
INTRODUCTION
Phosphatidylinositol-4,5-bisphosphate 3-kinases (PI3Ks) are belong to a family of enzymes, which are response to growth, cell proliferation, apoptosis, angiogenesis, DNA repair, motility, differentiation and survival [1, 2]. In cell signaling pathway, PI3Ks function as intermediate signaling molecules including PI3K/AKT/mTOR signaling pathway [1, 3, 4]. NVP-BKM120 (Buparlisib) is a small molecule orally-available compound, which has potent pan-class I PI3K inhibitory capability against the p110α/β/δ/γ catalytic subunit isoforms [5, 6]. Previously studies demonstrated that NVP-BKM120 exerts anti-proliferative and cytotoxic effects on solid tumor and hematological malignancies via selective AKT inhibition [7]. Several recent reports also demonstrated that NVP-BKM120 combine with other signaling pathways inhibitors promote its antitumor effects in mouse models [8-10].
P53 upregulated modulator of apoptosis (PUMA, also known as BBC3) belongs to proapototic protein, member of BH3-only Bcl-2 protein family. In cancer cells, PUMA play a key role in apoptosis regulation [11, 12]. PUMA can be induced by p53 -dependent and -independent manner. In p53-dependent transcription manner, DNA damage agents, such as common chemotherapeutic drugs, γ-irradiation, activate p53 and initiate apoptosis through PUMA induction [13]. Moreover, PUMA induction via p53-independent manner is modulated by the transcription factor such as p73, FoxO3a, E2F1, STAT1, or NF-κB [14, 15]. PUMA induction potently promotes apoptosis in cancer cells by binding to antiapoptotic Bcl-2 family members (Bcl-2 and Bcl-XL), which activates the proapoptotic Bcl-2 family members (Bax and Bak), resulting in dysfunction of mitochondrial leading to caspase cascade activity [16].
In this study, our results indicate that NVP-BKM120-induced PUMA induction through the AKT/FoxO3a pathway, and PUMA plays a key role in therapeutic response to NVP-BKM120 in CRC. Our results suggest that PUMA induction is indicative of the therapeutic efficacy of NVP-BKM120.
RESULTS
NVP-BKM120 induces apoptosis in CRC
To investigate the effective of NVP-BKM120 on CRC, six colon cancer cells were treated with different doses of NVP-BKM120 for 72 hours. MTS analysis was performed to quantify cell proliferation. Indeed, treatment of these cells with NVP-BKM120 led to decreased cell growth (Figure 1A). Apoptosis in HCT116, DLD1, Lim2405 and SW480 cells treated with NVP-BKM120 was analyzed by flow cytometry. As shown in Figure 1B, NVP-BKM120 increased does-dependent Annexin V-positive cells in these cell lines. Furthermore, it has been shown that NVP-BKM120 also induces caspases 3/7 activation in these cell lines (Figure 1C). In addition, HCT116, DLD1 and HT29 were treated with NVP-BKM120 for 24 hours, and then caspase activation was analyzed by Western blotting. As shown in Figure 1D, NVP-BKM120 induced caspase 3, 8 and 9 activation in these cells. These results suggest that NVP-BKM120 decreased cell proliferation and contributed to caspase-dependent apoptosis induction in CRC cells.
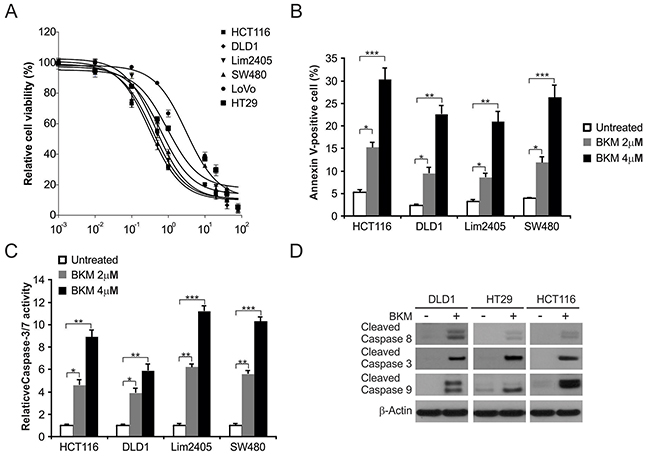
Figure 1: NVP-BKM120 induces apoptosis in CRC. (A) The indicated cell lines were treated with increasing dose of NVP-BKM120 for 72 hours. Cell proliferation was determined by MTS assay. (B) The indicated cell lines were treated with increasing NVP-BKM120 for 24 hours at indicated concentrations. Apoptosis was analyzed by Annexin V/PI staining followed by flow cytometry. (C) The indicated cell lines were treated with NVP-BKM120 for 24 hours at indicated concentrations. Caspase 3/7 activity was determined by fluorogenic analysis. (D) The indicated cell lines were treated with 4μmol/LNVP-BKM120 for 24 hours. Cleaved caspase 3, 8 and 9 were analyzed by Western blotting. Results in (A), (B) and (C) were expressed as means ± SD of 3 independent experiments. ***, P<0.001;**, P<0.01; *, P<0.05.
