
INTRODUCTION
CBF-AML, including AML with t(8;21) and AML with inv(16)/t(16;16), accounts for approximately 25% of pediatric and 15% of adult de novo AML patients. Compared to other AML subsets, CBF-AML is considered to have a good prognosis. Both alterations result in disruption of genes encoding subunits of the CBF (i.e. RUNX1 and CBFB), a heterodimeric transcription factor complex required for normal hematopoiesis [1]. Importantly, experiences from murine models [2], as well as the existence of preleukemic cells harboring a CBF rearrangement in healthy individuals [3, 4], have demonstrated that CBF disruption is insufficient to induce leukemia. CBF-AML is therefore considered as a model of multistep pathogenesis. Evidences supporting this model have been generated by the high frequency of cooperative events at time of diagnosis. Notably, mutations in genes encoding tyrosine kinase pathways effectors (especially KIT, FLT3 and RAS mutations) are found in up to 80% of CBF-AML patients [5–7]. Additional chromosomal aberrations are detected in approximately 70% of patients with t(8;21)-AML and 40% of patients with inv(16)-AML by conventional karyotype [5, 8–10]. These aberrations are nonrandom events and some of them are extremely rare in non-CBF-AML. In this context, the identification of recurrent events involved in CBF-AML pathophysiology and heterogeneity remains of great interest. We report here the SNP-array profiling of a large and well-annotated cohort of pediatric and adult patients with CBF-AML and the identification of new recurrent lesions in this particular subtype of leukemia.
RESULTS
CBF-AML genomes are characterized by a limited number of SNP-array-lesions
SNP-array analysis of 116 t(8;21)-AML and 82 inv(16)-AML revealed a total of 319 lesions, including 277 copy-number abnormalities (CNAs; 187 losses and 90 gains; median size: 26.1 Mb [range: 26 kb-155.1 Mb]) and 42 CN-LOH (Supplementary Table 1). Overall, 97 (84%) patients with t(8;21)-AML and 55 (64%) patients with inv(16)-AML had at least one genomic aberration (CNA and/or CN-LOH). There was no significant difference in the number of lesions between adult and pediatric patients (Supplementary Table 2) arguing for similar diseases as previously described [6]. Recurrent focal lesions associated with t(8;21) and inv(16) breakpoints were common events, occurring in 27 (14%) CBF-AML cases especially in the inv(16) subtype (22% vs. 7%, p=0.005). Considering them as part of the primary event, t(8;21) or inv(16), breakpoint-associated lesions (accounting for 41 of the 319 identified lesions) were excluded for subsequent descriptions. Finally, CBF-AML genomes exhibited a mean of 1.40 SNP-array aberrations per case (range: 0-7)(Table 1). CNAs were more numerous in t(8;21)-AML than in inv(16)-AML, mostly due to genomic deletions (0.98 vs. 0.44 losses/case respectively; p<0.001). Neither the presence of SNP-array lesions nor the number of lesions was a predictor of outcome (Supplementary Figure 1).
Table 1: Mean number of SNP-array lesions per CBF AML case according to genetic subtype
CBF AML |
t(8;21) AML |
inv(16) AML |
p-value |
|
|---|---|---|---|---|
Patients, n |
198 |
116 |
82 |
|
Number of CNAs†, mean (range) |
1.19 (0-6) |
1.35 (0-5) |
0.96 (0-6) |
0.004 * |
Gains†, mean (range) |
0.43 (0-4) |
0.37 (0-4) |
0.52 (0-3) |
0.084 |
Losses†, mean (range) |
0.76 (0-4) |
0.98 (0-3) |
0.44 (0-4) |
<0.001 * |
Number of CN-LOH†, mean (range) |
0.21 (0-6) |
0.17 (0-6) |
0.27 (0-4) |
0.372 |
Breakpoint lesions, mean (range) |
0.21 (0-2) |
0.09 (0-2) |
0.38 (0-2) |
0.002 * |
Total CNAs/CN-LOH†, mean (range) |
1.40 (0-7) |
1.53 (0-7) |
1.23 (0-7) |
0.020 * |
CNA: copy number abnormality; CN-LOH: copy neutral-loss of heterozygosity.
† excluding breakpoint-associated lesions.
SNP-array karyotyping in CBF-AML shows nonrandom copy number changes
Recurrent CNAs are listed in Table 2. Considering lesions that are non-associated with breakpoints, a large proportion of detected CNAs were broad aberrations or involved whole chromosomes (Figure 1) [11]. Most of them appeared to be nonrandom events and are usually seen by conventional karyotype [1]. Among t(8;21)-AML patients, loss of a sex chromosome (LOS) was by far the most common event, occurring in 62 (53%) patients, followed by interstitial deletion of the long arm of chromosome 9 [del(9q)] in 15 (13%) patients (p<0.001). Both aberrations were virtually absent among inv(16)-AML patients. All but one case with del(9q) shared a minimal deleted region (MRD) of 6.1Mb containing 19 genes (Supplementary Figure 2) in which TLE1 and TLE4 have been the most studied [12, 13]. By contrast, +22 was restricted to inv(16)-AML and occurred in 11 (13%) patients. Trisomy 8 and interstitial deletion of the long arm of chromosome 7 [del(7q)] were found in both genetic subtypes. Trisomy 8 was observed in 8 (10%) cases with inv(16) and 6 (5%) cases with t(8;21). Gain of the long arm of chromosome 8 (+8q) was seen is 2 additional cases with inv(16) (2 other cases with t(8;21) had +8q related to the rarely described duplication of the derivate chromosome der(21) t(8;21) [14]; Supplementary Figure 3). Del(7q) was found in 20 (10%) patients, including 10 (9%) cases with t(8;21) and 10 (12%) cases with inv(16). All cases with del(7q), whatever their genetic subtype t(8;21) or inv(16), shared a MDR of 4.2 Mb containing 71 genes in which the 2 epigenetics-related genes EZH2 and KMT2C (MLL3) were the more relevant and have already been studied by others [15–17] (Supplementary Figure 4). Overall, we did not find any association between these recurrent genetic aberrations and clinical outcome (Supplementary Figure 5). Other broad recurrent aberrations included a previously undescribed deletion 2q which appeared to be restricted to patients with t(8;21)-AML (n=5) as well as gains 1q (n=2), 4q (n=3) and 13q (n=3). All patients with gain(13q) had also del(7q) in SNP-array while conventional karyotype showed additional material of unknown origin on the long arm of chromosome 7 [add(7q)]. Whole chromosome 13 painting performed in one of them by fluorescent in situ hybridization confirmed transfer of material from chromosome 13 to chromosome 7 leading to both gain(13) and del(7q) (Supplementary Figure 6).
