
INTRODUCTION
T cell acute lymphoblastic leukaemia (T-ALL) is an aggressive haematopoietic malignancy that accounts for 15% of pediatric and 25% of adult ALL cases. Despite improvements in treatment over the years, approximately 25% of children and 50% of adults still fail to respond to intensive chemotherapy protocols or relapse [1]. Treatment options for T-ALL patients are limited. Standard combination chemotherapy regimens (e.g., CHOP) are relatively ineffective against T-ALL. A meta-analysis suggested that AZT therapy in combination with IFN may possess value, although morbidity and a lack of efficacy in patients with prior chemotherapy remain limitations of this regimen [2]. Antibody-based approaches, including those using daclizumab, alemtuzumab, and anti-CCR4, have shown value but are not curative [3–6]. Allogeneic bone marrow transplantation is an aggressive approach that is only curative in select patients [7]. Due to these limitations, new treatment strategies are clearly need for T-ALL.
Previous reports have demonstrated mutational activation in JAK3 in T lymphoblastic leukaemia, especially in NK/T cell lymphoma. Although the JAK2 mutation rate is low, a JAK2 fusion gene was found in T-ALL [8–11]. Last year, the Roncero AM group reported that 4 out of 16 patients with T lymphoblastic lymphoma contained a JAK2 mutation that contributed to T-ALL development. Thus, the use of JAK inhibitors may be considered for T-ALL treatment [12]. TG101209, a small-molecule Jak2-selective inhibitor, was identified by structure-based drug design and was found to be a potent inhibitor of the JAK2V617F and MPLW515L/K mutations commonly associated with primary myelofibrosis [13, 14]. To investigate the effectiveness of the JAK2 inhibitor strategy in T-ALL therapy, the current study analysed the effect of the JAK2 inhibitor TG101209 on 5 T-ALL cell lines. Flow cytometry and 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays showed that proliferation suppression, increased apoptosis and cell cycle arrest in the G2/M phase occurred in the T-ALL cell lines with a JAK2 gene copy gain. We chose 2 of the 5 cell lines that possessed a JAK2 gene copy gain (determined by fluorescence in situ hybridization, FISH) and were sensitive to TG101209 in subsequent experiments. Western blotting (WB) showed that the effect of TG101209 on the cell lines with the JAK2 gene copy number alteration occurred through the JAK-STAT pathway via the regulation of the expression of JAK and STAT family proteins. Interestingly, suppression of the BCL2, Beclin1 and Light Chain 3 (LC3) proteins was also observed in the TG101209-treated T-ALL cell lines, which indicated that crosstalk between apoptosis and autophagy might also be involved in the above phenomenon. The immunostaining results were consistent with the Western blotting results. To determine whether the JAK-STAT pathway and the autophagy status correlated with T-ALL development, we collected samples from patients with T-ALL and analysed the samples by Western blotting and FISH. The results implied that the expression levels of the JAK-STAT proteins and the autophagy-related proteins Beclin1 and LC3 were up-regulated in patients with T-ALL and that most of these patients showed the JAK2 gene copy gain. Therefore, JAK2 may be a potential target for T-ALL treatment. The results from the current study indicated that the JAK2 gene copy gain and the JAK-STAT pathway were highly correlated with T-ALL development. The use of the JAK2 inhibitor TG101209 suppressed T-ALL proliferation by regulating both the JAK-STAT pathway and the crosstalk between apoptosis and autophagy and ultimately inhibiting T-ALL cell proliferation.
RESULTS
Patients with T-ALL showed JAK-STAT pathway activity and up-regulated autophagy
The collected T-ALL patient samples were analysed by Real-time PCR and Western blotting to investigate JAK-STAT pathway activity and autophagy conditions. Comparing to normal control, the JAK/STAT pathway related genes (JAK1, JAK2, JAK3, STAT1, STAT2, STAT3 STAT5B, STAT6) were elevated in T-ALL patients (Supplementary Figure 1) The collected T-ALL patient samples were analysed by Western blotting to investigate JAK-STAT pathway activity and autophagy conditions. All patients with T-ALL showed up-regulated JAK2, JAK3, STAT3, Belclin1 and LC3 expression compared with the healthy controls; representative data are shown in Figure 1A. The JAK2 probe was applied, and the patient samples were analysed with FISH. Three patients showed a JAK2 copy gain; representative data are shown in Figure 1B. These results suggest that JAK-STAT pathway activity and autophagy may be involved in T-ALL development.
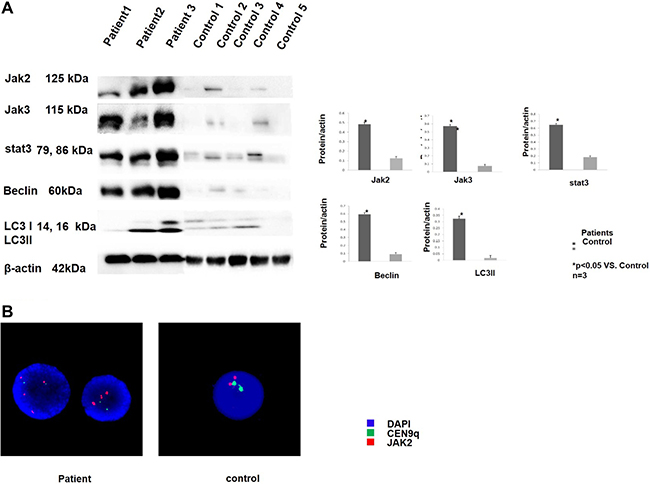
Figure 1: T-ALL patients showed JAK-STAT pathway activity and up-regulated autophagy. (A) The peripheral blood mononuclear cells were collected from 3 T-ALL patients and 5 healthy controls. The cells were lysed and analysed by western blotting. An increase in the JAK-stat pathway-related proteins was observed in all 3 of the patients compared to the healthy controls as shown in the 3 upper lanes. The expression of the autophagy-related proteins was also increased in all 3 of the patients compared to the healthy control as shown in the 2 middle lanes. All of the samples were normalized to β-actin (bottom lane) Bands of western blotting were quantified by densitometry with Scion Image software (Image J 1.48u). We used the LC3B-II/loading control ratio rather than the LC3B II/LC3B-I ratio for qualifcation of LC3-II expression levels according to a newly published guideline. All the results were analysed using SPSS11.0. The graphs were listed respectively. (B) Representative picture of the FISH analysis. The patient samples that possessed a JAK2 copy gain (red dots) are shown on the left, and the control sample that possessed a normal JAK2 copy number (red dots) is shown on the right. All of the samples were also analysed with the CEN9q probe as an internal reference (green dots). The nuclei were all counter-stained with DAPI (blue).