NVP-BKM120 induces p53-independent PUMA upregulation in CRC
We then analyzed the mechanism of NVP-BKM120 induced apoptosis in CRC cells. Treating HCT116 cells with 4 μmol/L NVP-BKM120 significantly increased the protein and mRNA level of PUMA in a time-dependent manner (Figure 2A-2C). NVP-BKM120 also increased PUMA protein and mRNA level in p53-Knockdown (p53-KD) HCT116 cells [17] (Figure 2A and 2D). Moreover, NVP-BKM120 upregulated PUMA expression in p53-WT Lim2405, LoVo CRC cells, as well as p53-mutant DLD1, HT29 and SW480 CRC cells (Figure 2E). In addition, as shown in Figure 2F, NVP-BKM120 treatment did not increased Bim and Noxa protein level, but decreased the anti-apoptotic Mcl-1 level. The above results demonstrate that PUMA is selective upregulated by NVP-BKM120 regardless of p53 status and may mediate its antitumor activity.
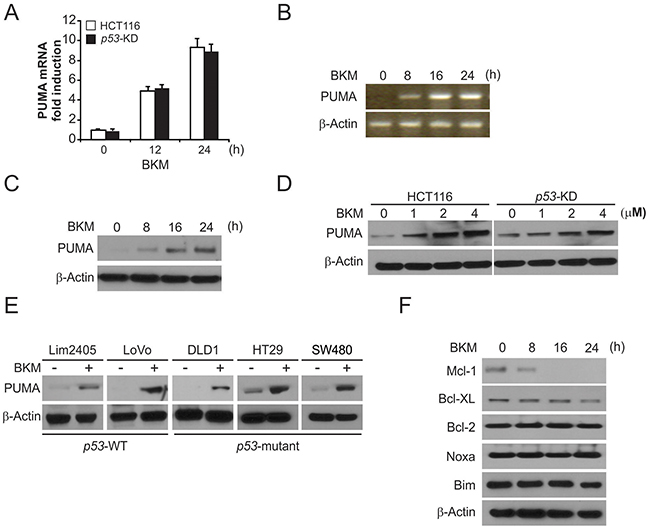
Figure 2: NVP-BKM120 induces p53-independent PUMA induction. (A) Parental and p53-KD HCT116 cells were treated with NVP-BKM120 at indicated time point. PUMA mRNA induction by NVP-BKM120 was analyzed by real-time reverse transcriptase (RT) PCR, with β-actin as a control. (B) HCT116 cells were treated with 4 μmol/L NVP-BKM120 at indicated time point. Total RNA was extracted, and PUMA mRNA expression was analyzed by semiquantitive reverse transcription PCR (RT-PCR). β-actin was used as a control. (C) HCT116 cells were treated with 4 μmol/L NVP-BKM120 at indicated time point. PUMA expression was analyzed by Western blotting. (D) Parental and p53-KD HCT116 cells were treated with NVP-BKM120 for 24 hours at indicated concentration. PUMA expression was analyzed by Western blotting. (E) Indicated colon cancer cell lines with different p53 status were treated with 4 μmol/L NVP-BKM120 for 24 hours. PUMA expression was analyzed by Western blotting. (F) HCT116 cells were treated with 4 μmol/L NVP-BKM120 at indicated time point. Indicated protein expression was analyzed by Western blotting.
PUMA mediates the anticancer effect of NVP-BKM120
Furthermore, to examine the function of PUMA in NVP-BKM120-induced apoptosis, we generated PUMA stable knockdown (PUMA-KD) HCT116 cells [17]. In PUMA-KD cells, apoptosis was significantly reduced, which was induced by 2-4 μmol/L NVP-BKM120 (Figure 3A). The reduction of NVP-BKM120-induced apoptosis in PUMA-KD HCT116 and DLD1 cells were confirmed by flow cytometry analysis following Annexin V/PI staining (Figure 3B). Consistently, the activation of caspase 3/7, which induced by NVP-BKM120, was reduced in PUMA-KD HCT116 cells (Figure 3C). The deficiency of PUMA abrogated NVP-BKM120-induced mitochondrial pathway apoptosis, which was observed in cleaved caspases 3 and 9 in HCT116 and DLD1 cells (Figure 3D) and cytochrome c release in HCT116 cells (Figure 3E). Notably, in the long-term colony formation assay, compare with parental cells, PUMA-KD cells had improved survival following NVP-BKM120 treatment (Figure 3F). These results indicate that apoptotic response of NVP-BKM120 is PUMA-dependent in CRC.