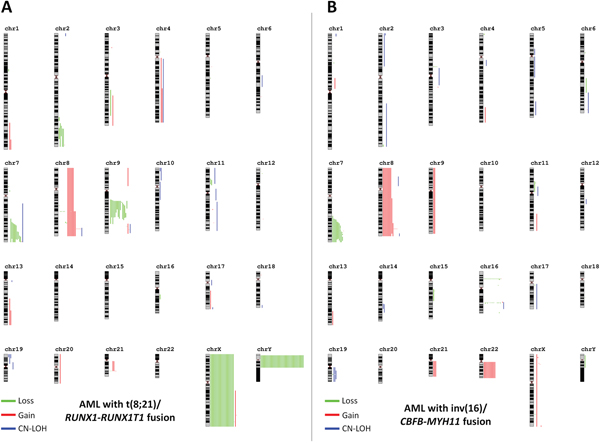
Figure 1: Karyograms of detected SNP-array lesions by genetic subtype. Each vertical line represents 1 event in 1 patient. Gains are in red, losses in green and CN-LOH in blue. Part (A) shows cases with t(8;21)-AML and part (B) shows cases with inv(16)-AML. Schematic representations were obtained using the Genomic Recurrent Event ViEwer (GREVE) web tool (http://www.well.ox.ac.uk/GREVE) [11].
Table 2: Recurrent copy number abnormalities in CBF AML patients
Recurrent SNP-array lesions |
Start ¥ |
End ¥ |
Size |
Gene count |
CBF AML |
inv(16) |
t(8;21) |
p-value |
|---|---|---|---|---|---|---|---|---|
gain(1)(q42.13q44) |
227 833 996 |
249 224 684 |
21391 kb |
197 genes |
2 (1%) |
0 (0%) |
2 (2%) |
0.512 |
del(2)(q33.2q35) |
204 563 014 |
220 260 561 |
15698 kb |
127 genes |
5 (3%) |
0 (0%) |
5 (4%) |
0.078 |
del(3)(p13) |
71 194 153 |
71 523 438 |
329 kb |
FOXP1 |
4 (2%) |
4 (5%) |
0 (0%) |
0.028 * |
gain(4)(q32.1q35.2) |
158 379 102 |
190 957 473 |
32578 kb |
124 genes |
3 (2%) |
1 (1%) |
2 (2%) |
1.000 |
del(7)(q35q36.1) |
147 660 930 |
151 908 681 |
4248 kb |
71 genes including EZH2 and KMT2C |
20 (10%) |
10 (12%) |
10 (9%) |
0.476 |
del(8)(q24.11) |
117 823 216 |
117 914 100 |
91 kb |
RAD21, RAD21-AS1, MIR3610 |
2 (1%) |
0 (0%) |
2 (2%) |
0.512 |
focal gain(8)(q24.21)† |
130 586 319 |
130 697 500 |
111 kb |
CCDC26 |
9 (5%) |
4 (5%) |
5 (4%) |
1.000 |
gain(8)(q24.11q24.3)† |
118 660 515 |
140 821 810 |
22161 kb |
92 genes including CCDC26 and MYC |
2 (1%) |
2 (2%) |
0 (0%) |
0.170 |
trisomy 8† |
whole chromosome |
- |
- |
14 (7%) |
8 (10%) |
6 (5%) |
0.264 |
|
del(9)(q21.2q21.33) |
80 806 493 |
86 951 615 |
6145 kb |
19 genes including TLE1 and TLE4 |
14 (7%) |
0 (0%) |
14 (12%) |
<0.001 * |
trisomy 9 |
whole chromosome |
- |
- |
2 (1%) |
2 (2%) |
0 (0%) |
0.170 |
|
del(11)(p13) |
31 972 741 |
32 633 735 |
661 kb |
RCN1, WT1, WT1-AS, EIF3M, CCDC73 |
4 (2%) |
2 (2%) |
2 (2%) |
1.000 |
gain(13)(q31.1q34) |
85 412 329 |
115 107 733 |
29695 kb |
135 genes |
3 (2%) |
1 (1%) |
2 (2%) |
1.000 |
del(17)(q11.2) |
29 357 586 |
29 520 056 |
162 kb |
MIR4733, NF1 |
2 (1%) |
2 (2%) |
0 (0%) |
0.170 |
trisomy 21 |
whole chromosome |
- |
- |
3 (2%) |
3 (4%) |
0 (0%) |
0.070 |
|
trisomy 22 |
whole chromosome |
- |
- |
11 (6%) |
11 (13%) |
0 (0%) |
<0.001 * |
|
del(X)(q26.1) |
129 129 272 |
129 211 954 |
83 kb |
BCORL1, ELF4 |
2 (1%) |
2 (2%) |
0 (0%) |
0.170 |
loss X or Y |
63 (32%) |
1 (1%) |
62 (53%) |
<0.001 * |
||||
loss X§ |
whole chromosome |
- |
- |
22 (24%) |
0 (0%) |
22 (42%) |
<0.001 * |
|
loss Y§ |
whole chromosome |
- |
- |
41 (39%) |
1 (2%) |
40 (63%) |
<0.001 * |
|
Breakpoint-associated lesions |
||||||||
del(8)(q21.3) |
93 096 598 |
93 128 271 |
32 kb |
RUNX1T1 |
5 (3%) |
0 (0%) |
5 (4%) |
0.078 |
del(16)(p13.11) |
15 828 494 |
16 056 322 |
228 kb |
MYH11, FOPNL, ABCC1 |
16 (8%) |
16 (20%) |
0 (0%) |
<0.001 * |
gain(16)(p13.11) |
15 725 039 |
15 814 747 |
90 kb |
KIAA0430, NDE1, MIR484, MYH11 |
1 (1%) |
1 (1%) |
0 (0%) |
0.414 |
del(16)(q22.1) |
67 132 654 |
67 176 123 |
43 kb |
CBFB, C16orf70 |
13 (7%) |
13 (16%) |
0 (0%) |
<0.001 * |
gain(16)(q21q22.1) |
65 352 347 |
67 131 638 |
1779 kb |
23 genes including CBFB |
1 (1%) |
1 (1%) |
0 (0%) |
0.414 |
del(21)(q22.12) |
36 183 871 |
36 210 100 |
26 kb |
RUNX1 |
3 (2%) |
0 (0%) |
3 (3%) |
0.268 |
gain(21)(q22.12) |
36 355 481 |
36 423 085 |
68 kb |
RUNX1, RUNX1-IT1 |
2 (1%) |
0 (0%) |
2 (2%) |
0.512 |
+der(21) t(8;21)(q22;q22) |
whole chromosome |
- |
- |
2 (1%) |
0 (0%) |
2 (2%) |
0.512 |
|
* p-value < 0.05.
