
INTRODUCTION
Thyroid carcinoma accounts for about 1% of all human cancers and its incidence has steadily increased over the last few decades. The majority of thyroid tumours arises from follicular thyroid cells and includes: papillary (PTC), follicular (FTC), poorly differentiated (PDTC) and anaplastic (ATC) carcinoma [1].
PTC is the most frequent thyroid cancer; it includes several histological variants characterized by different prognosis, from papillary thyroid microcarcinoma (PTMC) generally associated to an indolent course, to tall cell variant (TCV) associated to higher disease-related mortality and recurrence rates [2]. PTC is characterized by alternative driving genetic lesions converging on the RTK/RAS/BRAF/MAPK signaling pathway in about 70% of cases. Novel molecules and mechanisms relevant in PTC pathogenesis have been suggested by genes and miRNAs expression studies [3]. Nevertheless, the mechanisms underlying the onset and progression of this neoplasia have not been fully elucidated.
We have recently provided new insights into the molecular mechanisms of thyroid carcinogenesis by demonstrating, as first, the involvement of oncogene-induced senescence (OIS) [4].
OIS is a highly stable cell cycle arrest triggered by oncogenic stress in primary cells [5]. During OIS cells undergo multiple phenotypic changes, that include changes in morphology, metabolism, chromatin organization, an increase and reorganization in lysosome numbers (resulting in senescence-associated β-galactosidase (SA-β-Gal) activity), and the production of a complex secretome (referred as the SMS or SASP)[6;7]. Although there is debate on how best to define senescence there is an agreement that together with the stable cell cycle arrest several of the mentioned phenotypes (sometimes referred to as geroconversion) must occur, to distinguish senescence from cell cycle arrest [8].
One of the initial molecular triggers of OIS is the activation of a robust DNA-damage checkpoint response (DDR) caused by hyperproliferation and DNA hyper-replication resulting from oncogene expression [9;10]. Although DDR is not a universal feature of OIS [7;9;10] when present it contributes to activate p53-dependent reponses such as the upregulation of the cyclin-dependent kinase (CDK) inhibitor p21CIP1. The activation of the p53/p21CIP1 pathway, together with upregulation of p16INK4a are the key to implement the senescent growth arrest [7]. The relative contribution of p16 INK4a and p53/p21 CIP1 to senescence depends of the trigger and cell type where senescence occurs [11].
It is now appreciated that OIS represents an important barrier against tumour development in vivo: a number of studies have shown that premalignant lesions from human and mice are enriched in senescent cells. For instance, benign melanocytic nevi, which frequently carry the BRAFV600E oncogene, show features of senescence including SA-β-Gal activity and high p16INK4a expression [12]. Moreover, senescence markers were found in human dermal neurofibromas, murine lung adenomas, human and murine prostatic adenomas, murine pancreatic intraductal neoplasias and murine lymphomas [13-18].
Notably, OIS is tightly connected with inflammation. Senescent cells display the “senescence associated secretory phenotype” (SASP): they produce a wide range of inflammatory cytokines and growth factors which operate in a cell-autonomous manner, but also they communicate with and modify the microenvironment [6;19;20]. Through the SASP senescent cells can affect their microenvironment in opposing ways and reinforce senescence or promote carcinogenesis. The SASP can induce senescence in normal or low-grade premalignant cells but also can boost cancer progression programs in high-grade premalignant or malignant cells [21]. In addition, SASP factors can trigger senescence surveillance, an immuno-mediated clearance of senescent cells, recently proposed as an important extrinsic component of the senescence anti-tumour barrier [22].
We have previously demonstrated that OIS may represent a barrier to thyroid epithelial tumour progression. The expression of PTC-associated oncogenes (BRAF, RAS, RET/PTC and TRK) triggers senescence in human primary thyrocytes, as demonstrated by lack of proliferation, changes in cell morphology, induction of various OIS specific biomarkers including elevated SA-β-Gal activity, presence of senescence-associated heterochromatic foci (SAHF), as well as increased expression of p16INK4a, p21CIP1 and p53, and robust ERK1/2 activation. Furthermore, immunohistochemical analysis of a panel of thyroid tumours characterized by increasing aggressiveness showed that expression of OIS markers is upregulated at early tumour stages, and lost at more advanced ones, suggesting that OIS may influence the pathogenesis of thyroid carcinoma by constraining neoplastic transformation [4]. Of note, thyrocytes undergoing OIS model in vitro the early thyroid tumour stages.
Here we established an inducible system of OIS in thyrocytes. OIS could be induced in thyrocytes by adding 4-hydroxytamoxifen (4OHT) which induce the expression of an ER:RAS fusion protein. Using this system, we show that p16INK4a is a regulator of RAS-mediated senescence. We also report that upon RAS activation thyrocytes show increased levels of several proinflammatory components including IL-8 and its receptor CXCR2 that act in reinforcing cell cycle arrest in vitro. Importantly, we also observed that CXCR2 expression coexists with senescence markers in thyroid tumour samples, in line with the view that senescence is linked with inflammation.
RESULTS
Establishment and characterization of ER:RAS inducible system in thyrocytes
Our previous studies showed that PTC-associated oncogenes, including H-RASG12V, trigger senescence in human primary thyrocytes [4]. Even if BRAFV600E is the most common mutation in PTC, H-RASG12V is highly prevalent in PTC with follicular variant histology [23]. Thus, to better understand the molecular mechanisms and the players required for the induction and maintenance of senescence in our cellular setting we took advantage of the well established ER:RAS inducible retroviral vector carrying H-RASG12V oncogene fused to a 4-hydroxytamoxifen (4OHT)-responsive Estrogen Receptor ligand binding domain [24]. Primary thyrocytes were transduced with ER:RAS retroviral vector or empty vector, and two days later were selected with Geneticin (G418) for approximately 15 days. Upon 7 days of 4OHT treatment, we monitored the effects of RAS expression on primary thyrocytes by analyzing several senescence markers. 4OHT-treated ER:RAS cells displayed changes in morphology, becoming flat and enlarged. No morphology changes were detected in the untreated counterparts or in the cells transduced with the empty vector (Figure 1a, top panel). Moreover, activation of RAS induced growth arrest, whereas controls continued proliferating. For instance, as evident by crystal violet staining we observed a decrease in cellular density in 4OHT-treated ER:RAS thyrocytes compared with untreated ER:RAS and empty vector (Figure 1a ,middle panel). This was also corroborated with a BrdU incorporation assay: 4OHT-treated ER:RAS thyrocytes exhibited a stark reduction of S phase (2% of cells incorporated BrdU) when compared with the controls (25% and 13% of untreated and empty vector cells, respectively, incorporated BrdU) (Figure 1a, lower panel). To further prove that the growth arrest had characteristics of senescence, cells were stained for SA-β-Gal activity. Following RAS activation, a higher percentage (89%) of cells displayed SA-β-Gal activity compared with the percentage of positive cells in the untreated ER:RAS or empty vector (7% and 10% respectively) (Figure 1b). To address the mechanisms by which induction of RAS triggers senescence in primary thyrocytes, we analyzed by immunofluorescence the expression of some well-known senescence effectors. After 4OHT treatment RAS caused a marked increase in the expression of p16INK4a (80% vs 15% and 12% of control cells), p21CIP1 (45% vs 13% and 10% of control cells) and p53 (55% vs 17% and 10%) (Figure 1c). The upregulation of p16INK4a, p21CIP1 and p53 in senescent thyrocytes was also confirmed by western blot analysis (Figure 1d). Taking these results together we conclude that ER:RAS thyrocytes undergo senescence upon RAS activation, and thus they represent a powerful tool to get more insights into the molecular mechanisms and players involved in OIS thyrocytes.
