
INTRODUCTION
The guardian of the genome, p53, is a tumor suppressor and transcription factor that initiates cell cycle arrest and apoptosis in response to stress or insult and thereby prevents damaged cells from proliferating. At the same time, p53 transcriptional activity is tightly regulated in normal cells to prevent inappropriate activation of downstream anti-proliferative or cell death programs [1]. The two most prominent p53 inhibitors are Mdm2 and Mdm4 [2]. Mouse models have demonstrated that both Mdm2 and Mdm4 have essential roles in embryonic development through inhibition of p53 activity. Homozygous loss of either gene is embryonic lethal, a phenotype that is completely rescued by the concomitant deletion of p53 [3–5]. Additionally, transgenic overexpression of either is sufficient to promote tumorigenesis [6, 7]. In human cancers, increased levels of MDM2 and MDM4 abrogate the need for TP53 alterations [8]. However, many cancers retain wild type TP53 without alterations in known TP53 inhibitors suggesting that novel mechanisms of pathway inhibition remain to be identified [8].
Recently, digestive organ expansion factor (Diexf), was identified as a novel inhibitor of p53 in zebrafish [9–14]. In addition, in zebrafish and human cell lines, Def/DIEXF recruits Calpain3 in the nucleolus to regulate p53 stability through the Def-Capn3 pathway [15]. DIEXF is an evolutionarily conserved gene that was first identified in zebrafish and shown to be essential for expansion of digestive organs [9]. It is located on chromosome 1 in both human and mouse genomes coding for a 756 (772 in mouse) amino acid protein. DIEXF does not have any known functional domains except a glutamic acid repeat region in the amino-terminus which has been linked to RNA-processing [16]. Recent works in yeast, Arabidopsis, and zebrafish have demonstrated that Def is essential for pre-rRNA processing [16–18].
Loss of Def in zebrafish selectively up-regulates the expression of Δ113p53 isoform, transcribed from an alternative promoter in intron 4, in digestive organs [9, 19, 20]. Def-null fish overexpress Δ113p53 which results in increased p53 target gene expression and induction of cell cycle arrest in the digestive organs. This leads to under-expanded digestive organs and lethality by 8–11 days after fertilization [9]. Knock-down of both full-length p53 and Δ113p53 levels partially rescues the mutant phenotype indicating that Def acts as a negative regulator of p53 in zebrafish.
This study addresses the role of Diexf as a potential regulator of the p53 pathway in mammals. We demonstrate that DIEXF is amplified in many human cancers, and amplification is mutually exclusive with TP53 alterations in breast cancer. To directly evaluate the function of Diexf in vivo, we generated two independent knockout alleles in the mouse using the CRISPR/Cas9 technology. While heterozygous mice were normal, homozygous deletion of Diexf results in peri-implantation lethality. Concomitant deletion of p53 did not rescue the embryonic lethality in Diexf knockout animals. We additionally demonstrate that Diexf heterozygosity does not affect the radiation sensitivity of adult mice as observed in Mdm2 and Mdm4 heterozygous mice. Combined, these data suggest that while Diexf is an essential developmental gene in the mouse, it is not a significant regulator of the p53 pathway.
RESULTS
DIEXF is amplified in human cancers with an inverse correlation to TP53 alterations
Diexf is an evolutionarily conserved gene from yeast to humans, and recent studies in zebrafish have suggested that the Diexf protein is a negative regulator of p53 [9, 15]. In many different human cancers with wild type TP53, other TP53 inhibitors such as MDM2 and MDM4 are frequently amplified or overexpressed [8, 21]. To ascribe a role of DIEXF in human cancers, we exploited the data from cBioPortal and examined whether a similar inverse correlation between TP53 and DIEXF exists [22, 23]. We observed that DIEXF is indeed amplified in many human cancers (Figure 1A) including > 10% of the breast cancers, neuroendocrine prostate cancers (NEPCs), and cholangiosarcomas. In a breast cancer dataset of 2051 cases [24], 36% of total cases (n = 734) had TP53 alterations and 25% (n = 513) had Diexf amplification, but only 6% (n = 134) had both (Figure 1B). A significant mutual exclusivity (P < 0.001) was present between DIEXF amplification and TP53 alterations. Significant mutual exclusivity was also observed in other breast cancer datasets in Figure 1A. These results support the hypothesis that DIEXF is a negative regulator of the p53 pathway. However, it is important to note that DIEXF is located on chromosome 1q in close proximity (5.3Mbp) to the MDM4 locus. It is thus possible that DIEXF amplification is a consequence of co-amplification with MDM4. When we analyzed the same breast cancer datasets, we observed that amplification of DIEXF and MDM4 significantly overlapped (P < 0.001, Figure 1C). Similarly, when we examined other tumor types with DIEXF amplification, we found significant overlap with MDM4 amplification. We did not find any cancer where DIEXF amplification was significantly higher than MDM4 amplification. However, cases where either MDM4 or DIEXF amplification occurred independent of each other were also present in all tumor types examined.