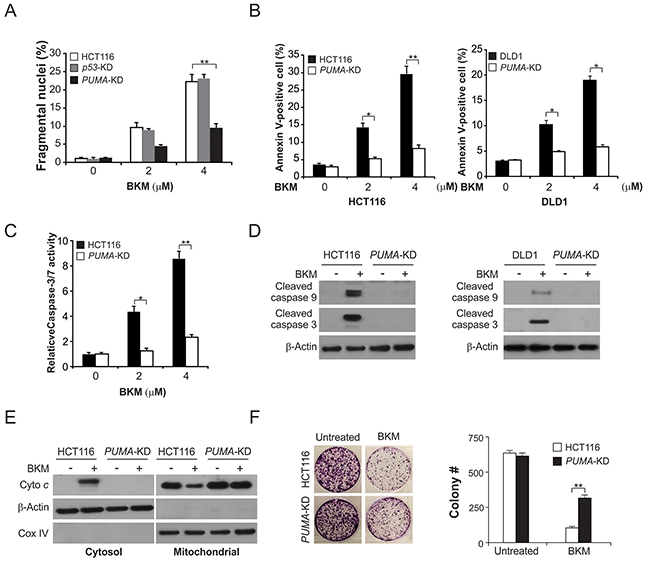
Figure 3: PUMA is required for the apoptotic activity of NVP-BKM120. (A) Parental, p53-KD and PUMA-KD HCT116 cells were treated with NVP-BKM120 at indicated concentration for 24 hours. Apoptosis was analyzed by a nuclear fragmentation assay. (B) Parental and PUMA-KD HCT116 or DLD1 cells were treated with NVP-BKM120 at indicated concentration for 24 hours. Apoptosis was analyzed by Annexin V/PI staining followed by flow cytometry. (C) Parental and PUMA-KO HCT116 cells were treated with NVP-BKM120 for 24 hours. Caspase 3/7 activity was determined by fluorogenic analysis. (D) Parental and PUMA-KD HCT116 or DLD1 cells were treated with 4 μmol/L NVP-BKM120 for 24 hours. Cleaved caspase 3 and 9 were analyzed by Western blotting. (E) The cytoplasm and mitochondria were fractionated from parental and PUMA-KO HCT116 cells treated with 4 μmol/L NVP-BKM120 for 24 hours. The distribution of cytochrome c was analyzed by Western blotting. β-Actin and cytochrome oxidase subunit IV (Cox IV) were analyzed as the control for loading and fractionation. (F) Parental and PUMA-KD HCT116 cells were treated with 4 μmol/L NVP-BKM120 for 24 hours. Colony formation assay was done by seeding an equal number of treated cells in 12-well plates, and then staining attached cells with crystal violet 14 days later. Left, representative pictures of colonies; Right, quantification of colony numbers. Results in (A), (B), (C) and (F) were expressed as means ± SD of 3 independent experiments.**, P<0.01; *, P<0.05.
FoxO3A is involved in NVP-BKM120 induced PUMA up-regulation
To further explore the mechanism of PUMA induction following NVP-BKM120 treatment in CRC cells, several transcription factors that can induce PUMA expression in p53-KD cells were examined. We exclude p73 and E2F1 due to lack of induction (Figure 4A). Interestingly, NVP-BKM120 treatment markedly dephosphorylation AKT at S473, also reduced FoxO3a phosphorylation, which is known to prevent its nuclear translocation and ensuing transactivation (Figure 4A and 4B). PUMA induction was suppressed by exogenous expression of active AKT following NVP-BKM120 treatment (Figure 4C). Furthermore, NVP-BKM120-induced PUMA upregulation was abrogated by FoxO3a knockdown in HCT116 cells (Figure 4D). Moreover, treating cells with pictilisib, which is another AKT inhibitor, was sufficient to induce the up-regulation of PUMA following AKT and FoxO3a dephosphorylation (Figure 4B). To further investigate if FoxO3a can directly binding to PUMA promoter, chromatin immunoprecipitation (ChIP) was performed. FoxO3a was found to be recruited to PUMA promoter following NVP-BKM120 treatment (Figure 4E). Together, these data suggest that in response to NVP-BKM120 treatment, FoxO3a directly binds to PUMA promoter in order to drive it transcriptional activation.
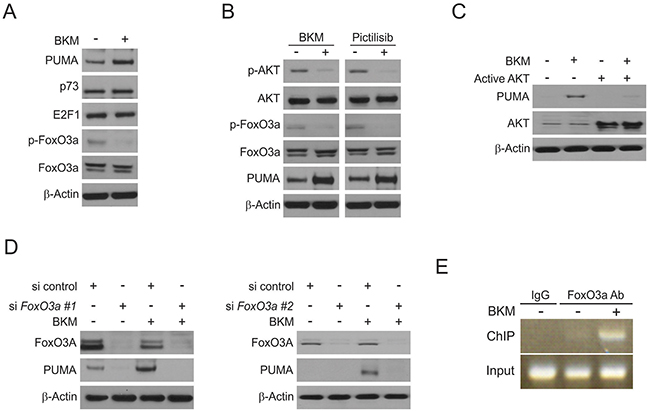
Figure 4: Induction of PUMA by NVP-BKM120 is mediated through the AKT inhibition. (A) HCT116 cells were treated with 4μmol/L NVP-BKM120 for 24 hours. PUMA, p73, E2F1, p-FoxO3a and FoxO3a expression was analyzed by Western blotting. (B) HCT116 cells were treated with 4μmol/L NVP-BKM120 or 4μmol/L Pictilisib for 24 hours. Indicated protein levels were analyzed by Western blotting. (C) HCT116 cells were transfected with active AKT plasmid for 6 hours, and then treated with 4μmol/L NVP-BKM120 for 24 hours. PUMA and AKT expression was analyzed by Western blotting. (D) HCT116 cells were transfected with either a control scrambled siRNA or FoxO3a siRNA for 24 hours, and then treated with 4μmol/L NVP-BKM120 for 24 hours. FoxO3a and PUMA expression was analyzed by Western blotting. siRNA FoxO3a #1 from Santa Cruz Biotechnology and siRNA FoxO3a #2 Thermo Fisher Scientific. (E) Chromatin immunoprecipitation (ChIP) was performed using anti-FoxO3a antibody on HCT116 cells following NVP-BKM120 treatment for 12 hours. ChIP with the control IgG was used as a control. PCR was carried out using primers surrounding the FoxO3a binding sites in the PUMA promoter.