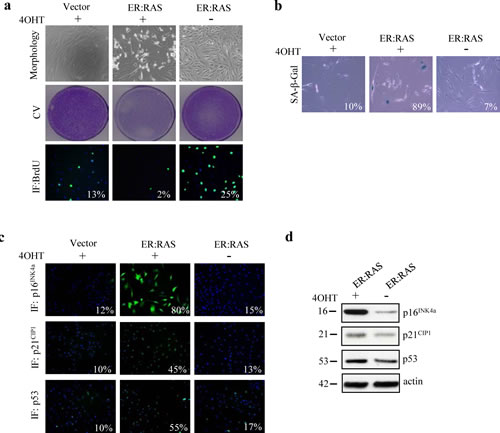
Figure 1: Oncogenic RAS triggers senescence in human primary thyrocytes. Cells carrying empty vector (Vector) or ER:RAS were left untreated or treated with 4OHT for 7 days. (a) Representative pictures showing cell morphology (upper panel, magnification 10x), Crystal violet staining (CV, middle panel) and BrdU incorporation detected by immunofluorescence (IF, bottom panel, magnification 10x). (b) SA-β-Gal staining (magnification 20x). (c) Expression of p16INK4a (upper panel), p21CIP1 (middle panel) and p53 (bottom panel) determined by IF; magnification 10x. The percentage of positive cells is indicated in each photograph. High Content Analysis (HCA) wais used for BrdU, p16INK4a, p21CIP1 and p53 quantification. For SA-β-Gal staining at least 200 cells were counted. (d) Determination by western blotting of protein levels as indicated in an independent experiment; β-actin serves as loading control. The molecular weight of each protein is indicated. Data are from a representative out of two independent experiments.
The relative contribution of p16INK4a, p53 and p21CIP1 in RAS-induced senescence in thyrocytes
To examine the requirement of p16INK4a or p53 to RAS-mediated thyrocytes senescence we applied gene silencing with shRNA delivered by retroviral vector. ER:RAS cells were transduced with shRNA targeting p16INK4a (shp16) or p53 (shp53) and selected for the integration of the construct with puromycin for approximately 10 days. Afterwards, cells were treated with 4OHT and analysed 7 days later (Figure 2a).
Regarding p16INK4a we observed a strong suppression of protein expression in shp16 transduced cells as judged by immunofluorescence (Figure 2b) and western blot analysis (Figure 2h). p16INK4a knockdown caused 4OHT-treated ER:RAS cells to recover proliferation capability, as evident by crystal violet staining (Figure 2c). To quantify such rescue we performed a BrdU incorporation assay (Figure 2d): the percentage of 4OHT-treated ER:RAS cells incorporating BrdU increased upon knockdown of p16INK4a and reached 17% compared with 2% of p16INK4a-intact ER:RAS cells. Similar results were also observed following transfection of p16INK4a siRNA or control siRNA (scramble) into ER:RAS thyrocytes (data not shown). Moreover, in shp16 transduced cells the ability to restart proliferation was accompanied by a clear reduction of SA-β-Gal activity: in fact only 5% of cells positively stained for SA-β-Gal versus 80% of p16INK4a -intact ER:RAS cells (Figure 2e). The depletion of p16INK4a did not affect the level of p21CIP1 and p53 proteins, as shown by immunofluorescence (Figure 2f-g) and western blot (Figure 2h). Hence, we conclude that disruption of p16INK4a allows primary thyrocytes to escape from RAS-induced senescence.
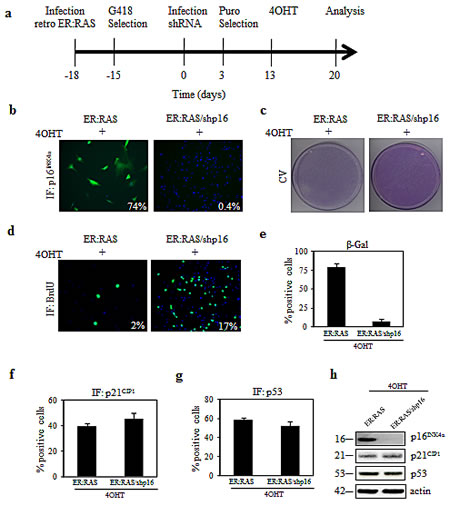
Figure 2: (a) Timeline of the experiments in this Figure and in Figures 3 and 6. ER:RAS cells retrovirally transduced with the indicated shRNA, and selected with puromycin, were treated with 4OHT for 7 days and then analyzed. (b-g) Knockdown of p16INK4a alleviates RAS-induced senescence in primary thyrocytes. ER:RAS cells with intact or null-p16INK4a were analyzed for: p16INK4a expression (b), proliferative potential through CV (c) and BrdU incorporation (d), SA-β-Gal activity (e), and expression of p21CIP1 (f) and p53 (g). High Content Analysis (HCA) was used to quantify the IF staining against p16INK4a, BrdU, p21CIP1 and p53. The percentage of SA-β-Gal positive cells was determined by counting at least 200 cells. (h) Determination by western blotting of protein levels as indicated, in an independent experiment; β-actin was used as loading control; the molecular weight of each protein is indicated. Images from a representative out of two independent experiments are shown; in the charts the mean ± s.d. of two independent experiments is reported. shp16: short hairpin targeting p16 INK4a; CV: crystal violet; IF: immunofluorescence.