TG101209 down-regulated the JAK-STAT pathway in T-ALL cell lines
The HSD2 and PEER T-ALL cell lines were chosen for the following investigations because both of these cell lines were sensitive to TG101209. The cells were treated with TG101209 for 48 h then collected and lysed for Western blotting. The cells treated with TG101209 showed reduced JAK-STAT pathway protein expression (JAK2, JAK3, STAT3, and STAT5) compared with the control group (Figure 2A), which implied that TG101209 effectively blocked the JAK-STAT signaling pathways. To observe the JAK2 gene activity of the T-ALL cell lines, we synthesized a JAK2 probe and performed FISH analysis. PEER cells, which were the most sensitive to TG101209, showed a karyotype abnormality indicative of a JAK2 gene copy gain (Figure 2B). The JAK2 gene copy gain and the karyotype abnormality occurred at chromosome 9, which might be an important clue for the identification of TG101209-sensitive cell lines.
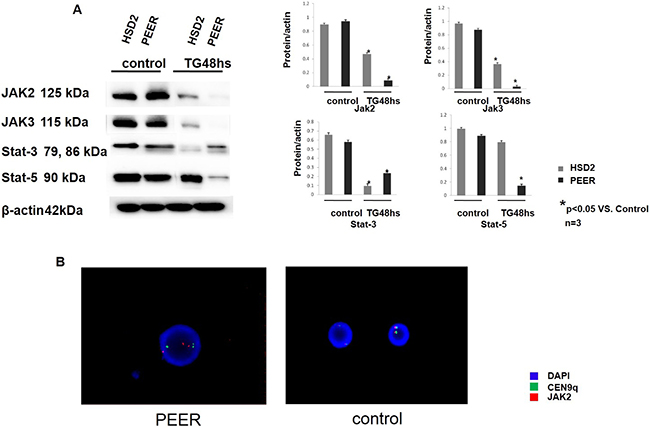
Figure 2: TG101209 down-regulated the JAK-STAT pathway in the T-ALL cell lines. (A) The T-All cell lines HSD2 and PEER were treated with TG101209 for 48 hours and lysed for western blotting. Untreated cells were used as the control group. The expression levels of the JAK-stat pathway related proteins JAK2 (1st lane), JAK3 (2nd lane), Stat3 (3rd lane) and Stat5 (4th lane) were all decreased after TG101209 treatment. All of the samples were normalized with β-actin (bottom lane). Bands of western blotting were quantified by densitometry with Scion Image software (Image J 1.48u). All the results were analysed using SPSS11.0. The graphs were listed respectively. (B) Representative image of the FISH analysis. The PEER, which possess a JAK2 copy gain (red dots), are shown in the left. The control samples, which possesses a normal JAK2 copy number (red dots), is shown on the right. All of the samples were also analysed with the CEN9q probe as an internal reference (green dots). The nuclei were all counter-stained with DAPI (blue).
TG101209 inhibits the proliferation of the T-ALL cell lines and primary bone marrow cells of T-ALL
The DU 528, HSD2, PEER, MOLT-4 and Jurkat T-ALL cell lines were treated with TG101209 (0, 1, 2, 4, 6, 8, or 10 μM), Ruxolitib (0, 1, 2, 4, 6, 8, 10, 20 um) or Dexamethasone (1 μM, 10 μM, 100 μM or 1000 μM), and cell proliferation was analysed using MTT assay. The IC50s of the cell lines were 2.542 μM (DU528), 0.329 μM (HSD2), 0.612 μM (PEER), 2.893 μM (MOLT-4) and 1.794 μM (Jurkat) (Supplementary Figure 2). Apoptosis was increased in the HSD2 and PEER cell lines in a TG101209 concentration-dependent manner, according to the flow cytometry analysis. The expression of apoptosis-related proteins (Bax, Cleaved PARP, caspase-3 and caspase-9) was determined by Western blotting. The expression levels of Bax and Cleaved PARP were up-regulated by TG101209, while caspase-3 and caspase-9 were down-regulated in both HSD2 and PEER cell lines (Figure 3A). The cell cycle of each cell line was analysed using flow cytometry. After treatment, the cell cycle was arrested mainly at the G2/M phase. The expression of the indicated cell cycle-related proteins (P21, P27, CDK4 and CDK6) was determined by Western blotting. The expression levels of P21 and P27 were up-regulated by TG101209, while those of CDK4 and CDK6 were down-regulated by TG101209 in both the HSD2 and PEER cell lines (Figure 3B). Primary bone marrow cells from T-ALL patients and healthy controls were treated with TG101209 (0, 1, 2, 4, 6, 8, or 10 μM), and cell proliferation was analysed using MTT assay. The IC50s were 0.755um and 1.565 um respectively. (Supplementary Figure 2). Apoptosis was increased in the Primary bone marrow cells from T-ALL patients in a TG101209 concentration-dependent manner, according to the flow cytometry analysis. (Figure 3A)
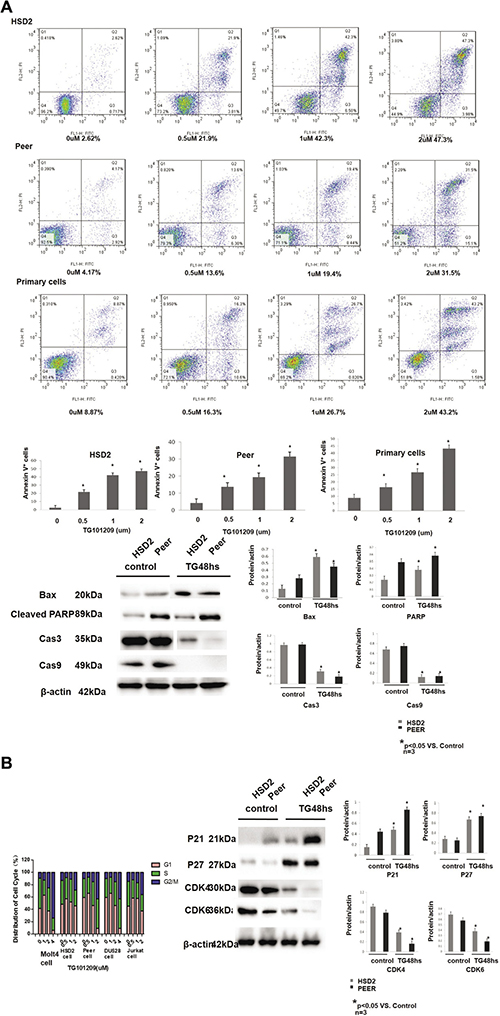