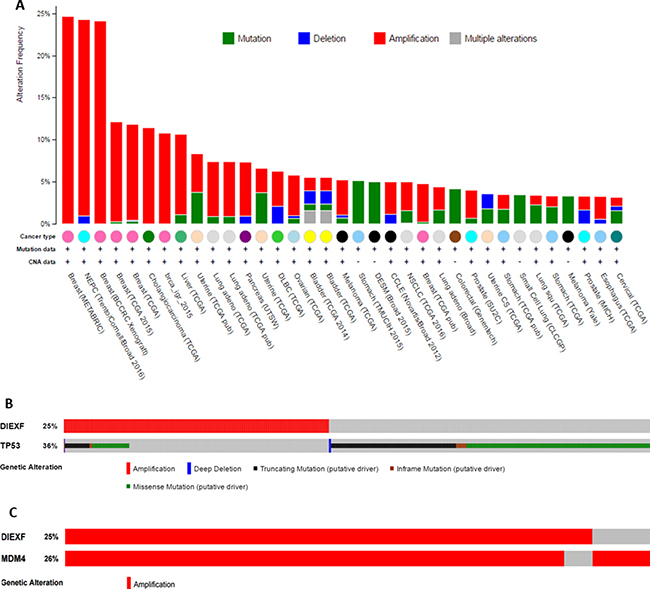
Figure 1: DIEXF amplification in human cancers. (A) Human cancer datasets from cBioPortal showing DIEXF alterations in human cancers. (B) A breast cancer dataset (METABRIC) of 2051 patients shows DIEXF amplification and TP53 alterations. There is a significant inverse correlation between DIEXF amplification and TP53 alterations (P < 0.001). Only cases with alteration in queried gene are shown. (C) A breast cancer dataset (METABRIC) of 2051 patients shows DIEXF and MDM4 co-amplifications. Only cases with amplification of at least one gene are shown.
Generation of Diexf knock-out mice
To directly evaluate the function of Diexf and its potential role in regulation of the p53 pathway in vivo, we generated a Diexf deletion in mice using CRISPR/Cas9 technology. Diexf is a 26 Kbp gene with 12 exons that code for a 772 amino acid protein (89 KDa) in mice. Exon 1 is 229 base pairs and contains the translational start site. We selected two sequences in Exon 1 downstream of the start codon to target with specific sgRNAs and the spCas9 endonuclease (Figure 2A) respectively. The two target sequences are located at the translational start site (bases 123–142) and further downstream (bases 188–207). Both sgRNAs have very few candidate off-target sites with scores of 98 and 93 out of 100 respectively per crispr.mit.edu analysis tool (Supplementary Table 1). When both sgRNAs and spCas9 endonuclease were used, only mice with disruption of the Diexf alleles at site 2 were obtained. Multiple alleles with both in-frame and frame-shift mutations were generated (Figure 2B). Of 16 animals born, 14 had alterations in one or both alleles of the Diexf gene (Table 1). Even though both alleles were targeted in 6 mice, no mice were born with two frame-shift alleles suggesting that homozygous loss may be lethal (Table 1). We selected two alleles with frame shift mutations for further experiments. The first allele DiexfΔ26 had 26 base pairs deleted (bases 194–219 in exon 1) resulting in a premature stop signal after 85 amino acids (23 endogenous and 62 novel amino acids). The second allele Diexf13ins had a 13 base pair insertion between bases 200–201 in exon 1 resulting in premature stop after 28 amino acids (26 endogenous and 2 novel amino acids).
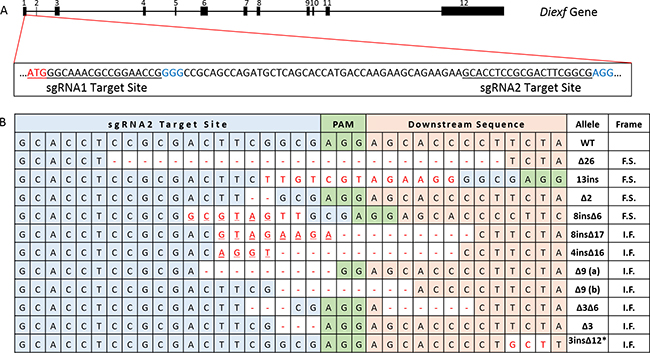
Figure 2: Diexf targeting using CRISPR/Cas9 system. (A) Genomic map of Diexf gene with 12 exons highlighting the sequence of exon 1 with the two target sites. The size of exons and introns are proportional to their width in the map. Translational start site (start codon) is in red, target sequence is underlined, and Protospacer adjacent motif (PAM) sequence is in blue. (B) Details of all alleles generated with alterations. Target site 2 is in blue cells, and alterations (deletion [-] or insertion) are in red. Underlined bold letters represent deletion and insertion at the same position. I.F., in-frame mutation; F.S., frame-shift mutation; *, an additional 12 base deletion (not shown) in this allele is 156 bases downstream of the 3 base insertion site.
Table 1: Genotype of Diexf alleles in all mice generated by CRISPR/Cas9 targeting
Mouse # |
Genotype |
|
|---|---|---|
Allele 1 |
Allele 2 |
|
1 |
13ins |
Δ9 (a) |
2 |
Δ26 |
Δ9 (a) |
3 |
Δ26 |
WT |
4* |
3insΔ12 |
Δ9 (b) and Δ26 |
5* |
Δ2 |
4insΔ16 and Δ9 (a) |
6 |
Δ3Δ6 |
8insΔ6 |
7 |
Δ9 (a) |
Δ9 (a) |
8* |
Δ9 (a) |
Δ3 and WT |
9* |
Δ9 (a) |
8insΔ17 and WT |
10–14 (n = 5) |
Δ9 (a) |
WT |
15–16 (n = 2) |
WT |
WT |
*, Mosaics. Of 16 mice, 14 had targeting in at least one allele. 6 animals had bi-allelic targeting (animals with no wild type allele, 2/6 mosaics). At least one allele in all animals are wild type or in-frame. Δ; deletion, ins; insertion, WT; wild type, (a) and (b); different Δ9 alleles.