† Partial chromosomal lesions were considered separately from abnormalities involving the whole chromosome. Gains 8q related to der(21) t(8;21) duplication (n=2) were considered apart from other lesions.
¥ Chromosomal locations were obtained from the Human Feb. 2009 (GRCh37/hg19) assembly of the UCSC Genome Browser (http://genome.ucsc.edu/).
§ Proportions are given within the female population for loss of X and the male population for loss of Y.
SNP-array identifies recurrent target genes involved in CBF-AML pathogenesis
One of the most common alterations was copy number gains at locus 8q24, which concerned 27 (13.5%) CBF-AML patients through several mechanisms (Supplementary Figure 7): 14 (7%) patients had +8, 4 (2%) patients had broad gains of the long arm of the chromosome 8 and the 9 (4.5%) remaining patients harbored focal gains that contained a single putative gene referred to as CCDC26 which has been recently linked to myeloid leukemia cell growth [18]. FOXP1 focal deletions were identified in 4 patients, all with inv(16)-AML. Interestingly, subsequent sequencing of all coding exons of FOXP1 by high-throughput sequencing (HTS) in the whole cohort identified 2 other inv(16) patients with FOXP1 truncating mutations (Figure 2). On the other hand, no FOXP1 deletion or mutation was found in t(8;21) patients.
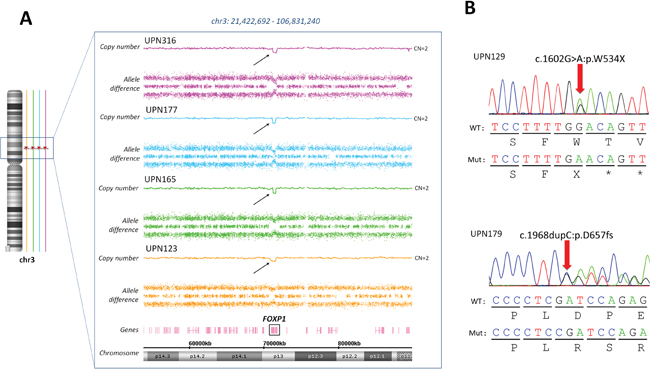
Figure 2: FOXP1 aberrations in inv(16) AML patients. (A) Focal deletions targeting the FOXP1 gene were detected in 4 patients harboring an inv(16). (B) Targeted sequencing of FOXP1 revealed to other inv(16) patients with FOXP1 truncating mutations.
Other recurrent focal aberrations included deletions of WT1 (n=4), BCORL1 (n=2), the cohesin core component RAD21 (n=2) and the RAS pathway modulator NF1 (n=2) whose mutations are recurrent in CBF-AML [6, 7]. Additionally, some focal unique CNAs involved highly relevant genes such as deletions of the transcription factors IKZF1 (n=1) and ETV6 (n=1), gain of MYB (n=1), deletions of the cohesin regulator PDS5A (n=1) or the potential tumor suppressor MGA (n=1). A gain of CNOT4, which amplification is expected to enhance JAK/STAT signaling [19], was seen in one patient as the sole secondary abnormality.
ZBTB7A is a target of CN-LOH in t(8;21)-AML
The short arm of chromosome 19 was recurrently affected by CN-LOH in 3 patients with restriction to the t(8;21) subtype. The minimal affected region was about 6 Mb and contained 209 genes. Notably, this region was previously reported by Kühn et al. in 2 t(8;21)-AML patients with paired samples [17]. This region contained the ZBTB7A gene recently described as highly mutated in patients with t(8;21)-AML but not in patients with inv(16)-AML [20, 21]. In order to validate ZBTB7A as a target of CN-LOH in 19p, we performed HTS of all coding exons of ZBTB7A in the whole cohort. We identified 23 ZBTB7A mutations in 19 patients (4 patients had 2 mutations) restricted to the t(8;21) subgroup (16%; Supplementary Table 3). Three patients harbored the same frameshift mutation at alanine 175. All patients with 19p CN-LOH harbored concomitant ZBTB7A mutation validating ZBTB7A as the target of this aberration (Figure 3A). Missense mutations clustered in the N-terminal BTB domain while frameshift mutations occurred through the whole protein as previously described by others [20, 21] (Figure 3B). We did not identify any association or exclusion with other known mutations (Figure 3C). There was no difference in age, sex or white blood cell count according the ZBTB7A mutational status. Finally, there was no impact of ZBTB7A mutations on overall survival (OS) and relapse-free survival (RFS) in t(8;21)-AML patients (Supplementary Figure 8).
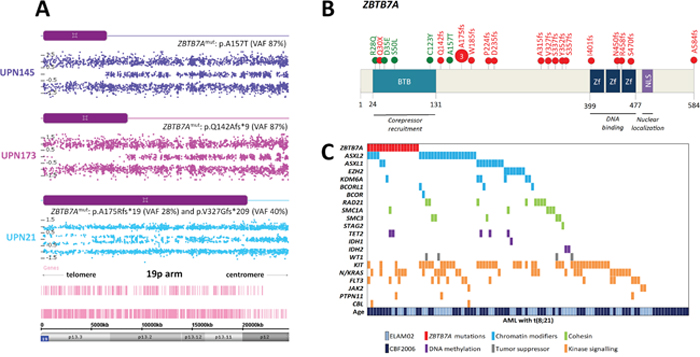
Figure 3: ZBTB7A aberrations in AML patients with t(8;21). (A) Concomitant mutation and CN-LOH in 3 patients with t(8;21)-AML. (B) ZBTB7A protein (NP_056982.1) and identified mutations (red = truncating; green = missense). BTB: BR-C ttk and bab; NLS: nuclear localization sequence; Zf: zinc finger. (C) Genomic landscape of t(8;21)-AML including ZBTB7A mutations.