Figure 3: TG101209 inhibits the proliferation of the T-ALL cell lines by inducing apoptosis and cell cycle arrest. (A) HSD2, peer and bone marrow primary T-ALLcells were treated with increasing concentrations of TG101209 for 48 h, which was followed by analysis of apoptosis by staining with PI and Annexin-V FITC. Annexin-V positive cells were measured by flow cytometry. Columns represent the average percent of Annexin V positive cells from more than 3 independent experiments, which are shown as the mean ± SD. Asterisks (*) represents statistically signifcant differences p < 0.05, respectively. Representative images are shown in the upper panel. The T-All cell lines HSD2 and PEER were treated with TG101209 for 48 hours and lysed for western blotting. Untreated cells were used as the control group. The expressions of apoptosis related proteins Bax (1st lane), CleavedPARP (2ND lane) were increased while Cas3 (3rd lane), and Cas9 (4th lane) were decreased after TG101209 treatment. All of the samples were normalized with β-actin (bottom lane). Bands of western blotting were quantified by densitometry with Scion Image software (Image J 1.48u). All the results were analysed using SPSS11.0. The graphs were listed respectively. (B) The Jurkat, HSD2 and PEER cell lines were treated with TG101209 (0, 0.5, 1, or 2 μM) for 48 hrs. The DU528 and MOLT-4 cell lines were treated with TG101209 (0, 1, 2, or 4 μM) for 48 hrs. The cells were collected and stained with PI, and the cell cycle distribution was analysed with flow cytometry (FACSCalibur, BD Biosciences). The data were summarized. The results showed that the cell cycle was mainly arrested in the G2/M phase in each cell line after treatment. The T-All cell lines HSD2 and PEER were treated with TG101209 for 48 hours and lysed for western blotting. Untreated cells were used as the control group. The expressions of cell cycle related proteins P21 (1st lane) , P27 (2nd lane) were increased while CDK4 (3rd lane), CDK6 (4th lane) were decreased after TG101209 treatment. All of the samples were normalized with β-actin (bottom lane). Bands of western blotting were quantified by densitometry with Scion Image software (Image J 1.48u). All the results were analysed using SPSS11.0. The graphs were listed respectively.
TG101209 regulates the crosstalk between apoptosis and autophagy
In addition to the JAK-STAT pathway, crosstalk between apoptosis and autophagy was also investigated using Western blotting analysis. The expression of the anti-apoptotic protein BCL2 was significantly decreased after TG101209 treatment, and the expression of the autophagy-related proteins Beclin 1 and LC3 was also decreased. These data suggest that crosstalk between apoptosis and autophagy might be involved in the TG101209-induced T-ALL cell line inhibition by the JAK/STAT/BCL2 pathway (Figure 4A). Immunofluorescence cytochemistry was performed to confirm our observations. HSD2 and PEER cells were treated with TG101209 or TG101209 + Dex, and their LC3 expression levels were compared with that of an untreated group. LC3 expression was dramatically decreased after TG or TG + Dex treatment, which was consistent with the Western blotting results (Figure 4B). The autophagy inhibition induced by TG101209 was compared with that of hydroxychloroquine sulfate (HCQ), a widely used autophagy inhibitor. We found that the effect of TG101209 (2 μM) was comparable to that HCQ (75–100 μM) in down-regulating the expression of autophagy-related proteins (BCL-2, Beclin and LC3) (Supplementary Figures 3 and 4).
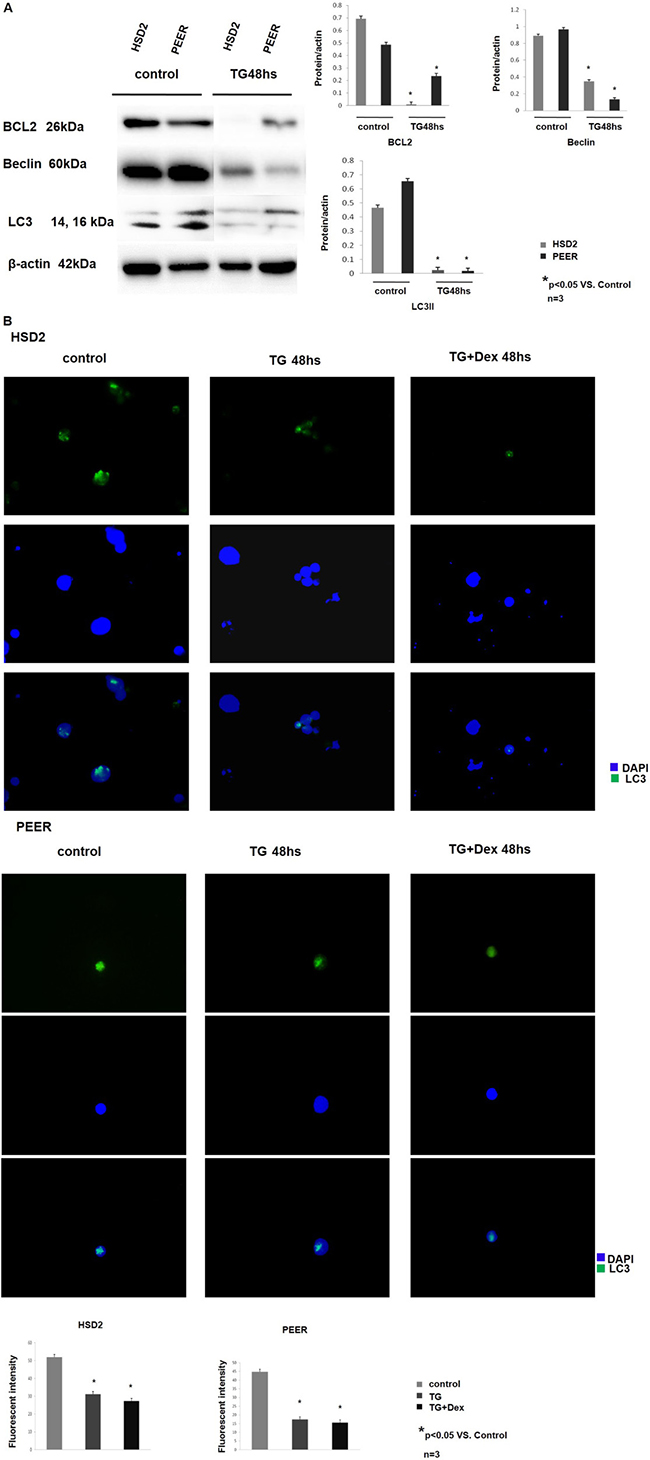
Figure 4: TG101209 regulates the crosstalk between apoptosis and autophagy. (A) The T-All cell lines HSD2 and PEER were treated with TG101209 for 48 hours and lysed for western blotting. Untreated cells were used as the control group. The expression levels of BCL2 (1st lane), Beclin (2nd lane), and LC3 (3rd lane) were decreased after TG101209 treatment. All of the samples were normalized with β-actin (bottom lane). Bands of western blotting were quantified by densitometry with Scion Image software (Image J 1.48u). All the results were analysed using SPSS11.0. The graphs were listed respectively. (B) The HSD2 and PEER cell lines were treated with TG101209 or TG101209+Dex, and their LC3 expression levels were compared with that of the untreated group (green) by immunofluorescence cytochemistry. Representative data showed that LC3 expression was dramatically decreased after TG or TG+Dex treatment. The nuclei were all counter-stained with DAPI (blue). The fluorescence intensity of single cell was quantifed by densitometry with Scion Image software (Image J 1.48u). All the results were analysed using SPSS11.0. The graphs were listed respectively.
DISCUSSION
