
Genomic profiling demonstrates a multitude of aberrations across a broad spectrum of cancer-related genes [1, 2] and represents a new way to classify cancer [3]. Many studies demonstrate that matching patients to treatment based on molecular characteristics can result in remarkable responses, even in advanced, refractory disease, despite heterogeneity in patients’ molecular landscapes [3]. However, most patients have short-lived responses.
Herein, we demonstrate that, amongst 57 patients with metastatic breast cancer, no two patients had the same molecular portfolio. The number of distinct aberrations driving advanced tumors may therefore be responsible for the remarkable resistance of metastatic malignancies to prolonged remission or cure, especially with our current therapeutic strategies [3]. These data suggest that traditional design paradigms governing diagnosis and management of metastatic cancer and the clinical research that informs them, all of which are predicated on grouping patients in order to evaluate targeted agents, may be inadequate to address the complexity unveiled by modern genomics.
We used next generation sequencing (NGS) of 182 to 236 genes to interrogate tumors from 57 women with metastatic breast cancer (26 with hormone receptor (HR)-positive and HER2-negative tumors; 7 with HR-positive/HER2-positive tumors; 4 with HR-negative/HER2-positive tumors; and, 20 with triple negative breast cancer (TNBC) (Supplemental Table 1) [4]. Importantly, if different variants in each gene were considered as distinct then each patient had a unique profile of genomic aberrations (Supplemental Table 1). Even if variants within a gene were considered identical, there were 54 different genomic landscape patterns amongst the 57 patients (Supplemental Table 1).
The most commonly altered genes (in order of decreasing frequency) were: TP53, PIK3CA, CCND1, MYC, HER2 (ERBB2), MCL1, PTEN, FGFR1, GATA3, NF1, PIK3R1, BRCA2, EGFR, IRS2, CDH1, CDKN2A, FGF19, FGF3 and FGF4 (Supplemental Table 2). The most frequent aberrations were mutations and amplifications, comprising 46% and 45% of abnormalities, respectively. The average number of molecular aberrations per patient was 4 (average = 4 in HR-positive/HER2-negative; 4 in HR-positive/HER2-positive; 3 in HR-negative /HER2-positive; and 4 in TNBC (Figure 1).
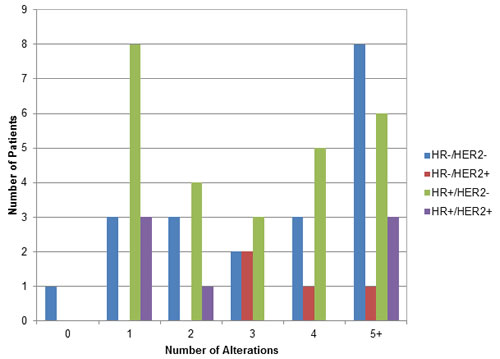
Figure 1: Number of Alterations seen in Patients Grouped by Hormone Receptor (HR) and HER2 Status
A total of 131 distinct somatic aberrations (216 aberrations in total) were identified: 80 mutations (including mutation variants in the same gene); 37 amplifications; five deletions; five splices; two truncations; one fusion; and, one rearrangement. Of the 80 distinct mutation variants we observed (Supplemental Table 3), 49 have not been previously reported for patients with breast cancer in COSMIC, The Cancer Genome Atlas (TCGA) [1, 2] and other large genomic databases [5, 6]. Thirty-seven distinct amplifications were found, the most common being CCND1 (12 patients), MYC (12 patients), MCL1 (7 patients), HER2 (ERBB2; 7 patients), and FGFR1 (6 patients). All amplifications had been previously reported in breast cancer.