With respect to p53, an efficient knockdown was obtained by shp53 transduction: following 4OHT treatment only 5% of p53-null ER:RAS thyrocytes displayed p53 expression compared with 41% of p53-intact ER:RAS cells, as assessed by immunofluorescence (Figure 3a). Such depletion was also confirmed by western blot analysis (Figure 3g). However, in response to 4OHT treatment, no difference in cell proliferation was observed between p53-intact and p53-null ER:RAS thyrocytes, as evident by crystal violet staining (Figure 3b) and BrdU incorporation (Figure 3c). Similar results were also obtained after p53 siRNA transfection. We again observed that depletion of p53 had no effect on thyrocytes growth inhibition induced by RAS activation (data not shown). Likewise, upon p53 depletion we did not observe a significant difference in SA-β-Gal activity, as well as in p16INK4a expression. In particular, in 4OHT-treated ER:RAS thyrocytes, SA-β-Gal activity was detected in 65% of p53-null and in 77% of p53-positive cells (Figure 3d). Similarly, the induction of p16INK4a was maintained in p53-null cells (69% vs 80% of control) (Figure 3e), whereas a slight decrease of p21CIP1 was detected in p53-null cells (Figure 3f). The same trend was also observed by western blot analysis, as shown in Figure 3g.
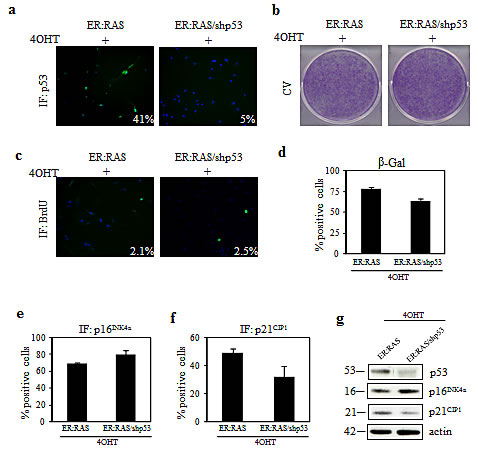
Figure 3: Effect of p53 knockdown on RAS-induced senescence in primary thyrocytes. (a-e) ER:RAS cells with intact or null p53 were analyzed for: p53 expression (a), proliferative potential through CV (b) and BrdU incorporation (c), SA-β-Gal activity (d), expression of p16INK4a (e) and p21CIP1 (f). High Content Analysis (HCA) was used to quantify IF for p53, BrdU, p16INK4a and p21CIP1. Data are from a representative out of two independents experiments. Error bars represent the standard deviation. The percentage of positive cells for SA-β-Gal staining is obtained by counting at least 200 cells. (g) Western blotting analysis for the expression of the indicated proteins performed in an independent experiment; β-actin was used as loading control; the molecular weight of each protein is shown. shp53: short hairpin targeting p53; CV: crystal violet; IF: immunofluorescence.
To investigate the role of p21CIP1 in mediating OIS, ER:RAS thyrocytes were reverse-transfected with siRNA targeting p21CIP1. After 24 hours cells were treated with 4OHT and analysed 4 days later (i.e.: 5 days post silencing). Cells untrasfected or transfected with scrambled siRNA served as negative controls. As shown in Figure 4a, an efficient silencing of p21CIP1 was observed. Such depletion did not influence the growth arrest induced by activation of RAS: only 2% of 4OHT-treated p21CIP1-null ER:RAS cells resulted positive for BrdU incorporation, an amount similar to the controls (Figure 4b). No effect of p21CIP1 silencing on p53 expression was observed: after 4OHT treatment, in p21CIP1-null ER:RAS cells the levels of p53 remained high and comparable to the ones observed in the control cells (Figure 4c).
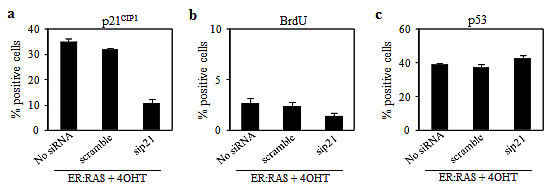
Figure 4: Effect of p21CIP1 depletion on RAS-induced senescence in primary thyrocytes. (a-c) Untrasfected ER:RAS cells (No siRNA) and ER:RAS cells transfected with a siRNA control (scramble) or with a siRNA targeting p21CIP1 (sip21) treated with 4OHT for 4 days, were subjected to IF for: p21CIP1 expression (a), BrdU incorporation (b), and p53 expression (c). High Content Analysis (HCA) is used for the quantification. Data are the mean ± s.d. of two independent experiments.
Together our data clearly demonstrate that p16INK4a is an essential effector of senescence in thyrocytes.
SASP components increase during RAS-induced senescence in thyrocytes
In addition to classical senescence players like p16INK4a, p21CIP1 and p53, a complex cocktail of factors including proinflammatory cytokines, chemokines, extracellular proteases, matrix components and growth factors, regulates the initiation and maintenance of senescence [25-28]. We investigated the expression of some SASP factors and cognate receptors. IL-8 and IL-6 are considered two of the most important cytokines secreted by senescent cells [25;26]. We analyzed the protein expression of IL-8, its receptor CXCR2, and of IL-6 via immunofluorescence. ER:RAS cells responded to RAS activation with high expression of IL-8/CXCR2 and IL-6, whereas the levels detected in the untreated counterparts were very low (Figure 5a, left panel). We also found that both cytokines are present in the conditioned medium of senescent thyrocytes, as shown by western blot analysis for IL-8 and ELISA for IL-6 (Fig.5a, middle and right panels).
We also quantified the mRNA levels of IL-1α, IL-1β, the CC chemokine CCL20, and the CC chemokine receptor CCR1 by quantitative real time PCR (qRT-PCR). As shown in Fig. 5b, transcripts encoding IL-1α and IL-1β, CCL20 and CCR1 increased when thyrocytes underwent senescent with respect to untreated ER:RAS cells. These results indicate that, as in others cellular models, a SASP-like response is activated in senescent thyrocytes.