CCDC26 disruption is likely to be associated with aberrant tyrosine kinase signaling in CBF-AML
Nine (4.5%) patients harbored focal gains confined to the CCDC26 locus (Figure 4). Interestingly, it has been recently suggested that CCDC26 could control myeloid leukemia cell growth through regulation of KIT expression [18]. Considering that hyperactive KIT mutations are highly prevalent in CBF-AML (about 35% of cases)[6] and that CCDC26 focal amplification (CCDC26amp) is found more frequently in CBF-AML than in non CBF-AML [22, 23], these findings could reveal a new lesion associated with aberrant tyrosine kinase pathway activation in CBF-AML patients. Importantly, all but one patient harboring CCDC26amp were KIT wild-type. In order to explore this hypothesis, expressions of the KIT receptor and the phosphorylated downstream effector AKT (AKTp473) were estimated on diagnostic blast cells by flow cytometry in patients with CCDC26amp (n=3). Results were compared with blast cells isolated from patient with normal CCDC26 copy number (CCDC26nor) and KIT mutation (KITmut; n=4), FLT3-ITD (n=2) or KIT/FLT3 wild-type (KITwt/FLT3wt; n=3). All patients were NRAS and KRAS wild-type (RASwt). Overall, there was no correlation between KIT expression and CCDC26 copy number or KIT/FLT3 mutational status. Median expression of AKTp473 showed a trend of higher expression in CCDC26amp-cells (+24%) compared with CCDC26nor/KITwt/FLT3wt/RASwt-cells (Figure 5). Although we could not directly linked AKTp473 and CCDC26amp, these data suggest an underlying mechanism leading to the activation of tyrosine kinase pathway in cells harboring CCDC26amp. On the other hand, median expression of AKTp473 clearly increased in CCDC26nor/KITwt/FLT3-ITD/RASwt-cells (+107%) compared with CCDC26nor/KITwt/FLT3wt/RASwt-cells while it was not observed for CCDC26nor/KITmut/FLT3wt/RASwt-cells.
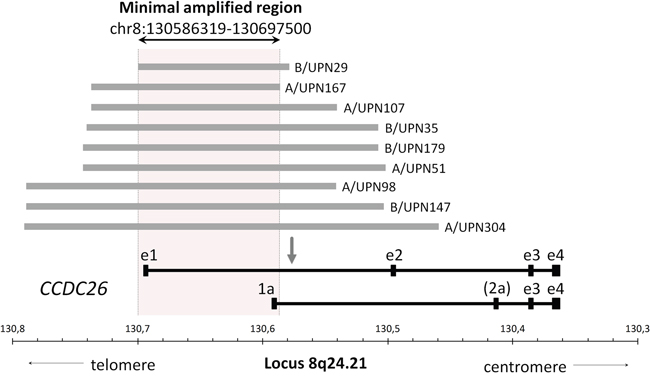
Figure 4: Focal CCDC26 amplifications. Horizontal grey lines illustrate focal recurrent amplifications detected in 9 CBF AML genomes across the CCDC26 locus. The area inside the dotted lines represents the minimal amplified region shared by all patients. Major variants of CCDC26 mRNA are shown in black below the figure. Exons are indicated by boxes. The long transcript consists of four exons (from e1 to e4) and the short transcripts comprise 3 or 4 exons (from 1a to e4, more or less exon 2a). The arrow shows the retrovirus insertion site defined by Yin et al [37].
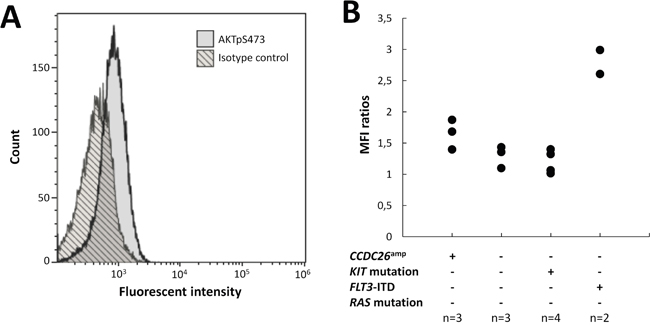
Figure 5: AKTp473 expression in blasts from CBF-AML patients. (A) AKTpS473 expression level in a patient with CCDC26 focal amplification (CCDC26amp). (B) AKTpS473 mean fluorescent intensity ratios according to CCDC26 focal amplification and KIT/FTL3/RAS mutational status. Ratios were calculated as (MFI of blast cells)/(MFI of isotype IgG control).
DISCUSSION
SNP-array karyotyping of 198 patients with CBF-AML highlight great differences between t(8;21)-AML and inv(16)-AML as described in most recent studies focused on cooperating mutations [6, 7, 20, 21, 24–26]. Notably, these studies identified frequent mutations in genes encoding epigenetic regulators and cohesin complex in t(8;21)-AML while they were absent in inv(16)-AML [6, 7, 20, 21]. Overall, we found more CNAs in t(8;21)-AML than in inv(16)-AML, mostly due to genomic deletions. Excluding breakpoint-associated lesions, the most common CNAs included large chromosomal lesions usually seen by conventional cytogenetics including LOS, del(9q), del(7q) and +8 in t(8;21)-AML compared with +22, del(7q), +8, +21 and +9 in inv(16)-AML. While it is clear that these events are nonrandom and contribute to the pathogenesis of CBF-AML, there was no association between clinical outcome and the number of SNP-array lesions nor the presence of these specific aberrations. Remarkably, there was no significant difference in the number of lesions between adult and pediatric patients suggesting they reflect the same entity and could be studied together in further biological experiments.
Concerning recurrent broad deletions, SNP-array led us to identify MDRs on chromosome 9 (involving TLE1 and TLE4) and chromosome 7 (containing EZH2 and KMT2C). In previous experiments, Dayyani et al shown that haploinsufficiency of TLE1 and TLE4 could overcome the negative survival and anti-proliferative effects of RUNX1-RUNXT1 on myeloid progenitors and promote leukemogenesis [13]. Using SNP-array profiling, Kühn et al previously identified a MDR on 7q containing only 4 genes including KMT2C [17]. By sequence analysis of 46 CBF-AML without KMT2C deletion, they identified a single somatic heterozygous frameshift mutation in this gene. More recently, it was shown that KMT2C act as a tumor suppressor gene in AML [16]. Together, these data suggest that KMT2C haploinsufficiency is likely to be a cooperating event in CBF-AML pathogenesis. While the MDR defined by Kühn et al did not contain EZH2, by contrast with the present study, the high frequency of polycomb mutations (ASXL1, ASXL2 and EZH2) in t(8;21)-AML suggest that EZH2 haploinsufficiency could be of interested, at least in t(8;21)-AML patients [6, 27].