Many, if not most, of the genomic aberrations represent actionable targets, either by approved drugs or by agents in clinical trials [7, 9-14]. Among the most common aberrations were those seen in the PI3K/AKT/mTOR pathway (Supplemental Table 2) [7, 8]: PIK3CA mutations with eight different variants were noted (15 patients, including 3 patients with more than one variant); PIK3CA amplifications (3 patients); PTEN deletions (6 patients); PIK3R1 mutation (5 patients with 4 different mutation variants); AKT3 amplifications (2 patients); and PTEN or AKT1 mutation (1 patient each). Other druggable aberrations include, but are not limited to, EGFR, HER2, CDKN2A, FGFR/FGF family, KRAS, MDM2, BRCA2, and more.
Our observations are consistent with the hypothesis that metastatic tumors, even when originating in the same organ (e.g., the breast), are mostly distinct at the molecular level (malignant snowflakes) [9]. Of special interest, many of the genomic aberrations we observed may be actionable, through either direct or indirect ‘cross talk’ signal suppression [7, 9-14]. For instance, prior literature indicates that PIK3CA is the oncogene showing the highest frequency of gain-of-function mutations in breast cancer [7, 10-12] (ranging from ~13% in TNBC to ~42% in HR positive+/HER2- subgroup) [10]. In our study, PIK3CA was the second most common aberrant gene, and was involved in 12% of abnormalities (Supplemental Table 2). Further, if patients with other molecular alterations in the PI3K/AKT/mTOR pathway are included (e.g. PTEN, AKT, PIK3R1, etc), the subset of patients expands. Of therapeutic interest, the BOLERO-2 study of the hormone modulator exemestane and the mTOR inhibitor everolimus showed that patients whose tumors had 0 or 1 genomic aberrations in PIK3CA or FGFR pathways or in CCND1 had better outcomes [13]. Further, we previously reported that heavily pretreated patients with PIK3CA-mutant breast and gynecologic tumors have higher than expected response rates (partial/complete response rates ~30%) when mTOR inhibitor-based combination regimens are administered [11, 14, 15]. However, despite the large percentage of aberrations (Supplemental Table 2) that can be targeted by drugs that are already approved or in clinical trials, our data illustrates why current clinical trials and practice generally provide only short-lived tumor regressions. In particular, the fact that most patients have multiple aberrations, and that the abnormalities differ from individual to individual, suggests that treating different patients with the same drug or drug combination may be insufficient to optimize success.
There are limitations to our data: the number of patients we analyzed was small and, in some cases, different aberrations in the same gene may have a similar effect on the downstream pathway. However, even if we considered all aberrations in the same gene as functionally equivalent (a highly unlikely situation), 95% (54 of 57) of our patients still had distinct genomic portfolios. The reality that NGS interrogation of these tumors has revealed is that, since most patients with metastatic breast malignancies have unique genomic landscapes, the standard methods deployed for therapeutic prosecution of molecular aberrations may need to be revised.
Classic clinical trial design mandates grouping patients by tumor site and histology (e.g., breast or colon cancer) and evaluating the efficacy of a drug or combination of agents. In the era of molecular therapeutics, the first generation of newer clinical trial designs stratify patients, often on the basis of a single molecular aberration as identified by a companion diagnostic, and test the efficacy of a cognate inhibitor to improve outcomes. The latter model represents a scientifically rational advancement that has been successful to an extent. Indeed, patients with HER2-positive breast cancer respond to trastuzumab, lapatinib, and pertuzumab [16], and ALK-rearranged advanced lung tumors respond to the ALK inhibitor crizotinib [17]. However, the majority of patients do not achieve durable responses. It is not surprising that these tactics fail to provide long-term survival benefit since our current observations suggest that each patient, despite the commonality of the anomaly that stratifies them for the clinical trial/treatment, has a distinct aberrant molecular backdrop. Indeed, these novel clinical trials using companion diagnostics to select patients so that an appropriately targeted agent can be tested are still retrofitting our new knowledge about metastatic cancer–at the molecular level, each patient has a unique and complex tumor–into a dated clinical trial paradigm where patients are grouped together to evaluate a drug.