PUMA mediates the chemo-sensitization effects of NVP-BKM120
NVP-BKM120 has been used in combination with other chemotherapeutic agents [18]. While the mechanism by which the chemo-sensitization effects of NVP-BKM120 is not well understood. From the above results, we reasoned that the chemo-sensitization effects of NVP-BKM120 were mediated by PUMA induction, due to concurrent PUMA induction following NVP-BKM120 and other agent treatment through different pathways. Importantly, we found that higher levels of PUMA was stimulated by NVP-BKM120 combined with 5-FU, compared to single agent alone treatment (Figure 5A). The result is consistent with parallel PUMA induction via p53-dependent and -independent mechanism by DNA damage and NVP-BKM120. Following the combination treatment, the apoptosis level and caspase 3 and 9 activation were markedly increased in parental cells, in contrary to PUMA-KD HCT116 and DLD1 cells (Figure 5A-5D). Furthermore, when HCT116 or DLD1 cells were treated with NVP-BKM120 and regorafenib, the PUMA-dependent sensitization effect was also detected (Figure 5E and 5G). Accordingly, we found that the combination treatment promote the level of apoptosis as well as caspase 3 activation, but not in PUMA-KD cells (Figure 5E-5H). These findings demonstrate that therapeutic efficacy of NVP-BKM120 can be markedly improved by PUMA-dependent chemo-sensitization effects of NVP-BKM120, and manipulating PUMA-mediated apoptosis.
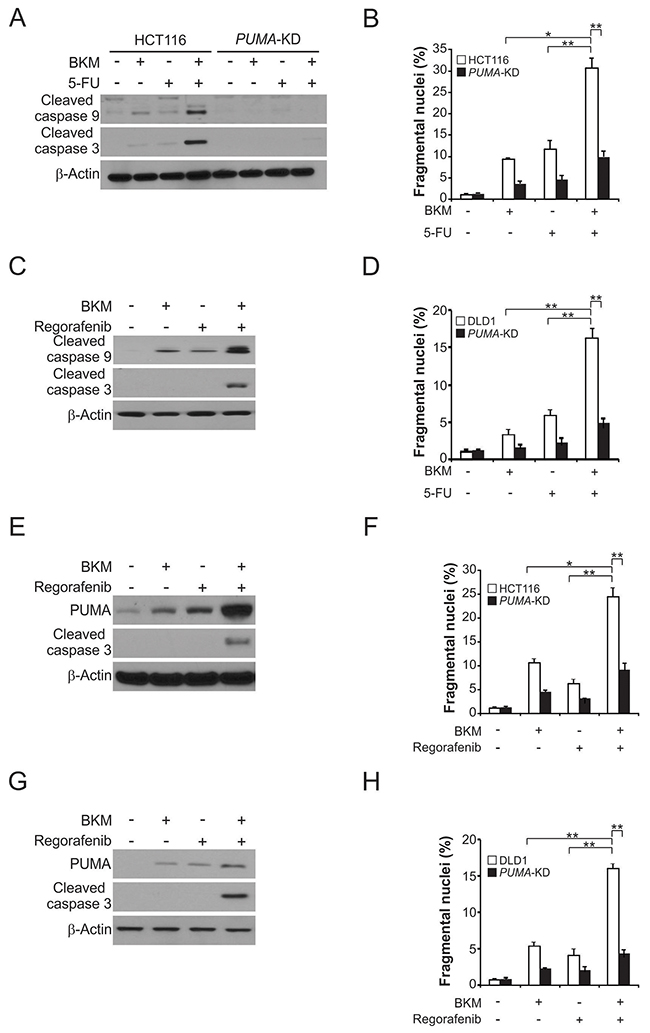
Figure 5: NVP-BKM120 synergizes with 5-FU or regorafenib to induce apoptosis via PUMA in CRC. (A) Parental and PUMA-KD HCT116 cells were treated with 2 μmol/L NVP-BKM120, 20 mg/L 5-fluorouracil (5-FU), or their combination for 24 hours. Cleaved caspase 3 and 9 were analyzed by Western blotting. (B) Parental and PUMA-KD HCT116 cells were treated 2 μmol/L NVP-BKM120, 20 mg/L 5-FU, or their combination for 24 hours. Apoptosis was analyzed by a nuclear fragmentation assay. (C) Parental and PUMA-KD DLD1 cells were treated with 2 μmol/L NVP-BKM120, 20 mg/L 5-FU or their combination for 24 hours. Cleaved caspase 3 and 9 were analyzed by Western blotting. (D) Parental and PUMA-KD DLD1 cells were treated 2 μmol/L NVP-BKM120, 20 mg/L 5-FU, or their combination for 24 hours. Apoptosis was analyzed by a nuclear fragmentation assay. (E) HCT116 cells were treated with 2 μmol/L NVP-BKM120, μmol/L regorafenib, or their combination for 24 hours. PUMA and cleaved caspase 3 were analyzed by Western blotting. (F) Parental and PUMA-KD HCT116 cells were treated 2 μmol/L NVP-BKM120, 20 μmol/L regorafenib, or their combination for 24 hours. Apoptosis was analyzed by a nuclear fragmentation assay. (G) DLD1 cells were treated with 2 μmol/L NVP-BKM120, 20 μmol/L regorafenib, or their combination for 24 hours. PUMA and cleaved caspase 3 were analyzed by Western blotting. (H) Parental and PUMA-KD DLD1 cells were treated 2 μmol/L NVP-BKM120, 20 μmol/Lregorafenib, or their combination for 24 hours. Apoptosis was analyzed by a nuclear fragmentation assay. Results in (B), (D), (F) and (H) were expressed as means ± SD of 3 independent experiments. **, P<0.01; *, P<0.05.