Although the number of CNAs was low, our analysis identified recurrent deletions and subsequent mutations in known and potentially new cancer genes. These included deletions in WT1, BCORL1, RAD21, EZH2 or NF1 whose mutations have been recurrently found in CBF-AML patients [6, 7, 20]. Interestingly, we identified FOXP1 aberrations (deletions or truncated mutations) in 7% of patients with inv(16)-AML, arguing for a pathogenic role in this particular subtype. FOXP1 (forkhead box P1) encodes one of the 4 members of the FOXP subfamily of forkhead transcription factors, known to be involved in human malignancies, cell survival and differentiation [28]. FOXP1 has been described as a target of chromosomal translocations and amplifications in B-cell lymphomas and prostate cancer [28]. By contrast, FOXP1 losses have been described in clear cell-type kidney cancer but also rarely in myeloproliferative neoplasms [29] and AML with normal [30] or complex karyotype [31]. FOXP1 has been shown to function as a transcriptional repressor in monocytic differentiation [32]. Thus, it is likely that FOXP1 loss-of-function could contribute to leukemogenesis especially in inv(16)-AML which is most often diagnosed as AML with a monocytic compartment [1].
Three patients with t(8;21)-AML had CN-LOH of 19p leading to homozygous ZBTB7A mutation. ZBTB7A (also known as LRF or Pokemon) encodes a transcription factor of the POK (poxvirus and zinc finger and Krüppel)/ZBTB (zinc finger and broad complex, tramtrack, and bric-a-brac) family involved in the hematopoietic development and the negative regulation of glycolysis [21]. Sequence analysis of ZBTB7A in the whole cohort identified mutations in 16% of t(8;21)-AML while no mutation was found in inv(16)-AML. Our results are in line with other studies previously reporting ZBTB7A mutations in 10% to 23% of t(8;21)-AML [7, 20, 21]. Somatic ZBTB7A mutations are also reported at low frequencies in various solid malignancies [33]. Missense mutations identified in our analysis clustered in the BTB domain which mediates the homodimerization and/or heterodimerization with other proteins [33]. Truncated mutations were distributed through the whole gene leading to the loss of the zinc-finger domain involved in DNA binding and/or nuclear localization signal. Previous experiments from Hartmann et al suggest that ZBTB7A act as a tumor suppressor in t(8;21)-AML [20]. Overexpression of ZBTB7A in Kasumi-1 cells (cell line harboring a t(8;21) rearrangement) leads to reduced proliferation while its haploinsufficiency should result in the induction of glycolysis promoting tumor progression [20, 33].
Finally, we identified CCDC26 (coiled-coil domain containing 26) focal amplifications in 4.5% of the total cohort, consistent with previous SNP-array investigations showing such lesions in 4.7% of CBF-AML genomes [17, 22]. The nature of CCDC26 remains ambiguous but it is more plausible that the CCDC26 locus encodes a long non-coding-RNA [34] involved in tumors, including low-grade gliomas [35] and pancreatic cancer [36]. This locus, also known as RAM (retinoic acid modulator), was initially reported as required for retinoic acid (RA)-induced myeloid differentiation. Retroviral DNA integration into this locus has been shown to generated RA-resistant cells [37]. Interestingly, Hirano et al showed that CCDC26-knockdown resulted in KIT up-regulation and enhanced survival in myeloid leukemia cell lines. First of all, these results appeared conflicting with our data showing CCDC26 amplification in CBF-AML. However, this paradox could be explicable by the fact that CCDC26 amplification does not extend to the whole gene. Partial amplification restricted to exons 1 and 1a could result in CCDC26 disruption leading to an abnormal mRNA structure without any activity or able to interfere with the remaining intact gene [38]. Thus, considering the high frequency class I mutations (especially in KIT, FLT3 and RAS genes) in CBF-AML [6], it is likely that CCDC26 disruption could highlight a new class I aberration leading to increased cell survival and proliferation in leukemia. In order to explore this hypothesis, we studied phosphoAKT expression by flow cytometry in CBF-AML cells harboring CCDC26 focal amplification. Although we were able to study only 3 patients with this lesion, blast cells from patients with CCDC26 disruption showed a subtle increased expression of phosphoAKT compared with blast cells from patients with normal CCDC26 copy number and no class I mutation (KIT, FLT3 and RAS wild-type). However, this was not observed blast cells from 4 KIT-mutated patients suggesting other activated pathways associated with KIT mutations. By contrast phosphoAKT expression were clearly increased in blast cells from 2 patients with FLT3-ITD. Unfortunately, we were not able to study other cytoplasmic effectors such as ERK, SRC or STAT proteins that could be deregulated in leukemia. Also, because we studied a very small number of patients, we were not able to give strong conclusions but our data suggest a mechanism leading to tyrosine kinase signaling in cells with CCDC26 disruption. Of course, further studies are needed to directly link CCDC26 disruption and aberrant tyrosine kinase signaling in CBF-AML.
In conclusion, we defined the landscape of SNP-array lesions in a cohort of 198 adult and pediatric CBF-AML at time of diagnosis. As no cell culture is required, we described the frequency of known cytogenetic abnormalities with an unbiased approach and found no association with clinical outcome. Although, the number of SNP-array lesions appeared very low in CBF-AML, when combining with sequence analyses, we were able to identify recurrent involvement of known and potentially new cancer genes including FOXP1 loss-of-function in inv(16)-AML, ZBTB7A homozygous mutations through CN-LOH in t(8;21)-AML and CCDC26 disruption in both genetic subgroups of CBF-AML. Because of the low frequency of recurrent events, further studies focused on specific genetic subgroups of AML are needed to specify the incidence and the role of these aberrations in leukemogenesis.
MATERIALS AND METHODS
Patients and samples
This study focused on diagnostic bone marrow (BM) samples from 198 CBF-AML patients including 116 AML with t(8;21) and 82 AML with inv(16). The cohort included 125 adults (aged from 18 to 60 years) and 73 children (aged from 1 to 17 years) enrolled in the French trials CBF2006 (ClinicalTrials.gov NCT00428558)[5] and ELAM02 (ClinicalTrials.gov NCT00149162) respectively. Median age was 30 years (range: 1-60). Patient’s characteristics are summarized in Supplementary Table 4. Studies were approved by the Ethics Committee of Nîmes University Hospital and by the Institutional Review Board of the French Regulatory Agency and were conducted in accordance with the Declaration of Helsinki. Details about treatment regimens are provided in Supplementary Figure 9 and Supplementary Figure 10.