Previously, we have shown that, using genomic aberrations to match patients with advanced, heavily-pretreated malignancies (including breast cancer) to drugs, in the context of phase I studies, resulted in improved outcomes as assessed by response rate, progression-free survival and overall survival, as well as progression-free survival on matched Phase I therapy as compared to last conventional therapy; in the case of melanoma, progression-free survival on matched phase I therapy was improved as compared to first-line treatment [8, 11, 18, 19]. Of interest, these studies used the first-generation molecular profiling technology available at the time (i.e. tumors were probed for gene aberrations one at a time, rather than with next generation sequencing). Further trials employing next generation sequencing, as well as additional advanced omic technologies, and randomized designs are warranted.
In summary, NGS may represent a disruptive technology that mandates a new approach to clinical research and cancer treatment. Even if there is some convergence of pathways with functional impact [20-23], introduction of additional omics tests–transcriptomics, proteomics, epigenomics etc.–may reveal yet more complexity and differences between patients’ tumors leading to major challenges in personalized treatment. The complexity of the molecular genetics of metastatic cancer is not conducive to the success of classic drug-centric clinical trial models, where patients are grouped together in order to test the efficacy of a drug or combination. We propose testing a new patient-centric, molecular matching strategy to find an optimal treatment regimen tailored to each patient’s genomic profile acquired from multi-assay molecular testing. Importantly, this approach would permit the therapy given to vary from individual to individual consistent with N-of-One customization. The framework for such a trial would utilize genomics and/or other omic technologies to navigate patients to individualized therapy. Forerunners of this design have been utilized in the IMPACT/PREDICT trials (NCT00851032) [8], as well as newer trials such as WINther (the latter utilizing both genomics and transcriptomics to type patients) (NCT01856296) and the planned NCI MATCH trial [24]. These trials concentrated on finding matches for single aberrations; next generation trials can exploit the same design, but deploy advanced multiplex technology, and focus on appropriate tailored combinations.
METHODS
Fifty-seven sequential consented patients with advanced, pathologically confirmed breast cancer who had next generation sequencing (182 to 236 genes) were analyzed. Tumor samples were evaluated for genomic alterations including base substitutions, short insertions and deletions, amplifications, homozygous deletions, gene fusions, truncations and rearrangements (Foundation Medicine, Cambridge, MA). DNA was extracted from 40 μm of FFPE tissue (minimum 20% tumor cells) using the Maxwell 16 FFPE Plus LEV DNA Purification kit (Promega) and quantified using a standardized PicoGreen fluorescence assay (Invitrogen). Library Construction was performed using 50-200 ng of DNA sheared by sonication to ~100-400 bp before end-repair, dA addition and ligation of indexed, Illumina sequencing adaptors. Enrichment of target sequences (all coding exons of 182 or 236 cancer-related genes and selected introns from 14 or 19 genes recurrently rearranged in cancer) was achieved by solution-based hybrid capture with custom biotinylated oligonucleotide baits. Enriched libraries were sequenced to an average median depth of >500X with 99% of bases covered >100X (Illumina HiSeq 2000 platform using 49 × 49 paired-end reads) and mapped to the reference human genome (hg19) using the Burrows-Wheeler Aligner and the publicly available SAMtools, Picard and Genome Analysis Toolkit. Point mutations were identified by a Bayesian algorithm; short insertions and deletions, determined by local assembly; gene copy number alterations (amplifications), by comparison to process matched normal controls; and gene fusions/rearrangements, by clustering chimeric reads mapped to targeted introns. Local site permissions to use clinical samples were also obtained.
ACKNOWLEDGEMENTS
Funded in part by CA016672 from the National Cancer Institute, Joan and Irwin Jacobs Fund and MyAnswerToCancer philanthropic fund.
REFERENCES
1. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, Teague JW, Campbell PJ, Stratton MR and Futreal PA. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011; 39(Database issue):D945-950.
2. Robbins DE, Gruneberg A, Deus HF, Tanik MM and Almeida JS. A self-updating road map of The Cancer Genome Atlas. Bioinformatics. 2013; 29(10):1333-1340.
3. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012; 366(10):883-892.
4. Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, Demeter J, Perou CM, Lonning PE, Brown PO, