 of KRAS were sequenced. For EGFR, we sequenced exons 3, 7, 15, and 18–21. Exons 1 and 3-7 of TP53 were sequenced. The primers for targeted sequencing covered approximately 50 nucleotides from each side of each exon. Sequencing and library quality were assessed for every run using MiSeq reporter, which calculates amplicon read coverage per sample and uniformity of coverage. Positive and negative control samples were also sequenced in parallel with each run to confirm the sensitivity and specificity of each run. Overall sequencing quality was also assessed with MiSeq Reporter software. The average sequencing coverage across the entire coding regions was 10,000 in 94% of the sequenced amplicons. Only clinically significant and biologically relevant mutations were considered. Mutations of unknown significance were not considered.
MET amplification
In this retrospective study, tissue sampled collected from 471 patients with various tumors, including 397 lung cancers, were tested for MET gene amplification by FISH by using a MET (7q31) probe and centromere 7 as a control (Agilent, Santa Clara, CA). Signals were quantified and ratios were calculated.
Statistics
Standard statistical tests were used to evaluate correlations between variables including: Chi-square and Kruskal-Wallis test.
Author contributions
Study conception and design – MA, SA, VF; Acquisition of data – SS, WA, SJ, WC; Analysis and interpretation of data – MA, VF, FB, SA; Drafting of manuscript – MA, FB, SA; Critical revision – FB, SA, VF.
CONFLICTS OF INTEREST
Maher Albitar, Wanlong Ma, Vincent Funari, Forrest Blocker, and Sally Agersborg are employed and own stocks in NeoGenomics.
FUNDING
All funding was provided by NeoGenomics Laboratories.
Ethical approval and informed consent
Work was performed after obtaining IRB approval.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017; 67:7-30.
2. Kobayashi K, Hagiwara K. Epidermal growth factor receptor (EGFR) mutation and personalized therapy in advanced nonsmall cell lung cancer (NSCLC). Target Oncol. 2013; 8:27-33.
3. Azzoli CG, Temin S, Aliff T, Baker S Jr, Brahmer J, Johnson DH, Laskin JL, Masters G, Milton D, Nordquist L, Pao W, Pfister DG, Piantadosi S, et al, and American Society of Clinical Oncology. 2011 focused update of 2009 american society of clinical oncology clinical practice guideline update on chemotherapy for stage IV non–small-cell lung cancer. J Clin Oncol. 2011; 29:3825-31.
4. Ji M, Liu Y, Li Q, Li X, Ning Z, Zhao W, Shi H, Jiang J, Wu C. PD-1/PD-L1 expression in non-small-cell lung cancer and its correlation with EGFR/KRAS mutations. Cancer Biol Ther. 2016; 17:407-13.
5. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012; 366:2455–65.
6. Herbst RS, Baas P, Kim DW, Felip E, Pérez-Gracia JL, Han JY, Molina J, Kim JH, Arvis CD, Ahn MJ, Majem M, Fidler MJ, de Castro G Jr, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. The Lancet. 2016; 387:1540-50.
7. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, Waterhouse D, Ready N, Gainor J, et al. Nivolumab versus docetaxel in advanced squamous-cell non–small-cell lung cancer. N Engl J Med. 2015; 373:123–35.
8. Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, Gadgeel SM, Hida T, Kowalski DM, Dols MC, Cortinovis DL, Leach J, Polikoff J, et al, and OAK Study Group. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. The Lancet. 2017; 389:255-65.
9. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, Carcereny E, Ahn MJ, Felip E, et al, and KEYNOTE-001 Investigators. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015; 372:2018–28.
10. Wang X, Teng F, Kong L, Yu J. PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther. 2016; 9:5023–39.
11. Yang H, Chen H, Luo S, Li L, Zhou S, Shen R, Lin H, Xie X. The correlation between programmed death-ligand 1 expression and driver gene mutations in NSCLC. Oncotarget. 2017; 8:23517-28. https://doi.org/10.18632/oncotarget.15627.
12. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, Kohrt HE, Horn L, Lawrence DP, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014; 515:563-7.
13. Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, Park K, Smith D, Artal-Cortes A, Lewanski C, Braiteh F, Waterkamp D, He P, et al, and POPLAR Study Group. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. The Lancet. 2016; 387:1837-46.
14. D’Incecco A, Andreozzi M, Ludovini V, Rossi E, Capodanno A, Landi L, Tibaldi C, Minuti G, Salvini J, Coppi E, Chella A, Fontanini G, Filice ME, et al. PD-1 and PD-L1 expression in molecularly selected non-small-cell lung cancer patients. Br J Cancer. 2015; 112:95–102.