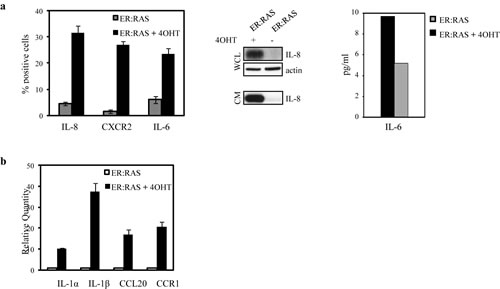
Figure 5: Upregulation of proinflammatory components during senescence in primary thyrocytes. ER:RAS cells were treated or not with 4OHT for 7 days. (a) IL-8, CXCR2 and IL-6 expression assessed by IF. High Content Analysis (HCA) is used for the quantification. Data are the mean ± s.d. of two independent experiments (left panel); determination of IL-8 protein level in whole cell lysates (WCL) and conditioned medium (CM) by western blotting (middle panel); determination of IL-6 levels in CM by ELISA (right panel). (b) qRT-PCR analysis of IL-1α, IL-1β, CCL20 and CCR1 transcript levels normalized with HPRT housekeeping gene and expressed respect to the control.
A critical role for IL-8/CXCR2 axis in mediating RAS-induced senescence in thyrocytes
To better understand the connection between senescence and inflammation in our cellular model we focused on IL-8. Notably, IL-8 is a well-known mediator of senescence in normal cells, and it has also been suggested as tumour progression factor in thyroid cancer, through increasing angiogenesis and vascularisation, metastatic spread and poor prognosis [29-31].
To investigate the IL-8 involvement in thyrocyte senescence we knocked down its expression with two independent retroviral shRNAs (#1, #2) which reduced by approximately 50% the number of IL-8 positive cells (Figure 6a). Such IL-8 suppression was sufficient to restore the proliferation capability in 4OHT-treated ER:RAS cells, as shown by crystal violet staining and BrdU incorporation (Figure 6b-c). For instance, the percentage of 4OHT-treated ER:RAS cells actively replicating DNA in the absence of IL-8 was 8% and 8.5% (shIL-8 #1 and #2, respectively) versus 3% of control cells (Figure 6c). Interestingly, upon IL-8 abrogation CXCR2 positive cells were 30% in IL-8 intact cells, and 13% and 9% in IL-8 knocked down cells (Figure 6d), suggesting that IL-8 suppression confers growth advantage by reducing CXCR2 expression.
In keeping with the well-known connection between IL-8 and its receptor CXCR2 for the induction and maintenance of OIS [26] and having shown that CXCR2 expression increased during senescence in our cellular model, we wondered whether CXCR2 is also required for the mediation of OIS. To this end, we used shRNA retroviral vector, which reduced CXCR2 protein level of approximately 60% (Figure 6e). This suppression led 4OHT-trated ER:RAS cells to resume proliferation, as demonstrated by crystal violet staining (Figure 6f). Such rescue was quantified by BrdU incorporation assay: following activation of RAS the percentage of positive cells increased by ∼4-fold upon CXCR2 knockdown (Figure 6g). Depletion of CXCR2 caused a decrease in IL-8 availability: upon 4OHT treatment 9% of CXCR2-null ER:RAS cells were positive for IL-8 staining compared with 25% of the CXCR2-intact cells (Figure 6h). These results suggest that IL-8/CXCR2 axis plays a role in mediating OIS in ER:RAS transduced thyrocytes, being involved in growth arrest triggered by oncogenic stress.
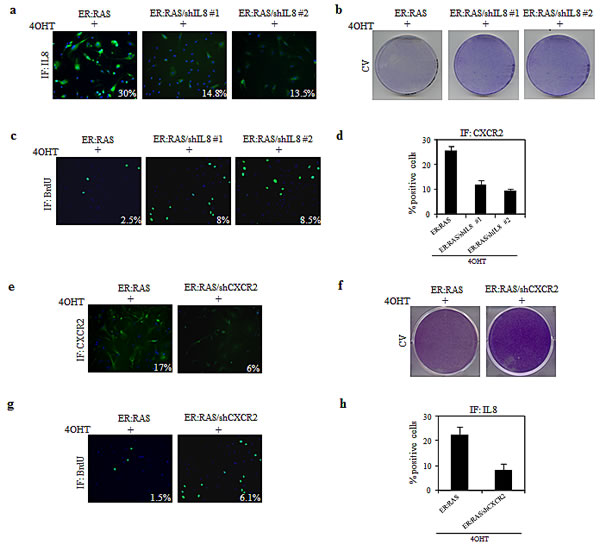
Figure 6: Role of IL-8/CXCR2 axis in mediating OIS in primary thyrocytes. (a-d) ER:RAS cells with intact or null IL-8(shIL8) were analyzed for: IL-8 expression (a), proliferative potential through CV (b) and BrdU incorporation (c), and CXCR2 expression (d). The levels of IL-8, BrdU and CXCR2 were analyzed by IF and quantified by High Content Analysis (HCA). (e-h) ER:RAS cells with intact or null CXCR2 (shCXCR2) were analyzed for: CXCR2 expression (e), proliferative potential through CV (f) and BrdU incorporation (g), and IL-8 expression (h). CXCR2, BrdU and IL-8 immunofluorescence was quantified by High Content Analysis (HCA). Images from a representative out of two independent experiments are shown; in the charts the mean ± s.d. of two independent experiments is reported. shIL8#1 and 2: short hairpins targeting IL8; shCXCR2: short hairpin targeting CXCR2; CV: cristal violet; IF: immunofluorescence.
CXCR2 expression coexists with OIS markers in thyroid tumour samples
We have recently shown that signs of OIS, such as p16INK4a upregulation and very low proliferation index are observed in PTMC, the early thyroid tumour stage, and then lost during tumour progression [4]. To investigate the trend of CXCR2 expression with respect to other OIS markers, a panel of 13 thyroid tumours was analyzed for the expression of p16INK4a, CXCR2 and Ki-67 by immunohistochemistry. As detailed in Table 1, tumour collection included 9 PTMCs (among which 3 bifocal and 1 trifocal) and 4 PTCs (3 not otherways specified (NOS) and one PDTC, the latter displaying both papillary and solid areas). All the PTMC samples showed a very low expression of the Ki-67 proliferation marker. Nine samples were scored positive for both CXCR2 and p16INK4a expression, and in most cases stretches of p16INK4a positivity largely overlapped with CXCR2 positive areas. Representative pictures from three samples are shown in Figure 7a. In the remaining 5 PTMC samples, focal CXCR2 positivity was also observed in tumour areas that were negative for p16INK4a staining.