To rule out the possibility of unintended mutations being incorporated into the genome by the CRISPR/Cas9 system, we evaluated off-target cleavage at the top six sites for sgRNA2 and top four sites for sgRNA1 based on scoring by crispr.mit.edu analysis tool (Supplementary Table 1). We did not observe any off-target mutations in these mice. DiexfΔ26 and Diexf13ins mice were crossed with wild type animals for three generations to further mitigate the potential of any off-target mutation being carried over to the experimental cohort.
Diexf null embryos are peri-implantation lethal
The two selected lines of Diexf knock-out alleles with frame-shift truncating mutations were used to evaluate if homozygous null animals were viable. For each line, we inter-crossed heterozygous animals and genotyped progeny at the time of weaning. Of 22 pups born from 3 litters in the Diexf13ins line, 7 animals were wild type, 15 were heterozygous mutants, and no animals were homozygous mutants (Table 2). Similarly, of 48 pups born from 7 litters in the DiexfΔ26 line; 19 animals were wild type, 29 were heterozygous, and none were homozygous mutants (Table 2). The deviations from the expected Mendelian ratios were significant in both lines (p = 0.02 and p = 0.0001 respectively). Failure to generate any viable homozygous knock-out animals clearly demonstrates an essential role of Diexf during embryonic development in mice.
Table 2: Homozygous deletion of Diexf is embryonic lethal at peri-implantation stage (E4.5-E5.5) in mice
Diexf+/− X Diexf+/− : Observed (Expected) |
|||||
|---|---|---|---|---|---|
Allele |
Age |
Diexf+/+ |
Diexf+/− |
Diexf−/− |
Empty Decidua |
Diexf13ins |
P21 |
7 (6) |
15 (11) |
0 (6) |
- |
DiexfΔ26 |
P21 |
19 (12) |
29 (24) |
0 (12) |
- |
DiexfΔ26 |
E14.5 |
4 (3) |
9 (7) |
0 (3) |
0 |
DiexfΔ26 |
E9.5 |
4 (4) |
10 (7) |
0 (4) |
4 |
DiexfΔ26 |
E7.5 |
9 (6) |
15 (12) |
0 (6) |
6 |
Heterozygous crosses between Diexf+/− mice (two lines) and observed vs expected numbers for all genotype at various stages. P21 = 21 days after birth. E = embryonic day. All expected numbers are rounded to the nearest integer.
In order to determine the timing of the lethality of homozygous mutants, we harvested and genotyped the embryos from Diexf+/Δ26 heterozygous crosses at various developmental time points. We screened 13 embryos from 2 females at E14.5 and did not find any homozygous mutants (Table 2). At E9.5, we screened 18 embryos from 3 females and observed 4 wild type, 10 heterozygous mutant embryos, and 4 empty decidua. Similarly, of 30 embryos from 4 females at E7.5, we observed 6 empty decidua while the remaining embryos were either wild type (n = 9) or heterozygous mutants (n = 15). The finding of empty decidua demonstrated that mutant embryos implanted but died soon thereafter and were likely resorbed by E7.5 [25]. This clearly demonstrates that the Diexf null embryos are peri-implantation lethal at E4.5-E5.5 stages. To further evaluate the viability of mutant embryos at pre-implantation stage, we flushed out blastocysts from heterozygous crosses, observed the morphology under microscope, and genotyped them. We did not observe obvious defects in dozens of blastocysts examined, and genotyping of 8 random blastocysts confirmed presence of homozygous mutants (Supplementary Figure 1). Combined, these results establish Diexf as an essential gene for early embryonic development.
Embryonic lethality of Diexf-/- mice is p53-independent
Early embryonic lethality occurs in knock-out mouse models of the p53 inhibitors Mdm2 and Mdm4. Lethality in these models is p53-dependent as the phenotype is rescued upon concomitant deletion of p53. Studies in zebrafish suggest that Diexf is a potential negative regulator of p53, and embryonic lethality of Def-null fish is partially p53-dependent. In order to address if embryonic lethality of Diexf-null mice is also p53-dependent, we crossed heterozygous animals carrying the DiexfΔ26 null allele with p53-/- mice. The resulting Diexf+/Δ26; p53+/- double heterozygous mice were inter-crossed and progeny were genotyped at weaning. We screened 108 animals from 15 litters and observed no DiexfΔ26/Δ26 homozygous mutants irrespective of p53 status (Table 3). Of the 16 mice with p53-/- genotype, 3 were wild type and 13 were heterozygous for DiexfΔ26. These results clearly demonstrate that loss of p53 fails to rescue the embryonic lethality due to homozygous Diexf loss. To evaluate the possibility of a partial rescue (delayed lethality) upon p53 loss in Diexf null embryos, we also screened embryos from Diexf+/Δ26; p53+/- double heterozygous mice crossed with Diexf+/Δ26; p53-/- animals at various time points. We failed to find any embryos with the DiexfΔ26/Δ26 genotype irrespective of p53 status at the time points examined, and empty decidua were observed at E7.5 and E9.5 as previously observed with the Diexf+/Δ26 heterozygous crosses (Table 3). These results indicate that embryonic lethality in mice due to Diexf loss is not a result of inappropriate activation of the p53 pathway.