15. Yang CY, Lin MW, Chang YL, Wu CT, Yang PC. Programmed cell death-ligand 1 expression in surgically resected stage I pulmonary adenocarcinoma and its correlation with driver mutations and clinical outcomes. Eur J Cancer. 2014; 50:1361-9.
16. Roengvoraphoj M, Tsongalis GJ, Dragnev KH, Rigas JR. Epidermal growth factor receptor tyrosine kinase inhibitors as initial therapy for non-small cell lung cancer: Focus on epidermal growth factor receptor mutation testing and mutation-positive patients. Cancer Treat Rev. 2013; 39:839-50.
17. Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor–resistant disease. J Clin Oncol. 2013; 31:1070-80.
18. Helena AY, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M, Riely GJ. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clinical Cancer Research. 2013; 19:2240-7.
19. Van Der Steen N, Pauwels P, Gil-Bazo I, Castañon E, Raez L, Cappuzzo F, Rolfo C. cMET in NSCLC: Can we cut off the head of the hydra? from the pathway to the resistance. Cancers. 2015; 7:556-73.
20. Cipriani NA, Abidoye OO, Vokes E, Salgia R. MET as a target for treatment of chest tumors. Lung Cancer. 2009; 63:169-79.
21. Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: Targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008; 7:504–16.
22. Schuler MH, Berardi R, Lim WT, Geel RV, Jonge MJD, Bauer TM, Azaro A, Gottfried M, Han JY, Lee DH, Wollner M, Hong DS, Vogel A, et al. Phase (Ph) I study of the safety and efficacy of the cMET inhibitor capmatinib (INC280) in patients (pts) with advanced cMET+ non-small cell lung cancer (NSCLC). J Clin Oncol. 2016; 34:9067.
23. Malhotra J, Jabbour SK, Aisner J. Current state of immunotherapy for non-small cell lung cancer. Transl Lung Cancer Res. 2017; 6:196–211.
24. Viticchiè G, Muller PA. c-MET and other cell surface molecules: Interaction, activation and functional consequences. Biomedicines. 2015; 3:46-70.
25. Organ SL, Tsao MS. An overview of the c-MET signaling pathway. Ther Adv Med Oncol. 2011; 3:S7-S19.
26. Solomon H, Brosh R, Buganim Y, Rotter V. Inactivation of the p53 tumor suppressor gene and activation of the RAS oncogene: Cooperative events in tumorigenesis. Discov Med. 2010; 9:448-54.
27. Beau-Faller M, Ruppert AM, Voegeli AC, Neuville A, Meyer N, Guerin E, Legrain M, Mennecier B, Wihlm JM, Massard G, Quoix E, Oudet P, Gaub MP. MET gene copy number in non-small cell lung cancer: Molecular analysis in a targeted tyrosine kinase inhibitor naive cohort. J Thorac Oncol. 2008; 3:331-9.
28. Cappuzzo F, Jänne PA, Skokan M, Finocchiaro G, Rossi E, Ligorio C, Zucali PA, Terracciano L, Toschi L, Roncalli M, Destro A, Incarbone M, Alloisio M, et al. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann Oncol. 2009; 20:298–304.
29. Schildhaus HU, Schultheis AM, Rüschoff J, Binot E, Merkelbach-Bruse S, Fassunke J, Schulte W, Ko YD, Schlesinger A, Bos M, Gardizi M, Engel-Riedel W, Brockmann M, et al. MET amplification status in therapy-naive adeno-and squamous cell carcinomas of the lung. Clinical Cancer Research. 2015; 21:907-15.
30. Lizotte PH, Ivanova EV, Awad MM, Jones RE, Keogh L, Liu H, Dries R, Almonte C, Herter-Sprie GS, Santos A, Feeney NB, Paweletz CP, Kulkarni MM, et al. Multiparametric profiling of non–small-cell lung cancers reveals distinct immunophenotypes. JCI Insight. 2016; 1:e89014.
31. Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, Piotrowska Z, Huynh TG, Zhao L, Fulton L, Schultz KR, Howe E, Farago AF, Sullivan RJ, et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non–small cell lung cancer: A retrospective analysis. Clinical Cancer Research. 2016; 22:4585-93.
32. Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, Mikse OR, Cherniack AD, Beauchamp EM, Pugh TJ, Wilkerson MD, Fecci PE, Butaney M, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013; 3:1355-63.