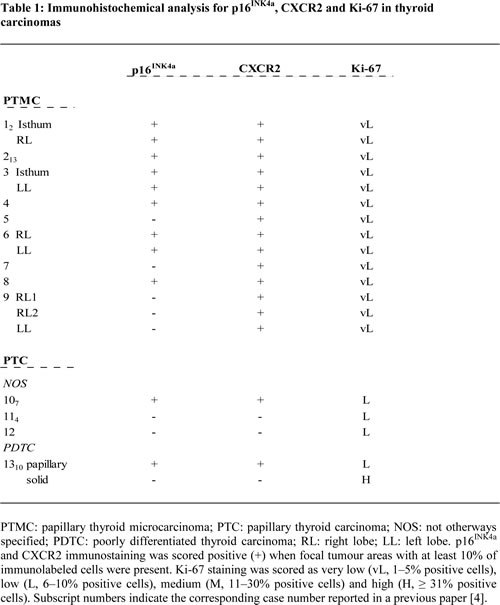
The three PTC NOS samples displayed low proliferation index. One sample expressing p16INK4a was also positive for CXCR2, whereas the others two samples were scored negative for both p16INK4a and CXCR2.
The co-existence of OIS markers and CXCR2 is strengthened by the analysis of a PDTC sample (#13), representing a model of in situ tumour progression: it displays areas with papillary and solid pattern of growth, previously shown, positive and negative for the expression of senescence markers [4]. As shown in Table 1 and Figure 7b the papillary areas, associated with low proliferation index, were positive for both p16INK4a and CXCR2, whereas the solid areas, with high Ki-67 expression, were negative. This finding corroborates the notion that loss of CXCR2 contributes to escape senescence.
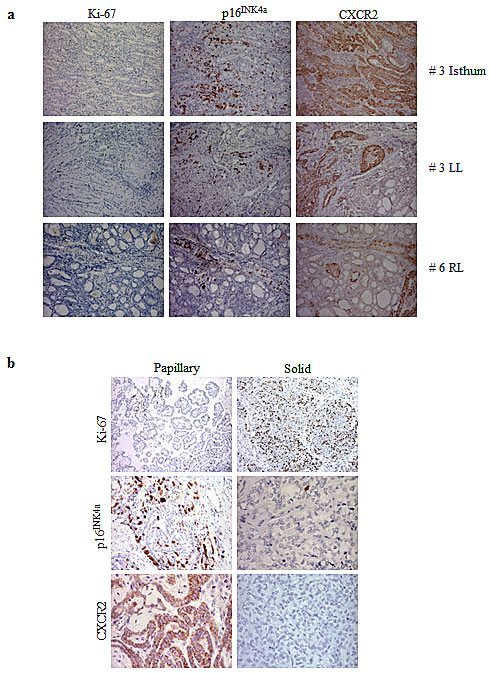
Figure 7: Immunohistochemical analysis for the expression of Ki-67, p16INK4a and CXCR2. (a) Representative pictures of PMTC #3 and #6. LL: left lobe; RL: right lobe. (b) Papillary and solid areas of PDTC #13.
DISCUSSION
We have recently proposed that OIS, a mechanism recognized as a barrier against tumor development, may be involved in the pathogenesis of thyroid carcinoma. In particular, we have reported that PTC-associated oncogenes, including H-RASG12V, induce senescence in human primary thyrocytes [4]. As tool for a deeper analysis of OIS thyrocytes, here we established and characterized an inducible model of thyrocytes senescence based on ER:RAS inducible system. Activation of RAS led to senescence-associated characteristics, as judged by the presence of growth arrest, changes in cell morphology, increased SA-β-Gal activity and upregulation of p16INK4a, p21CIP1 and p53.
As reported in several studies the critical role of p53 and p16INK4a in senescence is not universal across cells type and it differs between mouse and human cells. For instance, in mouse embryo fibroblasts disruption of p53 is sufficient to prevent senescence [32]. Likewise, inactivation of pRb gene together with other members of Rb family such as p107 and p130, but not pRb alone, is sufficient to escape senescence [33]. Unlike the behaviour of mouse cells, the inactivation of both p53 and pRb is essential to prevent the onset of replicative senescence in human cells [34], whereas disruption of only one of these proteins delays the onset of senescence.
To assess the relative contribution of the p16INK4a, p53 and p21CIP1 in the thyrocytes senescence program we used RNA interfering strategy. In particular, in our experimental setting, depletion of p53 had no impact on senescence upon oncogenic stress. For instance, no change in cell proliferation, BrdU incorporation or SA-β-Gal activity was observed between p53-intact and p53-null senescent thyrocytes. Interestingly, p53 depletion reduced the levels of p21CIP1, whereas the expression of p16INK4a was maintained. Similarly, p21CIP1 knockdown had no influence in the inhibition of proliferation induced by activation of RAS. On the contrary, the silencing of p16INK4a was sufficient to abrogate the onset of senescence. This was illustrated by increased proliferative potential as well as decreased in SA-β-Gal activity. Thus, in our experimental setting p16INK4a seems to be a relevant effector, supporting a model in which oncogenic RAS acts primarily through the p16INK4a pathway in thyrocytes.
It is important to note that these results are in contrast with our previous findings in BRAFV600E lentivirally transduced thyrocytes, where the inactivation of p16INK4a was not sufficient to prevent induction of senescence, suggesting that the critical role of p16INK4a in the abrogation of senescence may also depend on different genetic context, as previously proposed by others [5;12]. However, in thyrocytes infected with retroviral vector encoding BRAFV600E, p16INK4a silencing was able to overcome cell cycle arrest (unpublished results). Thus, the different oncogene expression levels due to the different methods of oncogene delivery may warrant the different response upon p16INK4a depletion.
In the last few years, growing body of evidence suggests that OIS is connected with inflammation. Cells undergoing senescence produce increased amount of inflammatory cytokines and chemokines that play an essential role in the initiation and maintenance of cellular senescence [25;26].
We found that several proinflammatory components, including IL-8/CXCR2, IL-6, IL-1, as well as CCL20 and CCR2 are upregulated in ER:RAS senescent thyrocytes, thus suggesting that a SASP-like phenomenon may occurr.
Our results suggest that IL-8/CXCR2 axis plays a role in mediating thyrocytes OIS, being involved in growth arrest triggered by oncogenic stress. Knockdown of IL-8 using two independent shRNAs vectors allowed 4OHT-treated ER:RAS cells to circumvent cell cycle arrest and resulted in decreased CXCR2 levels. Similarly, upon depletion of CXCR2 by shRNA vector, 4OHT-treated ER:RAS thyrocytes were able to resume proliferation. We also observed a drop of IL-8 expression after CXCR2 abrogation, suggesting a coordinated regulation of IL-8 and its receptor during senescence.