The JAK-STAT pathway plays an important role in lymphoid precursor cell proliferation, survival and differentiation [15–17]. Aberrant JAK-STAT signal transduction was first reported in T-ALLs exhibiting the t(9; 12)(p24; p13) translocation, which is a rare rearrangement resulting in a constitutively active TEL-JAK2 kinase fusion oncoprotein [18]. Activating JAK2 mutations in other haematological malignancies, such as myeloproliferative neoplasms, have demonstrated the clinical relevance of this gene and drawn more attention to its potential involvement in T-cell lymphoblastic leukaemia/lymphoma [19–21]. In a previous study, a combination of a JAK inhibitor and a Bcl-2/Bcl-xL inhibitor provided antitumour efficacy in ATL cell lines and a mouse model of human ATL [22].
Ramakrishnan’s study examined the pre-clinical activity of the JAK2 inhibitor TG101209, which induced dose- and time-dependent cytotoxicity in a variety of multiple myeloma cell lines. The induction of cytotoxicity was associated with the inhibition of cell cycle progression and the induction of apoptosis in the myeloma cell lines and patient-derived plasma cells [23]. In Pardanani’s study, TG101209 potently inhibited myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations [13]. TG101209 was also shown to enhance radiotherapy in lung cancer models by inhibiting JAK2 signaling [24].
In the current study, the JAK2 copy gain determined by FISH and real-time PCR occurred frequently in the T-ALL cell lines and patients (Figure 1B, Figure 2B, Supplementary Figure 1), suggesting that this copy gain was the most common type of JAK2 activation in T-ALL. The efficacy of the JAK2 inhibitor TG101209 was observed in 5 T-ALL cell lines. We found that 2 out of the 5 cell lines with a JAK2 gene copy number alteration responded to the inhibitor.
Cell cycle arrest and apoptosis were induced by TG101209 through the p21/p21-CDK4/CDK6 pathway and the BAX/BCL2-caspase3/caspase9 pathway. P21 and P27, cyclin-dependent kinase (CDK) inhibitors are negative regulators of cell cycle progression. These regulators inhibit CDK activity, resulting in decreased cell proliferation [25]. After TG101209 treatment, the protein level of p21 was significantly elevated in the corresponding group compared with the control group. A similar effect on the expression level of p27, which was accompanied by a decrease in the level of CDK4 and CDK6, was also observed. The caspase and Bcl-2 protein families play crucial roles in the initiation of apoptosis [26]. Caspase-3 has been considered the major executive caspase in apoptosis. It operates as the key enzyme in the mitochondria-dependent apoptosis pathway. Therefore, measurement of caspase-3 activity is the major and the most reliable determining factor for apoptosis [27]. The Bcl-2 family of proteins include both pro-apoptotic (e.g., Bax) and anti-apoptotic (e.g., Bcl-2) members [28]. The anti-apoptotic protein Bcl-2 can regulate apoptosis through caspase-9- and -3-dependent pathways [29]. Increased Bax and decreased Bcl-2 expression can stimulate the release of cytochrome c from the mitochondria, which activates caspase-9. Caspase-9 then catalyzes the activation of caspase-3, ultimately leading to apoptosis [30]. Poly (ADP-ribose) polymerase (PARP) is a family of proteins involved in a number of cellular processes, such as DNA repair, genomic stability, and programmed cell death. PARP is inactivated by caspase cleavage. It is believed that normal inactivation occurs in systems where DNA damage is extensive. In these cases, more energy would be invested in repairing damage than is feasible; instead, that energy is retrieved for other cells in the tissue through apoptosis [31].
The expression of the anti-apoptotic protein BCL-2 and the autophagy-related protein Beclin1 was also down-regulated in addition to the JAK-STAT pathway. The BCL-2 family regulates the mitochondrial apoptosis pathway through complex interactions that determine the integrity of the outer mitochondrial membrane [32, 33]. The BCL-2 family is divided into three groups according to their functions. The first group (BCL-X, BCL-2 BCL-W and MCL-1) contains three or four BCL-2 homology (BH) domains that are necessary for the anti-apoptotic function. The second group contains BAX and BAK, which induce mitochondrial permeabilization and the release of cell death-promoting proteins. The third group consists of proteins that contain only the short BH3 domain. These BH3-only proteins interact with both anti- and pro-apoptotic BCL-2 members to induce programmed cell death [34].