PUMA contributes to the antitumor activity of NVP-BKM120 in a mouse xenograft model
To determine the role of PUMA in anticancer effects of NVP-BKM120 in vivo, nude mice were injected with parental and PUMA-KD HCT116 cells to generate xenograft tumor model. After one week, mice with tumor were treated daily with 40 mg/kg NVP-BKM120 or control for 10 consecutive days by oral gavage. For the vehicle treatment, no significantly difference was found between parental and PUMA-KD tumors in growth (Figure 6A). The growth of parental tumors was suppressed with NVP-BKM120 treatment by 70-80% (Figure 6A). However, compared to parental tumors, PUMA-KD tumors were insensitive to NVP-BKM120 (Figure 6A), indicating that PUMA mediates the anti-tumor effects of NVP-BKM120. In NVP-BKM120-treated tumors, phosphorylation of FoxO3a and expression of PUMA were increased (Figure 6B). Our results showed that compare to control mice, significant apoptosis induction was found in tumor tissues in the mice treated with NVP-BKM120 by TUNEL staining. However, the tumor suppression by NVP-BKM120 treatment was largely abolished in the mice with PUMA-KD tumors (Figure 6C), which was also associated with decreased apoptosis detected by active caspase 3 staining (Figure 6D). These findings suggest that PUMA mediated the antitumor effect of NVP-BKM120 in vivo.
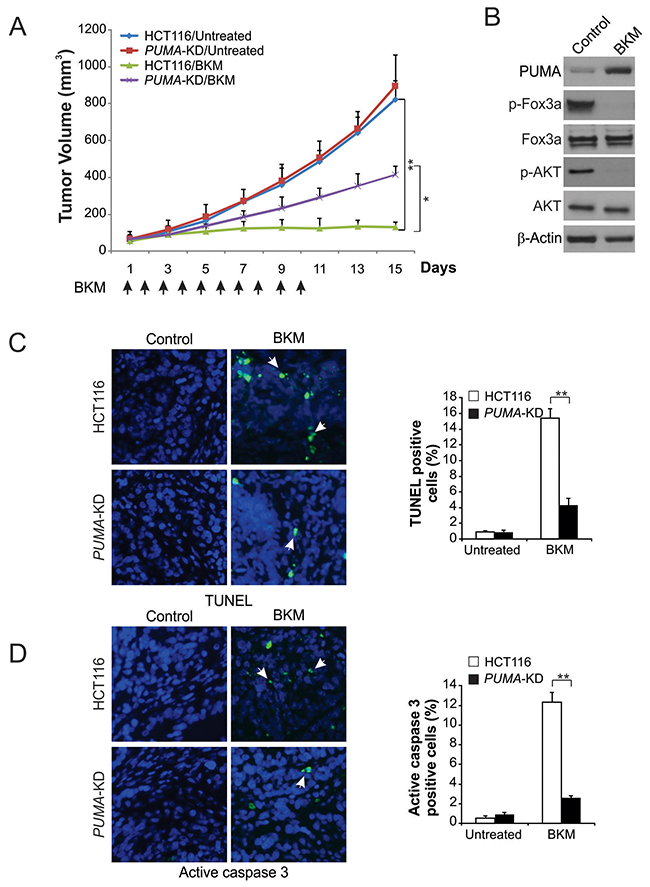
Figure 6: PUMA mediates the antitumor effects of NVP-BKM120 in a xenograft model. (A) Nude mice were injected s.c. with 4 × 106 parental and PUMA-KD HCT116 cells. After 1 week, mice were treated with 40 mg/kg NVP-BKM120 or buffer for 10 consecutive days. Tumor volume at indicated time points after treatment was calculated and plotted (n=6 in each group). Arrows indicate NVP-BKM120 injection. (B) Parental HCT116 xenograft tumors were treated with 40 mg/kg NVP-BKM120 or the control buffer as in (A) for 4 consecutive days. Indicated protein in representative tumors were analyzed by Western blotting. (C) Paraffin-embedded sections of tumor tissues from mice treated as in (B) were analyzed by TUNEL staining. Left, representative TUNEL staining pictures; Right, TUNEL-positive cells were counted and plotted. (D) Tissue sections from (C) were analyzed by active caspase 3 staining. Left, representative staining pictures; Right, active caspase 3-positive cells were counted and plotted. Results of (A), (C) and (D) were expressed as means ± SD of 3 independent experiments. **, P<0.01; *, P<0.05.
DISCUSSION
PI3K inhibitor NVP-BKM120, a well-tolerated agent, showed an effective induction of apoptosis in tumor cells and cancer xenograft models at clinically relevant doses [19, 20]. ALL four isoforms of Class I PI3K (α, β, γ, and δ) are selectively targeted by this oral PI3K inhibitor [8]. NVP-BKM120 has demonstrated proapoptotic, anti-proliferative, and anticancer effects in a variety of cell lines and animal models through cancers-dependent and -independent aberrant PI3K pathway activation [5, 21]. In the current study, we examined the effect of NVP-BKM120 on CRC. Our results demonstrated for the first time that the tumor suppression by NVP-BKM120 is at least in part mediated by the cell autonomous process of apoptosis induction, progressing from AKT inhibition, FoxO3a activation, leading to PUMA induction and onset of mitochondria-dependent apoptosis.