In parallel to these observations in vitro, we observed a correlation between increased CXCR2 expression and OIS markers immunopositivity in human thyroid samples, suggesting that IL-8/ CXCR2 axis limits thyroid tumour progression. These findings are in harmony with previously models [25;26] in which the activation of the inflammatory network plays an essential role in OIS in vitro and in vivo.
On the other hand, it is well known that cytokines and chemokines have also been implicated in promoting cancer, increasing angiogenesis, vascularisation and metastasis [35].
A link between thyroid cancer and inflammation has also been recognized; thyroid carcinomas are associated with the constitutive expression of several proinflammatory cytokines (IL-8, IL-6, GROα, IL-1α, G-CSF, GM-CSF) that can be able to maintain the invasive phenotype [36;37]. It has been shown that all PTC-associated oncogenes (BRAF, RAS and RET/PTC) induces the up-regulation of cytokines and chemokines in normal thyroid cells; these factors can promote proliferation, invasion and metastasis [30;38]. Interestingly, in this study we have shown that in senescent thyrocytes IL-8 acts in concert with its receptor CXCR2 to reinforce senescence. What switches the secretome components from prosenescence to protumorigenic activity is not well understood: stage and status of lesion and its genetic background could be crucial factors. To note, CXCR2 mutation or downregulation cancels its prosenescent action facilitating tumour progression [26]. Moreover, several studies have yielded a wealth of evidence that some of these secreted proteins reinforce senescence by acting in autocrine manner, whereas they promote malignant phenotype by paracrine activity. Interestingly, Acosta et al. [39] recently demonstrated that these factors can trigger senescence by paracrine fashion in vitro and in vivo models.
In this paper we have used an inducible model for the characterization of OIS in thyrocytes. Such model, which mimics in vitro thyroid tumour early stages, represents a useful tool for a further dissection of the mechanisms governing OIS in thyrocytes and the interplay between senescent thyrocytes and microenvironment. This kind of studies, coupled to analyses performed in thyroid tumour samples characterized by different aggressiveness, as well as in corresponding cell lines, will allow defining the role of OIS players, including chemokines and cytokines, at early and late tumour stages. This will assess whether prosenescence therapy may represent a strategy for thyroid cancer treatment.
METHODS
Cell culture and treatments
Normal thyroid samples were obtained from patients undergoing surgery at IRCCS Istituto Nazionale dei Tumori (Milan, Italy). All patients gave their written informed consent, and the study was approved by the Independent Ethical Committee of Fondazione IRCCS Istituto Nazionale dei Tumori. Primary thyrocyte cultures were established and maintained in nutrient mixture Ham’s F12 medium (custom made by Invitrogen, Carlsbad, CA, USA) containing 5% calf serum and bovine hypothalamus and pituitary extracts, as previously described [40].
4-hydroxytamoxifen (4OHT) (Sigma Aldrich, St Louis, Mo, USA) was used at 200 nM to activate RAS oncogene. Geneticin (G418, GIBCO, Carlsbad, CA, USA) and puromycin (Invitrogen, Carlsbad, CA, USA) were used for cell selection at 400 µg/ml and 2 µg/ml, respectively.
Constructs and siRNA
Vector encoding H-RASG12V has been described previously [41]. Knockdown of p16INK4a, p53, IL-8 and CXCR2 was achieved using validated shRNA retroviral vectors [26;41]. Silencing of p16INK4a, p53 and p21CIP1 was achieved using commercial siRNA (QIAGEN, Hilden, Germany), as described previously [26;41].
Retrovirus production and infection
Methods used for retrovirus production and infection have been previously described [41].
shRNA and siRNA transfection
For shRNA transduction ER:RAS cells were transduced with shRNA retroviral vector and maintained under puromycin selection for 10 days.
For siRNA transfection, ER:RAS cells were reverse-transfected with 30 nM siRNA using a 3.5% solution of Hiperfect (QIAGEN, Hilden, Germany). The Cy3-labeled siGLO cyclophilin B siRNA (Dharmacon, Buckinghamshire, UK) was used to monitor transfection efficiency. AllStars scrambled siRNA were used as negative controls.
Cristal violet staining
Cells (1x105) were plated in 10-cm dish. After 7 days cells were washed with PBS, fixed with 0.5% glutaraldehyde (w/v) in PBS for 30 minutes, and then stained with 0.2% crystal violet (w/v).
BrdU (5-Bromo-2’deoxyuridine) incorporation assay
Cells were plated in 96-well plates and subsequently incubated with 5-Bromo-2’ deoxyuridine (BrdU, 50 µM, Sigma Aldrich, St Louis, Mo, USA) for 24 hours. Cells were then washed with PBS, fixed for 30 minutes with 4% (w/v) paraformaldehyde (Sigma Aldrich, St Louis, Mo, USA), and subjected to immunofluorescence staining as previously described [26]. Briefly, after permeabilization for 5 minutes with 0.2 (v/v) Triton X-100 in PBS, and incubation for 30 minutes with 1X blocking solution (0.5% (w/v) BSA, 0.2% (w/v) fish skin gelatin), cells were incubated with anti-BrdU primary antibody (1:500, BD Becton Dickinson, NJ, USA) in the presence of DNase I (0.5U/µL; Sigma Aldrich, St Louis, Mo, USA) and 1 mM MgCl2 for 30 minutes. After washing with PBS cells were incubated with Alexa Fluor 488 mouse secondary antibody (1:500, Invitrogen, Carlsbad, CA, USA) for 30 minutes and then incubated with DAPI (1.5 µM, Invitrogen, Carlsbad, CA, USA) for 30 minutes. Plates were examined using In Cell Analyser and High Content Analysis was performed to discriminate positive BrdU nuclei and total nuclei (IN Cell Analyser 1000, GE Healthcare Buckinghamshire, UK).
Senescence-associated β-galactosidase (SA-β-Gal) assay
SA-β-Gal staining was performed as described previously [4]. Images were taken using the Olympus CKX41 inverted fluorescence microscope, supplied with a DP20 digital camera. The percentage of SA-β-gal positive cells were determined upon counting of at least 200 cells.
Quantitative RT-PCR
RNA was isolated with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and purified with RNeasy purification kit (QIAGEN, Hilden, Germany). One µg of RNA was retrotranscribed using SuperscriptIII (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. For each sample, 20 ng of retro-transcribed RNA were amplified by PCR carried out in triplicate on an ABI PRISM 7900 using the following TaqMan gene expression assays: Hs00174092_m1 for IL-1α expression; Hs00174097_m1 for IL-1β expression; Hs00171125_m1 for CCL20 and Hs00174298_m1 for CCR1 expression. Human HPRT (HPRT-Hs99999909_m1) was used as housekeeping gene for normalisation. Data analysis was performed by the Sequence Detection System (SDS) 2.2.2 software (Applied Biosystems, Foster City, CA, USA).