In addition to participating in apoptosis, BCl-2 also plays a role in the crosstalk between apoptosis and autophagy by interacting with Beclin 1, which plays a critical role in autophagosome formation. Beclin possesses a BCL-2 homology (BH) 3 region that physically interacts with BCL-2 proteins. The binding of BCL-2 to Beclin1 reduces the capacity of Beclin 1 to activate autophagy [35, 36]. In our study, the de-regulation of both BCL-2 and Beclin 1 after TG10129 treatment implied that TG10129 might function as not only a JAK2 inhibitor to reduce the proliferation of T-ALL cells but also as a regulator of apoptosis and autophagy.
The autophagy marker LC3 was originally identified as a subunit of microtubule-associated proteins 1A and 1B (termed MAP1LC3) and was subsequently found to contain similarities to the yeast protein Apg8/Aut7/Cvt5, which is critical for autophagy. Cleavage of LC3 at the carboxy terminus immediately following synthesis yields the cytosolic LC3-I form. During autophagy, LC3-I is converted to LC3-II through lipidation by a ubiquitin-like system involving Atg7 and Atg3 that allows LC3 to become associated with autophagic vesicles. The presence of LC3 in autophagosomes and the conversion of LC3 to the lower migrating form LC3-II are used as indicators of autophagy [37, 38]. We observed that LC3 II was down-regulated in Western blotting and immunofluorescence cytochemistry assays. In a previous study, inhibiting autophagy significantly enhanced rhArg-induced cell growth inhibition and apoptosis [39]. The autophagy inhibition induced by TG101209 was compared with that of HCQ, a widely used autophagy inhibitor [40]. HCQ could down-regulate autophagy-related proteins in a concentration-dependent manner. We also showed that TG101209 (2 μM) was comparable to HCQ (75–100 μM) in terms of down-regulating the expression of autophagy-related proteins (BCL-2, Beclin and LC3) (Supplementary Figures 3 and 4). We demonstrated that TG101209 down-regulated both anti-apoptosis- and autophagy-related proteins and thus might enhance cell growth inhibition and apoptosis.
Interestingly, by analysing the T-ALL patient samples and comparing them with healthy controls, we found JAK2 gene copy gains and high expression, as well as Beclin 1 and LC3 up-regulation, which indicated that JAK2 activation plays a very important role in the development of T-ALL and that it is a target for the treatment of T-ALL. The JAK inhibitor TG101209 decreased the JAK/STAT pathway and then down-regulated BCL2, which was the key effect in the crosstalk between apoptosis and autophagy; thus, this cascade inhibited the up-regulation of autophagy, decreased the proliferation of T-ALL cells and increased their apoptosis.
Our research provide the evdence of TG101209 anti-leukemic effection in vitro, we need firrther clinical trial to comfirm the curitive effect, and multiple pathogenesis and different targets may limite its application, TG101209 combind with chemotherapy may work out in the future.
MATERIALS AND METHODS
Patient samples
The study protocol was approved by the ethics committee of the Second Xiangya Hospital, Central South University, P.R. China. Informed consent was obtained in writing in accordance with the Declaration of Helsinki.Bone marrow/peripheral blood mononuclear cells of 21 patients novel diagnosed asT-ALL (9females, 12males, median age 32) and 23 healthy controls (10 females,13males, median age 29) were collected at the second Xiangya Hospital, Central South University, P.R. China. 3 patient samples and 5 healthy control samples were used for western blotting. 5 patient samples and 5 healthy control samples were used for MTT assay. 3 patient samples and 3 heathly control samples were used for FISH. 5 patient samples and 5 healthy control samples were used for apoptosis assay. 5 patient samples and 5 healthy control samples were used for PCR. The diagnosis of T-ALL was based on morphology, cytochemistry and immunophenotyping according to the World Health Organization [41].
T-ALL cell line maintenance
The T-ALL cell lines (HSD2, DU528, PEER, MOLT-4 and Jurkat) (Supplementary Table 1) were kindly provided by the A. Thomas Look lab at the Dana-Farber Cancer Institute at Harvard Medical School. All cell lines were maintained in RPMI 1640 medium with 10% FBS.
MTT assay
The T-ALL cell lines (HSD2, DU528, PEER, MOLT-4 and Jurkat) were treated with various concentrations of TG101209 (Selleck, S2692) (0, 1, 2, 4, 6, 8, or 10 μM), Ruxolitib (Dacogen, Xian Janssen, Johnson-Johnsons, USA) (0,1,2,4,6,8,10,20μM) and Dexamethasone (D4902, Sigma-Aldrich, Merck) (0,1,10,100,500,1000 μM) respectively for 48 h. Bone marrow cells collected from patients with T-ALL were treated with various concentrations of TG101209 (0, 1, 2, 4, 6, 8, or 10 μM) for 48h. MTT assay was conducted following the instructions provided by the manufacturer. The IC50 value was determined for each cell line. All experiments were repeated 3 times.
Flow-cytometry (cell cycle)
Jurkat, HSD2 and PEER cells were treated with TG101209 (0, 0.5, 1, or 2 μM) for 48 h. DU528 and MOLT-4 cells were treated with TG101209 (0, 1, 2, or 4 μM) for 48 h. The cells were collected and stained with propidium iodide (PI). The cell cycle distribution was determined using flow cytometry (FACSCalibur, BD Biosciences). All experiments were repeated 3 times.
Flow cytometry (apoptosis)
HSD2 cells , Peer cells and primary bone marrow cells derived from T-ALL patients were treated with TG101209 (0, 0.5, 1, or 2 μM) for 48 h respectively. Cells were double stained with Annexin V-FITC /PI and analysed by flow cytometry to determine the apoptosis status (FACSCalibur, BD Biosciences). All experiments were repeated at least 3 times.