PUMA induction in response to p53 and/or other apoptotic signals, which are associated with mitochondria and induce cell death when overexpressed in different kinds of cell lines, and the activity of apoptotic requires an complete BH3 domain [22]. PUMA has been shown involved in the critical process of tumorgenesis [23]. Previously study indicated that PUMA should be a potential chemotherapeutic target because activated PUMA inhibits tumor growth by restoring cell apoptosis in cancer cells [24]. In addition, PUMA can also be stimulated via a p53-independent way. In non-inflammatory toxin-stimulated conditions such as inflammatory cytokines, deprivation of growth factory and p53-independent PUMA induction can be mediated by various transcription factors such as NF-κB, E2F1, FoxO3a, and p73 [25-27]. PUMA induction strongly promoted cancer cell apoptosis by acting on other Bcl-2 family members, and triggered caspase cascade [28, 29]. These findings suggest that antitumor effect of NVP-BKM120 via p53-independent PUMA induction.
The current study indicates that PUMA-induced by NVP-BKM120 initiates apoptosis via the intrinsic apoptosis pathway in CRC. Several reports showed that PUMA induction plays a key role in apoptosis induction in response to numbers of chemotherapeutic agents, and is likely to be a useful biomarker of chemo-sensitivity [17, 22, 23]. The results demonstrate that the induction of PUMA can be used as a biomarker for predicting response of CRC to NVP-BKM120.
In summary, our results revealed a novel anticancer mechanism of NVP-BKM120 via PUMA-mediated apoptosis. AKT/FoxO3a signaling pathway was involved in NVP-BKM120-induced PUMA expression. Our results indicated PUMA induction can be used as a biomarker for clinical trials testing NVP-BKM120, and can help important implications for the future development and application.
MATERIALS AND METHODS
Cell culture and treatment
The human colon cancer cell lines including HCT116, DLD1, HT29, Lim2405, SW480 and LoVo were obtained from American Type Culture Collection. All colon cancer cell lines were cultured in DMEM medium supplemented with 10% newborn calf serum, 100 units/mL penicillin, and 100 μg/mL streptomycin (Invitrogen). The anticancer chemicals used including NVP-BKM120, regorafenib (Selleckchem), and 5-fluoreuracil (5-FU, Sigma) were diluted with DMSO. Constitutively active AKT was obtained from Addgene.
MTS assay
The indicated cell lines were seeded in 96-well plates at a density of 1×104 cells/well. After overnight incubation, cells were treated with NVP-BKM120 for 72 hours. 3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay was performed using the MTS assay kit (Promega) according to the manufacturer’s instructions. Luminescence was measured with a Wallac Victor 1420 Multilabel Counter (Perkin Elmer). Each assay was conducted in triplicate and repeated three times.
Real-time reverse transcriptase (RT) PCR
Total RNA was extracted using the TRIzol RNA Kit (Invitrogen) according to the manufacturer’s protocol. One μg of total RNA was used to generate cDNA using SuperScript II reverse transcriptase (Invitrogen). PCR was performed using SsoFasrTM Probes Supermix (Bio-Rad) in a final reaction volume of 20 μl with gene-specific primer/probe sets, and a standard thermal cycling procedure (35 cycles) on a Bio-Rad CFX96TM Real-time PCR System. PUMA and b-actin levels were assessed using TaqMan Gene Expression Real-Time PCR assays. Result was expressed as the threshold cycle (Ct). The relative quantification of the target transcripts was determined by the comparative Ct method (ΔΔCt) according to the manufacturer’s protocol. The 2-ΔΔCt method was used to analyze the relative changes in gene expression. Control experiments were conducted without reverse transcription to confirm that the total RNA was not contaminated with genomic DNA. β-Actin was used as an internal control gene in order to normalize.
Western blotting
Western blotting was performed as previously described [30], with antibodies for PUMA, Mcl-1 (Abcam), AKT, phospho-AKT, cleaved-caspase 3, cleaved-caspase 9, cleaved-caspase 8, phospho-FoxO3a, FoxO3a, cytochrome oxidase subunit IV (Cox IV), E2F1, p73, Bim, Bcl-2, Bcl-XL (Cell Signaling Technology), cytochrome c, β-actin, Noxa (Santa Cruz Biotechnology).
Apoptosis assays
Apoptosis was analyzed by nuclear staining with Hoechst 33258 (Invitrogen) [31]. Annexin V/propidium iodide (PI) staining was performed using annexin-Alexa 488 (Invitrogen) and PI. Caspase 3/7 activity was detected with a Cell Death Detection ELISAPlus Kit (Roche Molecular Biochemicals, Indianapolis). For analysis of cytochrome c release, cytosolic fractions were isolated by differential centrifugation, and probed by Western Blotting for cytochrome c. For colony formation assays, the treated cells were plated in 12-well plates at appropriate dilutions and allowing for cell growth for 2 weeks, followed by crystal violet staining of cell colonies.
Transfection and siRNA/shRNA knockdown
Cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Knockdown experiments were performed 24 hours prior to NVP-BKM120 treatment using 200 pmole of siRNA. The control scrambled siRNA and siRNA for human FoxO3a were from Santa Cruz Biotechnology and Thermo Fisher Scientific. For stable transfection a shRNA-expressing plasmid that containing PUMA-targeting sequence (CCTGGAGGGTCATGTACAATCTCTT), or a vector containing a scrambled sequence was transfected into HCT116 cells, followed transfection, cells were plated in 96-well plates in the presence of 5μg/mL puromycin. The protein expression of puromycin-resistant clones was then analyzed by western blotting.