SNP-array karyotyping
DNA was extracted from diagnostic cell pellets using the QIAamp Tissue Kit (Qiagen) according to the manufacturer’s instructions. Patient’s genomic DNA was processed and hybridized to Cytoscan HD array (Affymetrix) according to the manufacturer’s protocol. Data were analyzed using the Chromosome Analysis Suite (ChAS) software (Affymetrix). In a first step, only copy number variants with a size over 20 kb including at least 20 consecutive markers as well as CN-LOH over 3 Mb were considered for this analysis. In a second step, we adopted a stringent and conservative algorithm in order to distinguish somatic from constitutional SNP-array lesions. Variations were excluded as known copy number variants if there was more than 50% overlap with variants from the public Database of Genomic Variants. Based on previous studies [39–41], only interstitial CN-LOH over 10 Mb and CN-LOH extending to telomeres were considered to be acquired abnormalities. Remaining CN-LOH were considered as possibly constitutional and then rejected for subsequent analyses. Finally, all CNAs and CN-LOH fulfilling the above criteria were validated by visual inspection and annotated for size, position and location of genes based on the human genome version 19 (hg19) of the UCSC Genome Browser.
Mutational analysis
Considering data from SNP-array karyotyping, target sequencing was performed for all coding exons of FOXP1 (NM_001244810) and ZBTB7A (NM_015898). Libraries were prepared using the Ampliseq system according to the manufacturer’s instruction and run on Personal Genome Machine (PGM, Life Technologies). Raw HTS data from PGM sequencing were processed by Torrent Browser (Life Technologies) and SeqNext (JSI Medical System). The depth of coverage was more than 2000X for both genes. Variants were confirmed by direct Sanger Sequencing as previously described [27]. Frameshift and nonsense variants were always considered as relevant mutations. Single nucleotide variants were retained in the absence of description into public databases of human polymorphisms and effects on protein function were predicted with SIFT and Polyphen-2.
Multiparameter flow cytometry
Diagnostic blast cells were obtained from thawed cryopreserved BM samples after red blood cell lysis. Fixation, permeabilization and staining (with both intracellular and cell surface markers) were performed using the PerFix-no centrifuge assay kit (Beckman Coulter) according to the manufacturer’s instructions. The antibody panel contained: anti-AKTpS473-Vio515 (clone: REA359, Miltenyi Biotec), anti-CD33-PC5.5 (clone D3HL60.251, Beckman Coulter), anti-CD34-PC7 (clone 581, Beckman Coulter), anti-CD117/KIT-APC (clone 104D2D1, Beckman Coulter), anti-CD3-AA750 (clone UCHT1, Beckman Coulter), anti-CD4-PB (clone 13B8.2, Beckman Coulter) and anti-CD45-KO (clone J33, Beckman Coulter). Blast cells were gated as CD45dim, SSClow, CD33+, excluding lymphocytes (CD45bright, SSClow, CD33−), monocytes (CD45int/bright, SSCint, CD33bright) and mature myelomonocytic cells (CD45int, SSChigh, CD33dim/neg). Isotype control (clone REA293, Miltenyi Biotec) was used to better define the threshold of AKTpS473-positive cells. AKTpS473 expression levels were calculated as [mean fluorescent intensity (MFI) of blast cells/MFI of isotype IgG control]. Measurements were performed on a Navios flow cytometer and analyzed with Kaluza software (Beckman-Coulter).
Statistical methods
Failure time data were analyzed and compared after censoring at transplant for patients who received allogeneic stem cell transplantation in first complete remission (CR). RFS and OS were estimated by the Kaplan-Meier method. RFS was estimated taking into account death in first CR for competing risk. Comparisons between patient subgroups were performed by the Mann-Whitney test for continuous variables and by Fisher’s exact test for categorical variables. All statistical tests were performed with the SPSS Statistics software (IBM).
Abbreviations
AML: acute myeloid leukemia; BM: bone marrow; CBF: core binding factor; CNAs: copy-number abnormalities; CN-LOH: copy-neutral losses of heterozygosity; CR: complete remission; HTS: high-throughput sequencing; LOS: loss of a sex chromosome; MFI: mean fluorescent intensity; OS: overall survival; RA: retinoic acid; RFS: relapse-free survival; SNP: single nucleotide polymorphism.
Author contributions
EJ was the principal investigator of the CBF-2006 study. GL was the principal investigator of the ELAM02 study. EJ, NB, NI, HD, GL and AP enrolled patients in the studies. CL prepared samples. ND, EBL, SG, NH, PC and HL performed genetic analysis and analyzed mutational data. CT, IL and WC reviewed all cytogenetic data. OF performed cytologic examination. CR performed flow cytometry. HD, HL and CP created the patient database. KCL ensured database management. ND performed the research and wrote the paper. CP revised the manuscript which was approved by all the authors.
ACKNOWLEDGMENTS
The authors are grateful to Lamya Haddaoui (Tumour Bank for the GOELAMS group) and Christophe Roumier and Olivier Nibourel (Tumour Bank for the ALFA group) for handling, conditioning, and storing patient samples. The work of all clinical research assistants is also acknowledged here.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported by the French National Cancer Institute (PRT-K 2010-285 and PRT-K 2012-043).
REFERENCES
1. Duployez N, Willekens C, Marceau-Renaut A, Boudry-Labis E, Preudhomme C. Prognosis and monitoring of core-binding factor acute myeloid leukemia: current and emerging factors. Expert Rev Hematol. 2015; 8: 43–56. https://doi.org/10.1586/17474086.2014.976551.
2. Downing JR. The core-binding factor leukemias: lessons learned from murine models. Curr Opin Genet Dev. 2003; 13: 48–54. https://doi.org/10.1016/S0959-437X(02)00018-7.
3. Wiemels JL, Xiao Z, Buffler PA, Maia AT, Ma X, Dicks BM, Smith MT, Zhang L, Feusner J, Wiencke J, Pritchard-Jones K, Kempski H, Greaves M. In utero origin of t(8;21) AML1-ETO translocations in childhood acute myeloid leukemia. Blood. 2002; 99: 3801–5. https://doi.org/10.1182/blood.V99.10.3801.
4. Miyamoto T, Nagafuji K, Akashi K, Harada M, Kyo T, Akashi T, Takenaka K, Mizuno S, Gondo H, Okamura T, Dohy H, Niho Y. Persistence of multipotent progenitors expressing AML1/ETO transcripts in long-term remission patients with t(8;21) acute myelogenous leukemia. Blood. 1996; 87: 4789–96.
5. Jourdan E, Boissel N, Chevret S, Delabesse E, Renneville A, Cornillet P, Blanchet O, Cayuela JM, Recher C, Raffoux E, Delaunay J, Pigneux A, Bulabois CE, et al. Prospective evaluation of gene mutations and minimal residual disease in patients with core binding factor acute myeloid leukemia. Blood. 2013; 121: 2213–23. https://doi.org/10.1182/blood-2012-10-462879.