Table 3: Loss of p53 fails to rescue the embryonic lethality in Diexf null mice
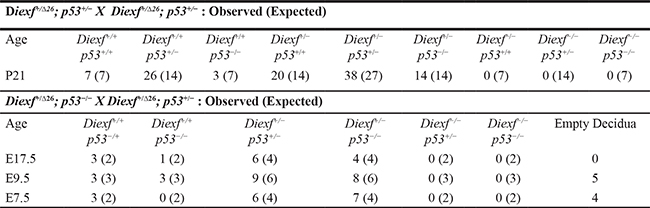
Inter-cross between Diexf+/Δ26; p53+/− double heterozygous animals and observed vs expected numbers for all genotypes. Bottom. Observed vs expected numbers for all genotypes in embryos derived from Diexf+/Δ26; p53+/− mice crossed with Diexf+/Δ26; p53−/− mice. All expected numbers are rounded to the nearest integer.
Diexf heterozygous animals are resistant to 6Gy ionizing radiation
We next wanted to explore the possibility that Diexf has different functions during development and in adult animals. To test whether Diexf is expressed in adults, we first examined Diexf protein expression in various tissues of 8 week old wild type mice by immunoblotting. We observed that Diexf was expressed in all mouse tissues that we examined, with high expression in the digestive organs and low expression in the heart (Figure 3A). Next, we wanted to evaluate the role of Diexf in p53 regulation under stress condition. p53 is stabilized and activated after exposure to stress conditions such as DNA damage [2]. Previous studies in our lab have shown that heterozygosity of Mdm2 and Mdm4 results in sensitivity to sub-lethal ionizing radiation (IR) in a p53-dependent manner [26]. To evaluate if Diexf regulates p53 activity in the adult mouse in response to DNA damage, we irradiated 5–9 week old Diexf+/Δ26 heterozygous animals (n = 14) along with Mdm2P2/P2 animals (n = 5) as a positive control [27], and wild type animals (n = 12) as a negative control. Mice were irradiated with sub-lethal IR (one dose of 6 Gy) and monitored daily for four weeks. As expected, radio-sensitive Mdm2P2/P2 animals died within three weeks after irradiation (Figure 3B). However, both Diexf heterozygous and wild-type mice survived the sub-lethal 6 Gy IR exposure and were alive by the end of 4 weeks monitoring period (Figure 3B). These results demonstrate that Diexf is not a potent negative regulator of p53 following DNA damage in adult mice.
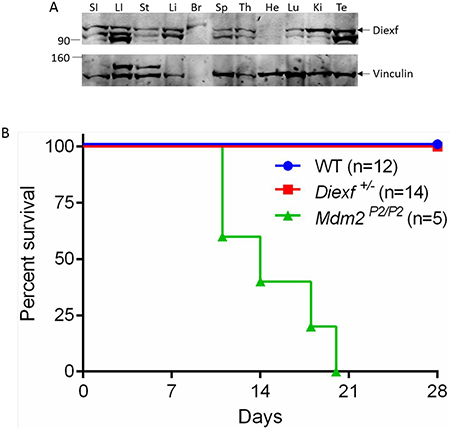
Figure 3: Diexf heterozygous animals are not sensitive to sub-lethal ionizing radiation. (A) Western blot showing Diexf expression in different mouse tissues. Vinculin was used to evaluate protein loading. SI, Small intestine; LI, Large Intestine; St, Stomach; Li, Liver; Br, Brain; Sp, Spleen; Th, Thymus; He, Heart; Lu, Lung; Ki, Kidney; Te, Testes. (B) Survival curve of Diexf+/Δ26 animals after 6 Gy radiation. Wild type and Mdm2P2/P2 animals were used as negative and positive controls respectively.
DISCUSSION
Understanding the mechanisms regulating the p53 pathway has important implications in cancer. MDM2 and MDM4 are critical TP53 inhibitors whose genetic loci are amplified in several tumor types that retain wild-type TP53. This knowledge has spurred the development of MDM2 inhibitors for cancer therapeutics. Recently, a novel gene Diexf was found to regulate the expression of a specific p53 isoform in zebrafish, and p53 loss was able to partially rescue the developmental defects in the mutant zebrafish digestive system. These data suggested a potential role of Diexf in regulating the p53 pathway in mammals.
Recent studies show that Diexf homologs in S. cerevisiae (Utp25p) and A. thaliana (Nof1) are nucleolus-localized components of small subunit processome that regulate pre-rRNA processing [16, 18, 28]. In addition to this function, Def also negatively regulates p53 activity in zebrafish via proteasome-independent degradation of p53 protein in the nucleolus [15, 17, 29]. Here, we first highlighted a significant mutually exclusive relationship between DIEXF amplification and TP53 mutation in human breast cancers. This relationship must, however, be considered in the context of the DIEXF genomic locus. DIEXF is in close proximity to MDM4 gene which encodes a potent p53-inhibitor that is frequently amplified in human cancers [30, 31]. Given the data presented herein, it is likely that this mutually exclusive relationship between DIEXF and TP53 is driven by amplification of MDM4. We cannot, however, exclude the possibility that overexpression of DIEXF contributes to p53 pathway attenuation in these cancers.
To directly evaluate the role of Diexf as a regulator of the p53 pathway in mammals, we used CRISPR/Cas9 technology to generate multiple independent Diexf knockout alleles in the mouse. The CRISPR/Cas9 system has revolutionized the way we generate animal models as it is efficient, fast, and less expensive than traditional techniques. One limitation of this system, however, is the difficulty in generation of knock-out alleles of genes that may be essential. If both alleles of an essential gene are disrupted, the embryos may not survive. Using only sgRNA1 and spCas9 endonuclease, we failed to generate any mice with disruption of Diexf gene. We could not determine whether the sgRNA failed to target the gene or if the sgRNA was so efficient that bi-allelic disruption of the gene in all targeted mice led to lethality. When both sgRNA1 and sgRNA2 were co-injected, only sgRNA2 generated mutations at its target site indicating that sgRNA1 failed. Moreover, even though both alleles were targeted by sgRNA2 in many animals, one or both alleles were always in-frame resulting in embryo survival. If most mutations had not been in-frame, more embryos would have died. We observed the same 9 bp deletion in 10/16 mice. The observation that non-homologous end joining (NHEJ) based repair can result in frequent recurrence of the same mutation in targeted alleles also presents a problem. If the recurring mutation is frame-shift, then more embryos would die. On the other hand, if the recurring mutation is in-frame, the possibility of getting a knock-out allele is diminished. Therefore, both lethality and sgRNA failure should be considered while targeting a possible essential gene. Another major concern of the CRISPR/Cas9 system is the modifications (insertions or deletions) at off-target sites. To our knowledge, there has been no report of high frequency targeting at non-targeted sites with 2 or more mismatches in mouse models. When we screened the top four and the top six predicted off-target sites for sgRNA1 and sgRNA2 respectively, we did not find any mutations.
Using two independent null-alleles, we demonstrate that homozygous loss of Diexf is peri-implantation lethal, establishing Diexf as an essential developmental gene in the mouse. Our observation that Diexf-null mouse embryos are peri-implantation lethal is very different from the phenotype observed in zebrafish. In fish, the lethality occurs due to digestive organ failure, while in mice, the embryos died before any of the digestive organs develop. In addition, the abnormalities in mutant fish are p53-dependent, as the phenotype can be partially rescued by concomitant p53 loss. However, embryonic lethality in mice due to Diexf loss was not rescued or even delayed by loss of p53 demonstrating that the phenotype is not a consequence of inappropriate activation of p53. The role of p53 regulators such as Mdm4 seem to have evolved from fish to mammals as well. A recent study has shown that Mdm4 is dispensable in zebrafish, while it is essential for murine embryonic development [4, 5, 7, 25]. We could hypothesize that as the role of Mdm4 to inhibit p53 became more potent in mammals, the role of other p53 regulators such as Diexf have diminished. We cannot rule out the possibility that Diexf may cooperate with other p53 regulators to attenuate the p53 pathway, but we can conclude that it does not have an essential role in p53 regulation on its own.
Finally, we find no evidence of Diexf as a significant functional regulator of the p53 pathway in response to ionizing radiation. Heterozygous loss of well-established p53 regulators results in sensitivity to a sub-lethal dose of ionizing radiation [26]. Diexf heterozygous animals do not exhibit enhanced radio-sensitivity, further supporting the argument that Diexf is not a potent inhibitor of the p53 pathway in mice under these conditions. In human cancers, negative regulators of the TP53 pathway are frequently overexpressed, so it remains possible that the overexpression of DIEXF may impact the TP53 pathway and have pathological consequences. Moreover, it may inhibit TP53 function under certain conditions that we have not tested.
Combined, this work identifies Diexf as an essential developmental gene in the mouse, and suggests that the function(s) of Diexf are largely independent of a role as a negative regulator of p53. It has been clearly demonstrated that Def is a component of the ribosomal processome, and the Def-Capn3 pathway possibly regulates proteins other than p53 in the nucleolus. Two previous publications with Capn3 deletion in mice show that the Capn3-/- mice are viable and fertile, and their digestive organs are not affected [32, 33]. These results indicate that embryonic lethality in Diexf-null mice is independent of both p53 and Calpain3, and suggests that the role of Diexf has evolved to have very diverse functions. Moreover, as DIEXF amplifications occur in cancers, future studies to better understand its functions will be instructive and important.
MATERIALS AND METHODS
Gene targeting strategy
The entire sequence of exon 1 (starting from 20 bases upstream of the start codon) was used to score all possible target sites using the crispr.mit.edu tool. The two sites with highest scores were selected for targeting. Four and six possible off-target sites with a PAM sequence for sgRNA1 and sgRNA2 respectively were also selected for screening.
sgRNA(s) synthesis by in-vitro transcription
All in-vitro transcription kits use T7 polymerase which requires a G at the transcription start site. For sgRNA1, N20 does not start with a G, and thus G (in parenthesis below) was added to the sgRNA. The T7 promoter sequence was underlined, and sgRNA target sites are in bold. Four random bases (GAAA) were added upstream of T7 promoter sequence to provide anchorage for T7 polymerase binding. The following sequence is the Forward oligo for the template. Reverse complement of this sequence will be the reverse oligo.
sgRNA1 forward oligo
GAAATTAATACGACTCACTATA(G)ATGGGCAAACGCCGGAACCGGTTTTAGAGCTAGAAA TAGCAAGTTAAAATAAGGCTAGTCCGTTATCAA CTTGAAAAAGTGGCACCGAGTCGGTGCTTTT
sgRNA2 forward oligo
GAAATTAATACGACTCACTATAGCACCTC CGCGACTTCGGCGGTTTTAGAGCT AGAAATAGCAAGTTAAAATAAGGCTAGTCC GTTATCAACTTGAAAAAGTGGCACCGAGTC GGTGCTTTT
Oligos were suspended in nuclease free water to a final concentration of 500 ng/μl. For each sgRNA, 5μg each of forward and reverse oligos (10 μl each) were mixed in 80μl nuclease-free water (100 μl total volume) and boiled for 5–10 minutes and then cooled at room temperature for 2 hrs to overnight. 200–400 ng of the prepared templates were used to synthesize sgRNAs using the MEGAshortscript Kit (Invitrogen AM1354) as per manufacturer’s protocol. sgRNAs were purified by acid phenol-chloroform extraction and ethanol precipitation as per manufacturer’s protocol (Invitrogen). sgRNAs were resuspended in 70 μl of RNase-free water and further purified by using Biospin P30 chromatography columns (#732–6223, Biorad) as per manufacturer’s protocol to remove any remaining free nucleotides. The concentration and quality of sgRNAs were determined by using Bio analyzer. If the concentration measured by Nanodrop was significantly higher than Bio analyzer reading, it indicated free nucleotide contamination. Column purification was repeated if necessary.
Zygote injection and implantation
A final injection solution containing 10 ng/μl Cas9 mRNA (PrecisionX hspCas9 SmartNuclease mRNA, System Biosciences) and 7.5 ng/μl of each sgRNA was prepared in Tris-EDTA buffer (5 mM Tris, 0.1 mM EDTA). The final solution was injected into the pronucleus of 200–250 zygotes. The zygotes were then implanted into pseudo-pregnant mice (20–25 per animal). All injections and implantations were done in the MD Anderson Genetically Engineered Mouse Facility.
Mouse breeding, maintenance, screening and genotyping
Mice were maintained in 100% C57BL/6 background. All mouse studies were conducted in compliance with an Institutional Animal Care and Use Committee protocol. Live mice were weaned at the age of three weeks, and ear biopsies were collected. The tissues were digested in Lysis buffer (1X PCR buffer, 1% Triton X, 250 μg/uL Proteinase K) at 55°C overnight, and heated at 95°C for 15 minutes to denature Proteinase K. 2 μL of the lysed tissue extract was used for PCR reaction to amplify 1 Kb region of the targeted site. The PCR product was gel purified and sanger-sequenced to identify any indels at the target site.
Screening primer Fwd: CGCATGCGTAGACAC GCCTATG; Screening primer Rev: GCACAAGGG CAGAGATGATCAG; DiexfΔ26 Genotyping primer Fwd: CGTTTCCGCTATGGGCAAACG; DiexfΔ26 Genotyping primer Rev: CTCAACTCGGCCGGA ACCAG.
Selection and screening of off-target sites
A list of all possible off-target sites was obtained from crispr.mit.edu site for both sgRNAs. The following criteria were used to select the sites for screening:
1. All sites with less than 3 mismatches were selected; 2. Sites with 3 or more mismatches should be followed by PAM sequence; 3. Intra-genic sites were given preference over inter-genic sites; 4. Sites with higher score were selected first; 5. For sites with the same score, one with fewest mismatches was selected; 6. For sites with the same score and the same number of mismatches, the average of the mismatch positions were considered (one with lowest average was selected).
Once the sites were selected, the 23 base sequence for each site was run on BLAT tool at UCSC Genome Browser website, and the genomic sequence was obtained. Primers were designed about 500 bases upstream and 500 bases downstream of the selected sites, and the region was PCR amplified and sanger-sequenced.
sgRNA1 OTS1 Fwd: CCTCCACCCCGCTC TAATTTC; sgRNA1 OTS1 Rev: CTGCCCTTCTCC TCTGTGGATC; sgRNA1 OTS2 Fwd: GGGAAGGAA GCTCAGGGGTTAG; sgRNA1 OTS2 Rev: CCACC TTGGAATTCCGTTCTTTC; sgRNA1 OTS3 Fwd: GGAGGCAGTGAAAGATAAAG; sgRNA1 OTS3 Rev: CGGATCACTCAGTTTGAATC; sgRNA1 OTS4 Fwd: CGGGTTGCTTTGGAAAAAATACAC ; sgRNA1 OTS4 Rev: CGGGCTGACCGTATTGAGGGAATC; sgRNA2 OTS1 Fwd: GGCTGTGGTAGGTGATTAC; sgRNA2 OTS1 Rev: GGTCACCAGCTAAGGAATG; sgRNA2 OTS2 Fwd: GACCCGGGTAAGAAGAAAAAG; sgRNA2 OTS2 Rev: CGGCGAAACAGACTGTTTC; sgRNA2 OTS3 Fwd: CACACAGGATGTCACATTCC; sgRNA2 OTS3 Rev: GCGAGCTGCCCTTTTAAG; sgRNA2 OTS4 Fwd: GCCATGCAACCTCCTAAG; sgRNA2 OTS4 Rev: CTCCAGGATCTTGCTTTTGG; sgRNA2 OTS5 Fwd: CTGGGACTAGTTTCTGGACTC; sgRNA2 OTS5 Rev: GCACACCCTGTATCTAACATTG; sgRNA2 OTS6 Fwd: GGGGCAGAGACAATCATG; sgRNA2 OTS6 Rev: GGCTGAGAATGGCTCAAG.