33. Mu CY, Huang JA, Chen Y, Chen C, Zhang XG. High expression of PD-L1 in lung cancer may contribute to poor prognosis and tumor cells immune escape through suppressing tumor infiltrating dendritic cells maturation. Med Oncol. 2011; 28:682-8.
34. Zhang Y, Wang L, Li Y, Pan Y, Wang R, Hu H, Li H, Luo X, Ye T, Sun Y, Chen H. Protein expression of programmed death 1 ligand 1 and ligand 2 independently predict poor prognosis in surgically resected lung adenocarcinoma. Onco Targets Ther. 2014; 7:567–73.
35. Kerr KM, Tsao MS, Nicholson AG, Yatabe Y, Wistuba II, Hirsch FR, and IASLC Pathology Committee. Programmed Death-Ligand 1 Immunohistochemistry in Lung Cancer: in what state is this art? J Thorac Oncol. 2015; 10:985–89.
36. Rimm DL, Han G, Taube JM, Yi ES, Bridge JA, Flieder DB, Homer R, West WW, Wu H, Roden AC, Fujimoto J, Yu H, Anders R, et al. A prospective, multi-institutional, pathologist-based assessment of 4 immunohistochemistry assays for pd-l1 expression in non–small cell lung cancer. JAMA Oncol. 2017; 3:1051–58.
37. Koh J, Go H, Keam B, Kim MY, Nam SJ, Kim TM, Lee SH, Min HS, Kim YT, Kim DW, Jeon YK, Chung DH. Clinicopathologic analysis of programmed cell death-1 and programmed cell death-ligand 1 and 2 expressions in pulmonary adenocarcinoma: comparison with histology and driver oncogenic alteration status. Mod Pathol. 2015; 28:1154–66.
38. Rangachari D, VanderLaan PA, Shea M, Le X, Huberman MS, Kobayashi SS, Costa DB. Correlation between classic driver oncogene mutations in EGFR, ALK, or ROS1 and 22C3–PD-L1≥ 50% expression in lung adenocarcinoma. J Thorac Oncol. 2017; 12:878-83.
39. Boch C, Kollmeier J, Roth A, Stephan-Falkenau S, Misch D, Grüning W, Bauer TT, Mairinger T. The frequency of EGFR and KRAS mutations in non-small cell lung cancer (NSCLC): Routine screening data for central europe from a cohort study. BMJ Open. 2013; 3:e002560.
40. Martín Martorell P, Huerta M, Compañ Quilis A, Abellán R, Seda E, Blesa S, Chaves FJ, Dualde Beltrán D, Roselló Keränen S, Franco J, Insa A. Coexistence of EGFR, KRAS, BRAF, and PIK3CA mutations and ALK rearrangement in a comprehensive cohort of 326 consecutive spanish nonsquamous NSCLC patients. Clin Lung Cancer. 2017; 18:e395-e402.
41. Wu CC, Hsu HY, Liu HP, Chang JWC, Chen YT, Hsieh WY, Hsieh JJ, Hsieh MS, Chen YR, Huang SF. Reversed mutation rates of KRAS and EGFR genes in adenocarcinoma of the lung in Taiwan and their implications. Cancer. 2008; 113:3199-208.
42. Sumimoto H, Takano A, Teramoto K, Daigo Y. RAS–mitogen-activated protein kinase signal is required for enhanced PD-L1 expression in human lung cancers. PLoS One. 2016; 11:e0166626.
43. Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, Behrens C, Kadara H, Parra ER, Canales JR, Zhang J, Giri U, Gudikote J, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015; 5:860-77.
44. Iwakawa R, Kohno T, Anami Y, Noguchi M, Suzuki K, Matsuno Y, Mishima K, Nishikawa R, Tashiro F, Yokota J. Association of p16 homozygous deletions with clinicopathologic characteristics and EGFR/KRAS/p53 mutations in lung adenocarcinoma. Clinical Cancer Research. 2008; 14:3746-53.
45. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014; 511:543-50.
46. Millar FR, Janes SM, Giangreco A. Epithelial cell migration as a potential therapeutic target in early lung cancer. Eur Respir Rev. 2017; 26:160069.
47. Roger L, Gadea G, Roux P. Control of cell migration: A tumour suppressor function for p53? Biol Cell. 2006; 98:141-52.
48. Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y, Araujo L, Carbone DP, Shilo K, Giri DK, Kelnar K, Martin D, Komaki R, et al. PDL1 Regulation by p53 via miR-34. J Natl Cancer Inst. 2015; 108: djv303.
49. Menendez D, Shatz M, Resnick MA. Interactions between the tumor suppressor p53 and immune responses. Curr Opin Oncol. 2013; 25:85-92.