Immunofluorescence and high content analysis
Immunofluorescence staining was performed as for BrdU assay using the following primary antibodies: anti-p16 INK4a (JC8, 1:100, Santa Cruz Biotechnology Inc, Santa Cruz, CA, USA); anti-p21CIP1 (P1484, 1:100, Sigma Aldrich, St Louis, Mo, USA); anti-p53 (DO-1, 1:100, Santa Cruz Biotechnolgy Inc, Santa Cruz, CA, USA), anti-IL-8 (1:100, BD, Becton Dickinson, NJ, USA), anti-CXCR2 (1:100, BD, Becton Dickinson, NJ, USA), anti-IL-6 (1:100, BD Becton Dickinson, NJ, USA). Anti-mouse Alexa488-conjugated antibody (1:500, Invitrogen, Carlsbad, CA, USA) was used as secondary antibody. Acquisition of immunofluorescence images was performed using the IN Cell Analyzer 1000 automated high-throughput microscope (GE Healthcare, Buckinghamshire, UK) with 10x objective. Image processing was performed using the IN Cell Investigator (v1.7) software (GE Healthcare, Buckinghamshire, UK). High Content Analysis (HCA) was used for quantification of immunofluorescence images and was performed as described elsewhere [26;39;41;42]. Briefly, two fluorescence images corresponding to DAPI and primary/antibody/Alexa Fluor 488-secondary were acquired for each field. No fewer than 1,000 cells were acquired. HCA was performed using the IN Cell Investigator (v1.7) software (GE Healthcare, Buckinghamshire, UK). For the analysis, DAPI staining of the nuclei was used to identify nuclear area and number of cells. The nuclei were defined by using top-hat segmentation, specifying a minimum nucleus area of 100 µm2. To determine the cellular expression of the analysed protein, the average intensity of pixels in the reference channel (Alexa Fluor 488) within the specified nuclear region (Object Nuclear Intensity) was measured. Each cell was assigned a nuclear intensity value for the specific protein expression and that value was used to set up a threshold filter, which determined high levels (positive) and low levels (negative) expressing cells. In order to set the filter cut-off, expression in the control cells was measured to define the negative population followed by the analysis of the positive control. As a result, the software classified each cell as either positive or negative for the expression of the analysed protein and generated a percentage of both cell population (positive and negative) per well. The mean of the nuclear intensity was also routinely analysed and equivalent results were obtained.
IL-8 and IL-6 detection in conditioned medium (CM)
IL-8 detection in CM was performed by western blot analysis. CM was obtained by incubating cells in serum-free medium for 24h. After concentration by centrifugation at 4000 r.p.m. using AgilentSpin Concentrators (Agilent Technologies Inc.,Wilmington, DE, USA), the CM was normalized to cells number, and processed for Western blot analysis.
IL-6 detection was performed by ELISA using the Human Inflammatory Cytokines Multi-Analyte ELISArray Kit (QIAGEN, Hilden, Germany) following manufacturer’s instructions. CM was obtained by incubating cells in complete medium for 24h.
Western Blotting analysis
Cell lysates were obtained by extraction in RIPA modified buffer (20mM Tris-HCl, pH 7.4, 150mM NaCl, 5mM EDTA, 1% NonidetP-40) supplemented with Complete Mini EDTA-free protease Inhibitor Cocktail (Roche, Manheim, Germany), 1mM Na3VO4 and 1mM PMSF. Protein samples were quantified by Bradford’s assay with BIO-RAD Protein Assay (Bio-Rad, Munchen, Germany). Conditioned medium protein samples were obtained as described above. Protein samples were boiled in NuPAGE LDS sample buffer (Invitrogen, Carlsbad, CA, USA) and separated on 4-12% NuPAGE Novex Gel (Invitrogen, Carlsbad, CA, USA) with MES running buffer. Proteins were transferred onto nitrocellulose filters and immunoblotted with the following primary antibodies to: p16INK4a (BD Becton Dickinson, NJ, USA); p21CIP1(Santa Cruz, Biotechnology, Inc, Santa Cruz, CA, USA ); p53 (DO-1, Santa Cruz Biotechnolgy Inc, Santa Cruz, CA, USA); β -Actin (Sigma Aldrich, St Louis, Mo, USA); IL-8 (Abcam, Cambridge, UK). The immunoreactive bands were visualized using horseradish peroxidase-conjugated secondary antibodies followed by enhanced chemiluminescence (GE Healthcare, Buckinghamshire, UK).
Tumour samples and immunohistochemical analysis
Tumour samples are from patients undergoing surgery at Fondazione IRCCS Istituto Nazionale dei Tumori (Milan, Italy). All patients gave their written informed consent, and the study was approved by the independent ethics committee of Fondazione IRCCS Istituto Nazionale dei Tumori.
Stainings were performed on formalin-fixed, paraffin-embedded tissue sections of 2 µm thickness. Antigen retrieval was performed using 1 mM citrate buffer (pH 6) in an autoclave at 95°C for 15 minutes. Incubation with primary antibodies was performed overnight at 4°C for anti-CXCR2 (1:100, BD, Becton Dickinson, NJ, USA) and for 1 hour at room temperature for anti-human Ki-67 Antigen (Mib1) (DakoCytomation, Glostrup, Denmark). Section were then incubated for 30 minutes with biotinylated anti-mouse, developed using 3,3’-diaminobenzidine (DakoCytomation, Glostrup, Denmark) as chromogen, and finally counterstained with hematoxylin.
p16INK4a staining was performed using CINtec Histologic Kit (mtm laboratories AG, Heidelberg, Germany) following manufacturer’s instructions.
ACKNOWLEDGEMENTS
We thank all the members of Dr. J. Gil’s laboratory for their unconditional help; Drs Valentina Mauro and Barbara Cortelazzi and Mrs. Francesca Dominoni (Pathology Department) for the IHC staining; Dr. Federica Perrone (Pathology Department) for assistance in the selection of archival thyroid tumour material; Dr. Silvia Veneroni and Tissue Bank of Fondazione IRCCS Istituto Nazionale dei Tumori for sample collection.
This work was supported by AIRC grant N. 11347 to A.Greco; M.G. Vizioli was supported by FIRC fellowship for abroad (June 2011-May 2012) and “Giavanna Ciani” AIRC fellowship (2013).