E7.5 and E9.5 embryo dissection
Heterozygous animals were mated with each other, and plugs were examined every morning. Females positive for plug (E0.5) were euthanized 7 days (E7.5) or 9 days (E9.5) from that date. Uteri were dissected, and each decidua was separated and dissected as previously described [34]. Embryos from each decidua were collected and genotyped.
Blastocyst harvest and genotyping
As described previously [34], 8–12 week old female mice were super-ovulated by intra-peritoneal injection of 5 IU of PMSG and 5 IU of HCG 48 hours later. Each female was set up in an individual mating cage with a male, and examined for plugs on the following morning. Plugged females were euthanized 3 days later (E3.5), and their uteri harvested. Buffer was injected into each uterine horn to flush out the blastocysts. Blastocysts were transferred into individual wells of a 96 well plate and were subjected to lysis and protein digestion with a lysis buffer. The entire sample was denatured, and used for PCR genotyping.
Irradiation of mice
5–9 week old mice were exposed to one dose of 6 Gy IR in a cesium-137 irradiator and monitored daily for four weeks. Moribund animals were sacrificed.
Western blot
Protein lysates were prepared by lysing tissues in SDS buffer. Protein estimation was carried out with BCA (Protein Assay kit, Pierce). Seventy micrograms of lysate was resolved on 8% SDS-PAGE and immunoblotted with antibodies against Diexf (1:500; A305–122A, BETHYL Labs) and Vinculin (1:1000; V9131, Sigma).
Statistical analysis
Student’s t-tests and Kaplan-Meier survival analyses were performed using Prism 5 software (GraphPad Software). P-values < 0.05 were considered statistically significant.
ACKNOWLEDGMENTS
The authors wish to thank Dr. David Lane for helpful suggestions and discussions.
CONFLICTS OF INTEREST
The authors do not have any conflict of interest to declare.
FUNDING
This study was supported by an NIH grant CA47296 (GL) and the MD Anderson Cancer Center Support Grant CA16672.
REFERENCES
1. Lane DP. Cancer: p53, guardian of the genome. Nature. 1992; 358:15–6. https://doi.org/10.1038/358015a0.
2. Pant V, Lozano G. Limiting the power of p53 through the ubiquitin proteasome pathway. Genes Dev. 2014; 28:1739–51. https://doi.org/10.1101/gad.247452.114.
3. Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995; 378:203–6. https://doi.org/10.1038/378203a0.
4. Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, Lozano G. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet. 2001; 29:92–5. https://doi.org/10.1038/ng714.
5. Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995; 378:206–8. https://doi.org/10.1038/378206a0.
6. Jones SN, Hancock AR, Vogel H, Donehower LA, Bradley A. Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proc Natl Acad Sci U S A. 1998; 95:15608–12.
7. Xiong S, Pant V, Zhang Y, Aryal NK, You MJ, Kusewitt D, Lozano G. The p53 inhibitor Mdm4 cooperates with multiple genetic lesions in tumourigenesis. J Pathol. 2017; 241:501–10. https://doi.org/10.1002/path.4854.
8. Wasylishen AR, Lozano G. Attenuating the p53 Pathway in Human Cancers: Many Means to the Same End. Cold Spring Harb Perspect Med. 2016; 6. https://doi.org/10.1101/cshperspect.a026211.
9. Chen J, Ruan H, Ng SM, Gao C, Soo HM, Wu W, Zhang Z, Wen Z, Lane DP, Peng J. Loss of function of def selectively up-regulates Delta113p53 expression to arrest expansion growth of digestive organs in zebrafish. Genes Dev. 2005; 19:2900–11. https://doi.org/10.1101/gad.1366405.
10. Allton K, Jain AK, Herz HM, Tsai WW, Jung SY, Qin J, Bergmann A, Johnson RL, Barton MC. Trim24 targets endogenous p53 for degradation. Proc Natl Acad Sci U S A. 2009; 106:11612–6. https://doi.org/10.1073/pnas.0813177106.
11. Collavin L, Lunardi A, Del Sal G. p53-family proteins and their regulators: hubs and spokes in tumor suppression. Cell Death Differ. 2010; 17:901–11. https://doi.org/10.1038/cdd.2010.35.
12. Jain AK, Barton MC. Regulation of p53: TRIM24 enters the RING. Cell Cycle. 2009; 8:3668–74. https://doi.org/10.4161/cc.8.22.9979.
13. Li L, Deng B, Xing G, Teng Y, Tian C, Cheng X, Yin X, Yang J, Gao X, Zhu Y, Sun Q, Zhang L, Yang X, et al. PACT is a negative regulator of p53 and essential for cell growth and embryonic development. Proc Natl Acad Sci U S A. 2007; 104:7951–6. https://doi.org/10.1073/pnas.0701916104.
14. Liu J, Zhu Y, Hu W, Feng Z. TRIM32 is a novel negative regulator of p53. Mol Cell Oncol. 2015; 2:e970951. https://doi.org/10.4161/23723548.2014.970951.