6. Duployez N, Marceau-Renaut A, Boissel N, Petit A, Bucci M, Geffroy S, Lapillonne H, Renneville A, Ragu C, Figeac M, Celli-Lebras K, Lacombe C, Micol JB, et al. Comprehensive mutational profiling of core binding factor acute myeloid leukemia. Blood. 2016; 127: 2451–9. https://doi.org/10.1182/blood-2015-12-688705.
7. Faber ZJ, Chen X, Gedman AL, Boggs K, Cheng J, Ma J, Radtke I, Chao JR, Walsh MP, Song G, Andersson AK, Dang J, Dong L, et al. The genomic landscape of core-binding factor acute myeloid leukemias. Nat Genet. 2016; 48: 1551–6. https://doi.org/10.1038/ng.3709.
8. Schlenk RF, Benner A, Krauter J, Büchner T, Sauerland C, Ehninger G, Schaich M, Mohr B, Niederwieser D, Krahl R, Pasold R, Döhner K, Ganser A, et al. Individual patient data-based meta-analysis of patients aged 16 to 60 years with core binding factor acute myeloid leukemia: a survey of the German Acute Myeloid Leukemia Intergroup. J Clin Oncol. 2004; 22: 3741–50. https://doi.org/10.1200/JCO.2004.03.012.
9. Marcucci G, Mrózek K, Ruppert AS, Maharry K, Kolitz JE, Moore JO, Mayer RJ, Pettenati MJ, Powell BL, Edwards CG, Sterling LJ, Vardiman JW, Schiffer CA, et al. Prognostic factors and outcome of core binding factor acute myeloid leukemia patients with t(8;21) differ from those of patients with inv(16): a Cancer and Leukemia Group B study. J Clin Oncol. 2005; 23: 5705–17. https://doi.org/10.1200/JCO.2005.15.610.
10. Appelbaum FR, Kopecky KJ, Tallman MS, Slovak ML, Gundacker HM, Kim HT, Dewald GW, Kantarjian HM, Pierce SR, Estey EH. The clinical spectrum of adult acute myeloid leukaemia associated with core binding factor translocations. Br J Haematol. 2006; 135: 165–73. https://doi.org/10.1111/j.1365-2141.2006.06276.x.
11. Cazier JB, Holmes CC, Broxholme J. GREVE: Genomic Recurrent Event ViEwer to assist the identification of patterns across individual cancer samples. Bioinformatics. 2012; 28: 2981–2.
12. Sweetser DA, Peniket AJ, Haaland C, Blomberg AA, Zhang Y, Zaidi ST, Dayyani F, Zhao Z, Heerema NA, Boultwood J, Dewald GW, Paietta E, Slovak ML, et al. Delineation of the minimal commonly deleted segment and identification of candidate tumor-suppressor genes in del(9q) acute myeloid leukemia. Genes Chromosomes Cancer. 2005; 44: 279–91. https://doi.org/10.1002/gcc.20236.
13. Dayyani F, Wang J, Yeh JR, Ahn EY, Tobey E, Zhang DE, Bernstein ID, Peterson RT, Sweetser DA. Loss of TLE1 and TLE4 from the del(9q) commonly deleted region in AML cooperates with AML1-ETO to affect myeloid cell proliferation and survival. Blood. 2008; 111: 4338–47. https://doi.org/10.1182/blood-2007-07-103291.
14. Mikulasovich M, LeBlanc A, Scalise A, Manwani D, Keyzner A, Najfeld V. Duplication and triplication of der(21) t(8;21)(q22;q22) in acute myeloid leukemia. Cancer Genet Cytogenet. 2009; 188: 83–7. https://doi.org/10.1016/j.cancergencyto.2008.10.004.
15. Wang X, Dai H, Wang Q, Wang Q, Xu Y, Wang Y, Sun A, Ruan J, Chen S, Wu D. EZH2 mutations are related to low blast percentage in bone marrow and -7/del(7q) in de novo acute myeloid leukemia. PLoS One. 2013; 8: e61341. https://doi.org/10.1371/journal.pone.0061341.
16. Chen C, Liu Y, Rappaport AR, Kitzing T, Schultz N, Zhao Z, Shroff AS, Dickins RA, Vakoc CR, Bradner JE, Stock W, LeBeau MM, Shannon KM, et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell. 2014; 25: 652–65. https://doi.org/10.1016/j.ccr.2014.03.016.
17. Kühn MW, Radtke I, Bullinger L, Goorha S, Cheng J, Edelmann J, Gohlke J, Su X, Paschka P, Pounds S, Krauter J, Ganser A, Quessar A, et al. High-resolution genomic profiling of adult and pediatric core-binding factor acute myeloid leukemia reveals new recurrent genomic alterations. Blood. 2012; 119: e67–75. https://doi.org/10.1182/blood-2011-09-380444.
18. Hirano T, Yoshikawa R, Harada H, Harada Y, Ishida A, Yamazaki T. Long noncoding RNA, CCDC26, controls myeloid leukemia cell growth through regulation of KIT expression. Mol Cancer. 2015; 14: 90. https://doi.org/10.1186/s12943-015-0364-7.
19. Grönholm J, Kaustio M, Myllymäki H, Kallio J, Saarikettu J, Kronhamn J, Valanne S, Silvennoinen O, Rämet M. Not4 enhances JAK/STAT pathway-dependent gene expression in Drosophila and in human cells. FASEB J. 2012; 26: 1239–50.
20. Hartmann L, Dutta S, Opatz S, Vosberg S, Reiter K, Leubolt G, Metzeler KH, Herold T, Bamopoulos SA, Bräundl K, Zellmeier E, Ksienzyk B, Konstandin NP, et al. ZBTB7A mutations in acute myeloid leukaemia with t(8;21) translocation. Nat Commun. 2016; 7: 11733. https://doi.org/10.1038/ncomms11733.
21. Lavallée VP, Lemieux S, Boucher G, Gendron P, Boivin I, Armstrong RN, Sauvageau G, Hébert J. RNA-sequencing analysis of core binding factor AML identifies recurrent ZBTB7A mutations and defines RUNX1-CBFA2T3 fusion signature. Blood. 2016; 127: 2498–501. https://doi.org/10.1182/blood-2016-03-703868.
22. Radtke I, Mullighan CG, Ishii M, Su X, Cheng J, Ma J, Ganti R, Cai Z, Goorha S, Pounds SB, Cao X, Obert C, Armstrong J, et al. Genomic analysis reveals few genetic alterations in pediatric acute myeloid leukemia. Proc Natl Acad Sci U S A. 2009; 106: 12944–12949.