Competing and financial interests
The authors declare no competing or financial interests.
REFERENCES
1. Delellis RA, Lloyd RV, Heitz PU, Eng C. Pathology and genetics of tumors of endocrine organs. In: IARC Press, editor. World Health Organization classification of tumours. Lyon: IARC Press, 2004.
2. Sheils O. Molecular classification and biomarker discovery in papillary thyroid carcinoma. Expert Rev Mol Diagn. 2005;5(6):927-946.
3. Greco A, Borrello MG, Miranda C, Degl’Innocenti D, Pierotti MA. Molecular pathology of differentiated thyroid cancer. Q J Nucl Med Mol Imaging. 2009;53(5):440-453.
4. Vizioli MG, Possik PA, Tarantino E, Meissl K, Borrello MG, Miranda C, Anania MC, Pagliardini S, Seregni E, Pierotti MA, Pilotti S, Peeper DS, Greco A. Evidence of oncogene-induced senescence in thyroid carcinogenesis. Endocr Relat Cancer. 2011;18:743-757.
5. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593-602.
6. Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. 2014;28:99-114.
7. Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463-2479.
8. Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging (Albany NY). 2012;4:159-165.
9. Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre’ M, Nuciforo PG, Bensimon A, Maestro R, Pelicci PG, dda di FF. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638-642.
10. Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, Takaoka M, Nakagawa H, Tort F, Fugger K, Johansson F, Sehested M, Andersen CL, Dyrskjot L, Orntoft T, Lukas J, Kittas C, Helleday T, Halazonetis TD, Bartek J, Gorgoulis VG. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633-637.
11. Gil J, Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. 2006;7:667-677.
12. Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600 -associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720-724.
13. Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dorken B, Jenuwein T, Schmitt CA. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660-665.
14. Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, Cordon-Cardo C, Pandolfi PP. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725-730.
15. Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51-57.
16. Courtois-Cox S, Genther Williams SM, Reczek EE, Johnson BW, McGillicuddy LT, Johannessen CM, Hollstein PE, MacCollin M, Cichowski K. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell. 2006;10:459-472.
17. Dankort D, Filenova E, Collado M, Serrano M, Jones K, McMahon M. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E -induced lung tumors. Genes Dev. 2007;21(4):379-384.
18. Ha L, Ichikawa T, Anver M, Dickins R, Lowe S, Sharpless NE, Krimpenfort P, DePinho RA, Bennett DC, Sviderskaya EV, Merlino G. ARF functions as a melanoma tumor suppressor by inducing p53-independent senescence. Proc Natl Acad Sci U S A. 2007;104(26):10968-10973.
19. Coppe JP, Kauser K, Campisi J, Beausejour CM. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem. 2006;281:29568-29574.
20. Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973-979.
21. Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009;9:81-94.
22. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656-660.
23. Suarez HG, du Villard JA, Severino M, Caillou B, Schlumberger M, Tubiana M, Parmentier C, Monier R. Presence of mutations in all three ras genes in human thyroid tumors. Oncogene. 1990;5:565-570.
24. Tarutani M, Cai T, Dajee M, Khavari PA. Inducible activation of Ras and Raf in adult epidermis. Cancer Res. 2003;63:319-323.
25. Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van DR, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019-1031.
26. Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da CM, Brown C, Popov N, Takatsu Y, Melamed J, dda di FF, Bernard D, Hernando E, Gil J. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006-1018.
27. Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8:877-884.
28. Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132:363-374.
29. Borrello MG, Alberti L, Fischer A, Degl’Innocenti D., Ferrario C, Gariboldi M, Marchesi F, Allavena P, Greco A, Collini P, Pilotti S, Cassinelli G, Bressan P, Fugazzola L, Mantovani A, Pierotti MA. Induction of a proinflammatory programme in normal human thyrocytes by the
RET/PTC1 oncogene. Proc Natl Acad Sci U S A. 2005;102:14825-14830.
30. Guarino V, Castellone MD, Avilla E, Melillo RM. Thyroid cancer and inflammation. Mol Cell Endocrinol. 2010;321:94-102.
31. Kobawala TP, Patel GH, Gajjar DR, Patel KN, Thakor PB, Parekh UB, Patel KM, Shukla SN, Shah PM. Clinical utility of serum interleukin-8 and interferon-alpha in thyroid diseases. J Thyroid Res. 2011;2011:270149.
32. Dirac AM, Bernards R. Reversal of senescence in mouse fibroblasts through lentiviral suppression of p53. J Biol Chem. 2003;278:11731-11734.
33. Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B, Theodorou E, Jacks T. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev. 2000;14:3037-3050.
34. Smogorzewska A, de LT. Different telomere damage signaling pathways in human and mouse cells. EMBO J. 2002;21:4338-4348.
35. Mantovani A, Pierotti MA. Cancer and inflammation: a complex relationship. Cancer Lett. 2008;267:180-181.
36. Borrello MG, Degl’Innocenti D., Pierotti MA. Inflammation and cancer: the oncogene-driven connection. Cancer Lett. 2008;267:262-270.
37. Muzza M, Degl’Innocenti D, Colombo C, Perrino M, Ravasi E, Rossi S, Cirello V, Beck-Peccoz P, Borrello MG, Fugazzola L. The tight relationship between papillary thyroid cancer, autoimmunity and inflammation: clinical and molecular studies. Clin Endocrinol (Oxf). 2010;72:702-708.
38. Melillo RM, Castellone MD, Guarino V, De F, V, Cirafici AM, Salvatore G, Caiazzo F, Basolo F, Giannini R, Kruhoffer M, Orntoft T, Fusco A, Santoro M. The RET/PTC-RAS-BRAF linear signaling cascade mediates the motile and mitogenic phenotype of thyroid cancer cells. J Clin Invest. 2005;115:1068-1081.
39. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, Pascual G, Morris KJ, Khan S, Jin H, Dharmalingam G, Snijders AP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978-990.
40. Curcio F, Ambesi-Impiombato FS, Perrella G, Coon HG. Long-term culture and functional characterization of follicular cells from adult normal human thyroids. Proc Natl Acad Sci U S A. 1994;91:9004-9008.
41. Barradas M, Anderton E, Acosta JC, Li S, Banito A, Rodriguez-Niedenfuhr M, Maertens G, Banck M, Zhou MM, Walsh MJ, Peters G, Gil J. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 2009;23:1177-1182.
42. Banito A, Rashid ST, Acosta JC, Li S, Pereira CF, Geti I, Pinho S, Silva JC, Azuara V, Walsh M, Vallier L, Gil J. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009;23:2134-2139.