Chromatin immunoprecipitation (ChIP)
ChIP with FoxO3a antibody (Cell Signaling Technology) was performed using the Chromatin Immunoprecipitation Assay Kit (Millipore) according to the manufacturer’s instructions. The precipitates were analyzed by PCR using primers 5’-GCGCACAGGTGCCTCGGC-3’ and 5’-TGGGTGTGGCCGCCCT-3’.
Animal tumor experiments
All the experimental procedures were carried out following the guidelines of The People’s Hospital of Liaoning Province and have been approved by the Committee for animal experimentation of The People’s Hospital of Liaoning Province. Parental and PUMA-KD HCT116 cells were harvested, and 4 x 106 cells in 0.1 mL of medium were implanted subcutaneously on the back of athymic nude female mice. After tumor growth for 7 days, mice were treated with daily with NVP-BKM120 at 40 mg/kg by oral gavage for 10 consecutive days. Tumor growth was monitored by calipers, and tumor volumes were calculated according to the formula 1/2 × length × width2. Mice were euthanized when tumors reached ~1.0 cm3 in size. Tumors were dissected and fixed in 10% formalin and embedded in paraffin. TUNEL and active caspase 3 immunostaining was performed on 5 μM paraffin-embedded tumor sections, by using an Alexa Fluor 488-conjugated secondary antibody (Invitrogen) for signal detection.
Statistical analysis
Statistical analyses were carried out using GraphPad Prism V software. P values were calculated by the student’s t-test and were considered significant if p < 0.05. The means ± standard deviation (SD) is displayed in the figures.
Abbreviations
ChIP, chromatin immunoprecipitation; Cox IV, cytochrome oxidase subunit IV; CRC, colorectal cancer; E2F1, E2F transcription factor 1; FoxO3a, Forkhead Box O3a; 5-FU, 5-fluoreuracil; PUMA, p53 upregulated modulator of apoptosis; RT-PCR, reverse transcriptase-PCR; STAT1, signal transducer and activator of transcription 1; TUNEL, terminal deoxynucleotidyl transferase mediated dUTP nick end labeling.
Author contributions
SY, XL, WG, MQ, XY and ZY conceived and performed the experiments. SY, ZY and HZ analyzed the data and wrote the manuscript.
ACKNOWLEDGMENTS
We thank our lab members for critical reading. This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
None.
REFERENCES
1. Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012; 13: 195-203. https://doi.org/10.1038/nrm3290.
2. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014; 13: 140-56. https://doi.org/10.1038/nrd4204.
3. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009; 9: 550-62. https://doi.org/10.1038/nrc2664.
4. Bunney TD, Katan M. Phosphoinositide signalling in cancer: beyond PI3K and PTEN. Nat Rev Cancer. 2010; 10: 342-52. https://doi.org/10.1038/nrc2842.
5. Speranza MC, Nowicki MO, Behera P, Cho CF, Chiocca EA, Lawler SE. BKM-120 (Buparlisib): a phosphatidyl-inositol-3 kinase inhibitor with anti-invasive properties in glioblastoma. Sci Rep. 2016; 6: 20189. https://doi.org/10.1038/srep20189.
6. Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, Schnell C, Guthy D, Nagel T, Wiesmann M, Brachmann S, Fritsch C, Dorsch M, et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther. 2012; 11: 317-28. https://doi.org/10.1158/1535-7163.MCT-11-0474.
7. Han B, Yao W, Oh YT, Tong JS, Li S, Deng J, Yue P, Khuri FR, Sun SY. The novel proteasome inhibitor carfilzomib activates and enhances extrinsic apoptosis involving stabilization of death receptor 5. Oncotarget. 2015; 6: 17532-42. https://doi.org/10.18632/oncotarget.3947.
8. Brachmann SM, Kleylein-Sohn J, Gaulis S, Kauffmann A, Blommers MJ, Kazic-Legueux M, Laborde L, Hattenberger M, Stauffer F, Vaxelaire J, Romanet V, Henry C, Murakami M, et al. Characterization of the mechanism of action of the pan class I PI3K inhibitor NVP-BKM120 across a broad range of concentrations. Mol Cancer Ther. 2012; 11: 1747-57. https://doi.org/10.1158/1535-7163.MCT-11-1021.
9. Netland IA, Forde HE, Sleire L, Leiss L, Rahman MA, Skeie BS, Miletic H, Enger PO, Goplen D. Treatment with the PI3K inhibitor buparlisib (NVP-BKM120) suppresses the growth of established patient-derived GBM xenografts and prolongs survival in nude rats. J Neurooncol. 2016; 129: 57-66. https://doi.org/10.1007/s11060-016-2158-1.
10. Geuna E, Milani A, Martinello R, Aversa C, Valabrega G, Scaltriti M, Montemurro F. Buparlisib, an oral pan-PI3K inhibitor for the treatment of breast cancer. Expert Opin Investig Drugs. 2015; 24: 421-31. https://doi.org/10.1517/13543784.2015.1008132.
11. Yu J, Zhang L. PUMA, a potent killer with or without p53. Oncogene. 2008; 27: S71-83. https://doi.org/10.1038/onc.2009.45.
12. Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001; 7: 683-94.
13. Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014; 13: 217-36. https://doi.org/10.1038/nrd4236.