15. Tao T, Shi H, Guan Y, Huang D, Chen Y, Lane DP, Chen J, Peng J. Def defines a conserved nucleolar pathway that leads p53 to proteasome-independent degradation. Cell Res. 2013; 23:620–34. https://doi.org/10.1038/cr.2013.16.
16. Charette JM, Baserga SJ. The DEAD-box RNA helicase-like Utp25 is an SSU processome component. RNA. 2010; 16:2156–69. https://doi.org/10.1261/rna.2359810.
17. Tao T, Sondalle SB, Shi H, Zhu S, Perez-Atayde AR, Peng J, Baserga SJ, Look AT. The pre-rRNA processing factor DEF is rate limiting for the pathogenesis of MYCN-driven neuroblastoma. Oncogene. 2017; 36:3852–3867. https://doi.org/10.1038/onc.2016.527.
18. Harscoet E, Dubreucq B, Palauqui JC, Lepiniec L. NOF1 encodes an Arabidopsis protein involved in the control of rRNA expression. PLoS One. 2010; 5:e12829. https://doi.org/10.1371/journal.pone.0012829.
19. Bourdon JC, Fernandes K, Murray-Zmijewski F, Liu G, Diot A, Xirodimas DP, Saville MK, Lane DP. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005; 19:2122–37. https://doi.org/10.1101/gad.1339905.
20. Fujita K, Mondal AM, Horikawa I, Nguyen GH, Kumamoto K, Sohn JJ, Bowman ED, Mathe EA, Schetter AJ, Pine SR, Ji H, Vojtesek B, Bourdon JC, et al. p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat Cell Biol. 2009; 11:1135–42. https://doi.org/10.1038/ncb1928.
21. Pant V, Larsson CA, Aryal N, Xiong S, You MJ, Quintas-Cardama A, Lozano G. Tumorigenesis promotes Mdm4-S overexpression. Oncotarget. 2017; 8:25837–47. https://doi.org/10.18632/oncotarget.15552.
22. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–4. https://doi.org/10.1158/2159-8290.CD-12-0095.
23. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1. https://doi.org/10.1126/scisignal.2004088.
24. Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ, Tsui DW, Liu B, Dawson SJ, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016; 7:11479. https://doi.org/10.1038/ncomms11479.
25. Papaioannou VE, Behringer RR. Early embryonic lethality in genetically engineered mice: diagnosis and phenotypic analysis. Vet Pathol. 2012; 49:64–70. https://doi.org/10.1177/0300985810395725.
26. Terzian T, Wang Y, Van Pelt CS, Box NF, Travis EL, Lozano G. Haploinsufficiency of Mdm2 and Mdm4 in tumorigenesis and development. Mol Cell Biol. 2007; 27:5479–85. https://doi.org/10.1128/MCB.00555-06.
27. Pant V, Xiong S, Jackson JG, Post SM, Abbas HA, Quintas-Cardama A, Hamir AN, Lozano G. The p53-Mdm2 feedback loop protects against DNA damage by inhibiting p53 activity but is dispensable for p53 stability, development, and longevity. Genes Dev. 2013; 27:1857–67. https://doi.org/10.1101/gad.227249.113.
28. Goldfeder MB, Oliveira CC. Utp25p, a nucleolar Saccharomyces cerevisiae protein, interacts with U3 snoRNP subunits and affects processing of the 35S pre-rRNA. FEBS J. 2010; 277:2838–52. https://doi.org/10.1111/j.1742-4658.2010.07701.x.
29. Nuaaman MM, Benchimol S. Proteasome-independent p53 degradation. Cell Res. 2013; 23:597–8. https://doi.org/10.1038/cr.2013.38.
30. Danovi D, Meulmeester E, Pasini D, Migliorini D, Capra M, Frenk R, de Graaf P, Francoz S, Gasparini P, Gobbi A, Helin K, Pelicci PG, Jochemsen AG, et al. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol Cell Biol. 2004; 24:5835–43. https://doi.org/10.1128/MCB.24.13.5835-5843.2004.
31. Riemenschneider MJ, Knobbe CB, Reifenberger G. Refined mapping of 1q32 amplicons in malignant gliomas confirms MDM4 as the main amplification target. Int J Cancer. 2003; 104:752–7. https://doi.org/10.1002/ijc.11023.
32. Richard I, Roudaut C, Marchand S, Baghdiguian S, Herasse M, Stockholm D, Ono Y, Suel L, Bourg N, Sorimachi H, Lefranc G, Fardeau M, Sebille A, et al. Loss of calpain 3 proteolytic activity leads to muscular dystrophy and to apoptosis-associated IkappaBalpha/nuclear factor kappaB pathway perturbation in mice. J Cell Biol. 2000; 151:1583–90.
33. Kramerova I, Kudryashova E, Tidball JG, Spencer MJ. Null mutation of calpain 3 (p94) in mice causes abnormal sarcomere formation in vivo and in vitro. Hum Mol Genet. 2004; 13:1373–88. https://doi.org/10.1093/hmg/ddh153.
34. Behringer R, Gertsenstein M, Vintersten Nagy K, Nagy A. Manipulating the Mouse Embryo: A Laboratory Manual, Fourth Edition. New York, NY: CSH Press; 2014. 814 pp.