Western blotting
The Western blotting analysis was performed as previously described [42]. Briefly, the cells were treated with TG101209 (2 μM) or hydroxychloroquine sulfate (HCQ) (Selleck, S4430) (25, 50, 75, 100 μM) for 48 h or were collected from the clinical T-ALL patients. The cells were harvested, and an equivalent amount of the cell lysate was subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to polyvinylidene fluoride (PVDF) membranes for the Westsern blotting assay. The membranes were incubated with the JAK2 (D2E12) XP® Rabbit monoclonal antibody (mAb) (Cell Signaling Technology, #3230), STAT3 (124H6) mouse mAb (Cell Signaling Technology, #9139), STAT5 antibody (Cell Signaling Technology, #9363), Bcl-2 (50E3) rabbit mAb (Cell Signaling Technology, #2870), Bax (D2E11) rabbit mAb (Cell Signaling Technology, #5023S ) , PARP (46D11) Rabbit mAb (Cell Signaling Technology, #9532S), caspase-3 (8G10) Rabbit mAb (Cell Signaling Technology, #9665S) and ,caspase-9 (D2D4) Rabbit mAb, #7237S Beclin-1 antibody (Cell Signaling Technology, #3738), and LC3 (Cell Signaling Technology, #12741) antibody and the appropriate horseradish peroxidase (HRP)-linked anti-rabbit/mouse IgG secondary antibodies (Cell Signaling Technology, #7074), according to the species of the primary antibody. The membranes were treated with an enhanced chemiluminescent detection kit (Thermo Fisher Scientific, #17097), and the signals were collected and photographed (Bio-Rad, Gel Doc, XR). All experiments were repeated 3 times. These bands were quantified by densitometry with Scion Image software (ImageJ 1.48u). We used the LC3B-II/loading control ratio rather than the LC3B II/LC3B-I ratio to quantify LC3-II expression levels according to a newly published guideline [43].
Immunofluorescence cytochemistry
The Peer and HSD-2 cell lines were treated with TG101209 (2 μM), HCQ (50 μM, 100 μM) or TG101209 (1 μM) + Dex (100 μM) for 48 h and then collected and fixed in ice-cold 100% methanol for 15 min at -20°C. Then, the cells were smeared onto an amino silane (APS)-coated slide. After air dying, the samples were blocked in blocking buffer (1X PBS, 5% normal serum, and 0.3% Triton™ X-100) for 60 min at room temperature. LC3 was diluted in antibody dilution buffer (1X PBS, 1% bovine serum albumin (BSA), and 0.3% Triton X-100) at a 1:100 ratio and incubated at 4°C overnight, followed by incubation with a fluorochrome-conjugated secondary antibody diluted in antibody dilution buffer for 1 h at room temperature in the dark. The slides were also incubated with the ProLong Gold Antifade reagent with DAPI (Cell Signaling Technology, #8961). The samples were observed and imaged using a Zeiss AX10 Scope A1. All experiments were repeated 3 times. The fluorescence intensity of single cell was quantified by densitometry with Scion Image software (ImageJ 1.48u).
Fluorescence in situ hybridization (FISH)
The JAK2/CEN9q FISH probe (Cat No. FA0225, Lot No. GC158-GBCBI) was purchased from the ABNOVA (Taiwan) Corporation. The size of the JAK2 probe was approximately 290 kb, the fluorophore was Texas red, and the location was 9q24. The size of the CEN9q probe was approximately 470 kb, the fluorophore was FITC, and the location was 9p21. FISH was performed following the manufacturer’s instructions. Cell lines treated with TG101209 or patient samples were seeded and treated with colchicine (20 μg/ml); then, the cells were centrifuged, resuspended in hypotonic solution and incubated in a 37°C water bath for 1 h. The cells were fixed with fixing solution (methanol:triacetate = 3:1). After dehydration with 95% EtOH, the cells were smeared onto a slide, and the FISH probe was applied. The samples were denatured at 75°C for 5 min, and hybridization was performed in a humidified box at 37°C for 24 h. After hybridization, the samples were dipped into 2X SSC at room temperature for 5 min, followed by 2X SSC/0.3% NP-40 at 75°C for 2 min, and then finally transferred into 2X SSC and incubated at room temperature for 1 min. DAPI was applied as a counter-stain, and the target area was covered with a glass cover slip. The samples were observed and imaged using an Olympus BX50 fluorescence microscope. All experiments were repeated 3 times.
CONCLUSIONS
Based on the above evidence, JAK2 inhibitor -TG101209 may be an alternative choose for the therapy of T-ALL in the near future.
Ethics approval and consent to participate
This study was performed in strict accordance with recommendations of the regulations on human and animal experimentation of Central South University (China). All experimental protocols were approved by the ethics committee of the Second Xianya Hospital of Central South University (China).
Consent for publication
Not applicable.
Availability of data and material
All data generated or analysed during this study are included in this published article (and its supplementary information files).
Abbreviations
T-ALL: T cell acute lymphoblastic leukemia; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay; FISH: fluorescence in situ hybridization; EtOH: ethanol CH3CH2OH; Dex: Dexamethasone, DXMS; TG: TG101209.
Author contributions
Zhao Cheng: Data acquisition, analysis of the data and writing of the paper; YiFang Yi: Data acquisition; Sisi Xie: Data acquisition; Haizhi Yu: Data acquisition; Hong-ling Peng: Study design and manuscript revisions; Guangsen Zhang: Manuscript revisions and comments reaction.
ACKNOWLEDGMENTS
Dr. A. Thomas Look of the Dana-Farber Cancer Institute at Harvard Medical School kindly provided the T-ALL cell lines (HSD2, DU528, PEER, MOLT-4 and Jurkat).
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
FUNDING
This work was supported by the National Natural Science Foundation 81200368. This work was supported by the National Natural Science Foundation 81400093. This work was supported by the National Natural Science Foundation 81670160.
REFERENCES
1. Vitale A, Guarini A, Ariola C, Mancini M, Mecucci C, Cuneo A, Pane F, Saglio G, Cimino G, Tafuri A, Meloni G, Fabbiano F, Recchia A, et al. Adult T-cell acute lymphoblastic leukemia: biologic profile at presentation and correlation with response to induction treatment in patients enrolled in the GIMEMA LAL 0496 protocol. Blood. 2006; 107:473–9. https://doi.org/10.1182/blood-2005-04-1754.
2. Gill PS, Harrington W Jr, Kaplan MH, Ribeiro RC, Bennett JM, Liebman HA, Bernstein-Singer M, Espina BM, Cabral L, Allen S, Kornblau S, Pike MC, Levine AM. Treatment of adult T-cell leukemia-lymphoma with a combination of interferon alfa and zidovudine. N Engl J Med. 1995; 332:1744–8. https://doi.org/10.1056/NEJM199506293322603.