14. Gurzov EN, Germano CM, Cunha DA, Ortis F, Vanderwinden JM, Marchetti P, Zhang L, Eizirik DL. p53 up-regulated modulator of apoptosis (PUMA) activation contributes to pancreatic beta-cell apoptosis induced by proinflammatory cytokines and endoplasmic reticulum stress. J Biol Chem. 2010; 285: 19910-20. https://doi.org/10.1074/jbc.M110.122374.
15. Shaltouki A, Freer M, Mei Y, Weyman CM. Increased expression of the pro-apoptotic Bcl2 family member PUMA is required for mitochondrial release of cytochrome C and the apoptosis associated with skeletal myoblast differentiation. Apoptosis. 2007; 12: 2143-54. https://doi.org/10.1007/s10495-007-0135-z.
16. Tong J, Wang P, Tan S, Chen D, Nikolovska-Coleska Z, Zou F, Yu J, Zhang L. Mcl-1 degradation is required for targeted therapeutics to eradicate colon cancer cells. Cancer Res. 2017; 77: 2512-21. https://doi.org/10.1158/0008-5472.CAN-16-3242.
17. Yang S, Zhu Z, Zhang X, Zhang N, Yao Z. Idelalisib induces PUMA-dependent apoptosis in colon cancer cells. Oncotarget. 2017; 8: 6102-13. https://doi.org/10.18632/oncotarget.14043.
18. Yu F, Zhao J, Hu Y, Zhou Y, Guo R, Bai J, Zhang S, Zhang H, Zhang J. The combination of NVP-BKM120 with trastuzumab or RAD001 synergistically inhibits the growth of breast cancer stem cells in vivo. Oncol Rep. 2016; 36: 356-64. https://doi.org/10.3892/or.2016.4799.
19. Hu Y, Guo R, Wei J, Zhou Y, Ji W, Liu J, Zhi X, Zhang J. Effects of PI3K inhibitor NVP-BKM120 on overcoming drug resistance and eliminating cancer stem cells in human breast cancer cells. Cell Death Dis. 2015; 6: e2020. https://doi.org/10.1038/cddis.2015.363.
20. Ren H, Zhao L, Li Y, Yue P, Deng X, Owonikoko TK, Chen M, Khuri FR, Sun SY. The PI3 kinase inhibitor NVP-BKM120 induces GSK3/FBXW7-dependent Mcl-1 degradation, contributing to induction of apoptosis and enhancement of TRAIL-induced apoptosis. Cancer Lett. 2013; 338: 229-38. https://doi.org/10.1016/j.canlet.2013.03.032.
21. Zheng Y, Yang J, Qian J, Zhang L, Lu Y, Li H, Lin H, Lan Y, Liu Z, He J, Hong S, Thomas S, Shah J, et al. Novel phosphatidylinositol 3-kinase inhibitor NVP-BKM120 induces apoptosis in myeloma cells and shows synergistic anti-myeloma activity with dexamethasone. J Mol Med (Berl). 2012; 90: 695-706. https://doi.org/10.1007/s00109-011-0849-9.
22. Yu J, Zhang L. No PUMA, no death: implications for p53-dependent apoptosis. Cancer Cell. 2003; 4: 248-9.
23. Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L. PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci U S A. 2003; 100: 1931-6. https://doi.org/10.1073/pnas.2627984100.
24. Yang J, Zhao X, Tang M, Li L, Lei Y, Cheng P, Guo W, Zheng Y, Wang W, Luo N, Peng Y, Tong A, Wei Y, et al. The role of ROS and subsequent DNA-damage response in PUMA-induced apoptosis of ovarian cancer cells. Oncotarget. 2017; 8: 23492-506. https://doi.org/10.18632/oncotarget.15626.
25. Hikisz P, Kilianska ZM. PUMA, a critical mediator of cell death--one decade on from its discovery. Cell Mol Biol Lett. 2012; 17: 646-69. https://doi.org/10.2478/s11658-012-0032-5.
26. Li Y, Liu YJ, Lv G, Zhang DL, Zhang L, Li D. Propofol protects against hydrogen peroxide-induced apoptosis in cardiac H9c2 cells is associated with the NF-kappaB activation and PUMA expression. Eur Rev Med Pharmacol Sci. 2014; 18: 1517-24.
27. Al-Bahlani S, Fraser M, Wong AY, Sayan BS, Bergeron R, Melino G, Tsang BK. P73 regulates cisplatin-induced apoptosis in ovarian cancer cells via a calcium/calpain-dependent mechanism. Oncogene. 2011; 30: 4219-30. https://doi.org/10.1038/onc.2011.134.
28. Chipuk JE, Green DR. PUMA cooperates with direct activator proteins to promote mitochondrial outer membrane permeabilization and apoptosis. Cell Cycle. 2009; 8: 2692-6. https://doi.org/10.4161/cc.8.17.9412.
29. Vavrova J, Rezacova M. Importance of proapoptotic protein PUMA in cell radioresistance. Folia Biol (Praha). 2014; 60: 53-6.
30. Tong J, Tan S, Zou F, Yu J, Zhang L. FBW7 mutations mediate resistance of colorectal cancer to targeted therapies by blocking Mcl-1 degradation. Oncogene. 2017; 36: 787-96. https://doi.org/10.1038/onc.2016.247.
31. Peng R, Tong JS, Li H, Yue B, Zou F, Yu J, Zhang L. Targeting Bax interaction sites reveals that only homo-oligomerization sites are essential for its activation. Cell Death Differ. 2013; 20: 744-54. https://doi.org/10.1038/cdd.2013.4.