23. Renneville A, Abdelali RB, Chevret S, Nibourel O, Cheok M, Pautas C, Dulery R, Boyer T, Cayuela JM, Hayette S, Raffoux E, Farhat H, Boissel N, et al. Clinical impact of gene mutations and lesions detected by SNP-array karyotyping in acute myeloid leukemia patients in the context of gemtuzumab ozogamicin treatment: results of the ALFA-0701 trial. Oncotarget. 2013; 5: 916–32. https://doi.org/10.18632/oncotarget.1536.
24. Eisfeld AK, Kohlschmidt J, Schwind S, Nicolet D, Blachly JS, Orwick S, Shah C, Bainazar M, Kroll KW, Walker CJ, Carroll AJ, Powell BL, Stone RM, et al. Mutations in the CCND1 and CCND2 genes are frequent events in adult patients with t(8;21)(q22;q22) acute myeloid leukemia. Leukemia. 2017; 31: 1278–85. https://doi.org/10.1038/leu.2016.332.
25. Krauth MT, Eder C, Alpermann T, Bacher U, Nadarajah N, Kern W, Haferlach C, Haferlach T, Schnittger S. High number of additional genetic lesions in acute myeloid leukemia with t(8;21)/RUNX1-RUNX1T1: frequency and impact on clinical outcome. Leukemia. 2014; 28: 1449–58. https://doi.org/10.1038/leu.2014.4.
26. Paschka P, Du J, Schlenk RF, Gaidzik VI, Bullinger L, Corbacioglu A, Späth D, Kayser S, Schlegelberger B, Krauter J, Ganser A, Köhne CH, Held G, et al. Secondary genetic lesions in acute myeloid leukemia with inv(16) or t(16;16): a study of the German-Austrian AML Study Group (AMLSG). Blood. 2013; 121: 170–7. https://doi.org/10.1182/blood-2012-05-431486.
27. Micol JB, Duployez N, Boissel N, Petit A, Geffroy S, Nibourel O, Lacombe C, Lapillonne H, Etancelin P, Figeac M, Renneville A, Castaigne S, Leverger G, et al. Frequent ASXL2 mutations in acute myeloid leukemia patients with t(8;21)/RUNX1-RUNX1T1 chromosomal translocations. Blood. 2014; 124: 1445–9. https://doi.org/10.1182/blood-2014-04-571018.
28. Katoh M, Igarashi M, Fukuda H, Nakagama H, Katoh M. Cancer genetics and genomics of human FOX family genes. Cancer Lett. 2013; 328: 198–206. https://doi.org/10.1016/j.canlet.2012.09.017.
29. Klampfl T, Harutyunyan A, Berg T, Gisslinger B, Schalling M, Bagienski K, Olcaydu D, Passamonti F, Rumi E, Pietra D, Jäger R, Pieri L, Guglielmelli P, et al. Genome integrity of myeloproliferative neoplasms in chronic phase and during disease progression. Blood. 2011; 118: 167–76. https://doi.org/10.1182/blood-2011-01-331678.
30. Bullinger L, Krönke J, Gaidzik V, Döhner H, Döhner K. Comment on ‘integrative genomic profiling of human prostate cancer.’ Leukemia. 2010; 24: 1970–2. https://doi.org/10.1038/leu.2010.194.
31. Milosevic JD, Puda A, Malcovati L, Berg T, Hofbauer M, Stukalov A, Klampfl T, Harutyunyan AS, Gisslinger H, Gisslinger B, Burjanivova T, Rumi E, Pietra D, et al. Clinical significance of genetic aberrations in secondary acute myeloid leukemia. Am J Hematol. 2012; 87: 1010–6. https://doi.org/10.1002/ajh.23309.
32. Shi C, Sakuma M, Mooroka T, Liscoe A, Gao H, Croce KJ, Sharma A, Kaplan D, Greaves DR, Wang Y, Simon DI. Down-regulation of the forkhead transcription factor Foxp1 is required for monocyte differentiation and macrophage function. Blood. 2008; 112: 4699–711.
33. Liu XS, Liu Z, Gerarduzzi C, Choi DE, Ganapathy S, Pandolfi PP, Yuan ZM. Somatic human ZBTB7A zinc finger mutations promote cancer progression. Oncogene. 2016; 35: 3071–8. https://doi.org/10.1038/onc.2015.371.
34. Hirano T. Is CCDC26 a Novel Cancer-Associated Long-Chain Non-Coding RNA? In: Siregar Y, editor. Oncogene and Cancer - From Bench to Clinic. InTech; 2013. p. 415–34.
35. Shete S, Hosking FJ, Robertson LB, Dobbins SE, Sanson M, Malmer B, Simon M, Marie Y, Boisselier B, Delattre JY, Hoang-Xuan K, Hallani SE, Idbaih A, et al. Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet. 2009; 41: 899–904. https://doi.org/10.1038/ng.407.
36. Peng W, Jiang A. Long noncoding RNA CCDC26 as a potential predictor biomarker contributes to tumorigenesis in pancreatic cancer. Biomed Pharmacother. 2016; 83: 712–7. https://doi.org/10.1016/j.biopha.2016.06.059.
37. Yin W, Rossin A, Clifford JL, Gronemeyer H. Co-resistance to retinoic acid and TRAIL by insertion mutagenesis into RAM. Oncogene. 2006; 25: 3735–44.
38. Hirano T. The role of the CCDC26 long noncoding RNA as a tumor suppressor. RNA Dis. 2015. https://doi.org/10.14800/rd.1022.
39. Tiu RV, Gondek LP, O’Keefe CL, Huh J, Sekeres MA, Elson P, McDevitt MA, Wang XF, Levis MJ, Karp JE, Advani AS, Maciejewski JP. New lesions detected by single nucleotide polymorphism array–based chromosomal analysis have important clinical impact in acute myeloid leukemia. J Clin Oncol. 2009; 27: 5219–26. https://doi.org/10.1200/JCO.2009.21.9840.
40. O’Keefe C, McDevitt MA, Maciejewski JP. Copy neutral loss of heterozygosity: a novel chromosomal lesion in myeloid malignancies. Blood. 2010; 115: 2731–9. https://doi.org/10.1182/blood-2009-10-201848.
41. Heinrichs S, Li C, Look AT. SNP array analysis in hematologic malignancies: avoiding false discoveries. Blood. 2010; 115: 4157–61. https://doi.org/10.1182/blood-2009-11-203182.