3. Berkowitz JL, Janik JE, Stewart DM, Jaffe ES, Stetler-Stevenson M, Shih JH, Fleisher TA, Turner M, Urquhart NE, Wharfe GH, Figg WD, Peer CJ, Goldman CK, et al. Safety, efficacy, and pharmacokinetics/pharmacodynamics of daclizumab (anti-CD25) in patients with adult T-cell leukemia/lymphoma. Clin Immunol. 2014; 155:176–87. https://doi.org/10.1016/j.clim.2014.09.012.
4. Yamamoto K, Utsunomiya A, Tobinai K, Tsukasaki K, Uike N, Uozumi K, Yamaguchi K, Yamada Y, Hanada S, Tamura K, Nakamura S, Inagaki H, Ohshima K, et al. Phase I study of KW-0761, a defucosylated humanized anti-CCR4 antibody, in relapsed patients with adult T-cell leukemia-lymphoma and peripheral T-cell lymphoma. J Clin Oncol. 2010; 28:1591–8. https://doi.org/10.1200/JCO.2009.25.3575.
5. Ishida T, Joh T, Uike N, Yamamoto K, Utsunomiya A, Yoshida S, Saburi Y, Miyamoto T, Takemoto S, Suzushima H, Tsukasaki K, Nosaka K, Fujiwara H, et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: a multicenter phase II study. J Clin Oncol. 2012; 30:837–42. https://doi.org/10.1200/JCO.2011.37.3472.
6. Marcais A, Suarez F, Sibon D, Bazarbachi A, Hermine O. Clinical trials of adult T-cell leukaemia/lymphoma treatment. Leuk Res Treatment. 2012; 2012:932175. https://doi.org/10.1155/2012/932175.
7. Hishizawa M, Kanda J, Utsunomiya A, Taniguchi S, Eto T, Moriuchi Y, Tanosaki R, Kawano F, Miyazaki Y, Masuda M, Nagafuji K, Hara M, Takanashi M, et al. Transplantation of allogeneic hematopoietic stem cells for adult T-cell leukemia: a nationwide retrospective study. Blood. 2010; 116:1369–76. https://doi.org/10.1182/blood-2009-10-247510.
8. Elliott NE, Cleveland SM, Grann V, Janik J, Waldmann TA, Dave UP. FERM domain mutations induce gain of function in JAK3 in adult T-cell leukemia/lymphoma. Blood. 2011; 118:3911–21. https://doi.org/10.1182/blood-2010-12-319467.
9. Cornejo MG, Kharas MG, Werneck MB, Le Bras S, Moore SA, Ball B, Beylot-Barry M, Rodig SJ, Aster JC, Lee BH, Cantor H, Merlio JP, Gilliland DG, et al. Constitutive JAK3 activation induces lymphoproliferative syndromes in murine bone marrow transplantation models. Blood. 2009; 113:2746–54. https://doi.org/10.1182/blood-2008-06-164368.
10. Koo GC, Tan SY, Tang T, Poon SL, Allen GE, Tan L, Chong SC, Ong WS, Tay K, Tao M, Quek R, Loong S, Yeoh KW, et al. Janus kinase 3-activating mutations identified in natural killer/T-cell lymphoma. Cancer Discov. 2012; 2:591–7. https://doi.org/10.1158/2159-8290.CD-12-0028.
11. Atak ZK, Gianfelici V, Hulselmans G, De Keersmaecker K, Devasia AG, Geerdens E, Mentens N, Chiaretti S, Durinck K, Uyttebroeck A, Vandenberghe P, Wlodarska I, Cloos J, et al. Comprehensive analysis of transcriptome variation uncovers known and novel driver events in T-cell acute lymphoblastic leukemia. PLoS Genet. 2013; 9:e1003997. https://doi.org/10.1371/journal.pgen.1003997.
12. Roncero AM, Lopez-Nieva P, Cobos-Fernandez MA, Villa-Morales M, Gonzalez-Sanchez L, Lopez-Lorenzo JL, Llamas P, Ayuso C, Rodriguez-Pinilla SM, Arriba MC, Piris MA, Fernandez-Navarro P, Fernandez AF, et al. Contribution of JAK2 mutations to T-cell lymphoblastic lymphoma development. Leukemia. 2016; 30:94–103. https://doi.org/10.1038/leu.2015.202.
13. Pardanani A, Hood J, Lasho T, Levine RL, Martin MB, Noronha G, Finke C, Mak CC, Mesa R, Zhu H, Soll R, Gilliland DG, Tefferi A. TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations. Leukemia. 2007; 21:1658–68. https://doi.org/10.1038/sj.leu.2404750.
14. Wang Y, Fiskus W, Chong DG, Buckley KM, Natarajan K, Rao R, Joshi A, Balusu R, Koul S, Chen J, Savoie A, Ustun C, Jillella AP, et al. Cotreatment with panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood. 2009; 114:5024–33. https://doi.org/10.1182/blood-2009-05-222133.
15. Pike KA, Tremblay ML. TC-PTP and PTP1B: Regulating JAK-STAT signaling, controlling lymphoid malignancies. Cytokine. 2016; 82:52–7. https://doi.org/10.1016/j.cyto.2015.12.025.
16. Mace TA, King SA, Ameen Z, Elnaggar O, Young G, Riedl KM, Schwartz SJ, Clinton SK, Knobloch TJ, Weghorst CM, Lesinski GB. Bioactive compounds or metabolites from black raspberries modulate T lymphocyte proliferation, myeloid cell differentiation and Jak/STAT signaling. Cancer Immunol Immunother. 2014; 63:889–900. https://doi.org/10.1007/s00262-014-1564-5.
17. Muthian G, Bright JJ. Quercetin, a flavonoid phytoestrogen, ameliorates experimental allergic encephalomyelitis by blocking IL-12 signaling through JAK-STAT pathway in T lymphocyte. J Clin Immunol. 2004; 24:542–52. https://doi.org/10.1023/B:JOCI.0000040925.55682.a5.
18. Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffe M, Berthou C, Lessard M, Berger R, Ghysdael J, Bernard OA. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. 1997; 278:1309–12.
19. Baran-Marszak F, Magdoud H, Desterke C, Alvarado A, Roger C, Harel S, Mazoyer E, Cassinat B, Chevret S, Tonetti C, Giraudier S, Fenaux P, Cymbalista F, et al. Expression level and differential JAK2-V617F-binding of the adaptor protein Lnk regulates JAK2-mediated signals in myeloproliferative neoplasms. Blood. 2010; 116:5961–71. https://doi.org/10.1182/blood-2009-12-256768.
20. Passamonti F, Maffioli M, Caramazza D, Cazzola M. Myeloproliferative neoplasms: from JAK2 mutations discovery to JAK2 inhibitor therapies. Oncotarget. 2011; 2:485–90. https://doi.org/10.18632/oncotarget.281.
21. Akada H, Akada S, Hutchison RE, Sakamoto K, Wagner KU, Mohi G. Critical role of Jak2 in the maintenance and function of adult hematopoietic stem cells. Stem Cells. 2014; 32:1878–89. https://doi.org/10.1002/stem.1711.
22. Zhang M, Mathews Griner LA, Ju W, Duveau DY, Guha R, Petrus MN, Wen B, Maeda M, Shinn P, Ferrer M, Conlon KD, Bamford RN, O'Shea JJ, et al. Selective targeting of JAK/STAT signaling is potentiated by Bcl-xL blockade in IL-2-dependent adult T-cell leukemia. Proc Natl Acad Sci U S A. 2015; 112:12480–5. https://doi.org/10.1073/pnas.1516208112.
23. Ramakrishnan V, Kimlinger T, Haug J, Timm M, Wellik L, Halling T, Pardanani A, Tefferi A, Rajkumar SV, Kumar S. TG101209, a novel JAK2 inhibitor, has significant in vitro activity in multiple myeloma and displays preferential cytotoxicity for CD45+ myeloma cells. Am J Hematol. 2010; 85:675–86. https://doi.org/10.1002/ajh.21785.
24. Sun Y, Moretti L, Giacalone NJ, Schleicher S, Speirs CK, Carbone DP, Lu B. Inhibition of JAK2 signaling by TG101209 enhances radiotherapy in lung cancer models. J Thorac Oncol. 2011; 6:699–706. https://doi.org/10.1097/JTO.0b013e31820d9d11.
25. Sherr CJ. Cancer cell cycles. Science. 1996; 274:1672–7.
26. Yang HL, Dong YB, Elliott MJ, Liu TJ, McMasters KM. Caspase activation and changes in Bcl-2 family member protein expression associated with E2F-1-mediated apoptosis in human esophageal cancer cells. Clin Cancer Res. 2000; 6:1579–89.
27. Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999; 6:99–104. https://doi.org/10.1038/sj.cdd.4400476.
28. Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004; 116:205–19.
29. Marsden VS, O'Connor L, O'Reilly LA, Silke J, Metcalf D, Ekert PG, Huang DC, Cecconi F, Kuida K, Tomaselli KJ, Roy S, Nicholson DW, Vaux DL, et al. Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature. 2002; 419:634–7. https://doi.org/10.1038/nature01101.
30. Cain K, Bratton SB, Cohen GM. The Apaf-1 apoptosome: a large caspase-activating complex. Biochimie. 2002; 84:203–14.
31. Dale Rein I, Solberg Landsverk K, Micci F, Patzke S, Stokke T. Replication-induced DNA damage after PARP inhibition causes G2 delay, and cell line-dependent apoptosis, necrosis and multinucleation. Cell Cycle. 2015; 14:3248–60. https://doi.org/10.1080/15384101.2015.1085137.
32. Adachi S, Cross AR, Babior BM, Gottlieb RA. Bcl-2 and the outer mitochondrial membrane in the inactivation of cytochrome c during Fas-mediated apoptosis. J Biol Chem. 1997; 272:21878–82.
33. Tan C, Dlugosz PJ, Peng J, Zhang Z, Lapolla SM, Plafker SM, Andrews DW, Lin J. Auto-activation of the apoptosis protein Bax increases mitochondrial membrane permeability and is inhibited by Bcl-2. J Biol Chem. 2006; 281:14764–75. https://doi.org/10.1074/jbc.M602374200.
34. Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010; 37:299–310. https://doi.org/10.1016/j.molcel.2010.01.025.
35. Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, Hickman JA, Geneste O, Kroemer G. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007; 26:2527–39. https://doi.org/10.1038/sj.emboj.7601689.
36. Maiuri MC, Criollo A, Tasdemir E, Vicencio JM, Tajeddine N, Hickman JA, Geneste O, Kroemer G. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L). Autophagy. 2007; 3:374–6.
37. Kouno T, Mizuguchi M, Tanida I, Ueno T, Kanematsu T, Mori Y, Shinoda H, Hirata M, Kominami E, Kawano K. Solution structure of microtubule-associated protein light chain 3 and identification of its functional subdomains. J Biol Chem. 2005; 280:24610–7. https://doi.org/10.1074/jbc.M413565200.
38. Fass E, Amar N, Elazar Z. Identification of essential residues for the C-terminal cleavage of the mammalian LC3: a lesson from yeast Atg8. Autophagy. 2007; 3:48–50.
39. Li Y, Zeng X, Wang S, Fan J, Wang Z, Song P, Mei X, Ju D. Blocking autophagy enhanced leukemia cell death induced by recombinant human arginase. Tumour Biol. 2016; 37:6627–35. https://doi.org/10.1007/s13277-015-4253-x.
40. Ye H, Chen M, Cao F, Huang H, Zhan R, Zheng X. Chloroquine, an autophagy inhibitor, potentiates the radiosensitivity of glioma initiating cells by inhibiting autophagy and activating apoptosis. BMC Neurol. 2016; 16:178. https://doi.org/10.1186/s12883-016-0700-6.
41. Bene MC, Castoldi G, Knapp W, Ludwig WD, Matutes E, Orfao A, van't Veer MB. Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL). Leukemia. 1995; 9:1783–6.
42. Cheng Z, Ito S, Nishio N, Thanasegaran S, Fang H, Isobe K. Characteristics of cardiac aging in C57BL/6 mice. Exp Gerontol. 2013; 48:341–8. https://doi.org/10.1016/j.exger.2013.01.005.
43. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, Adhihetty PJ, Adler SG, Agam G, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016; 12:1–222. https://doi.org/10.1080/15548627.2015.1100356.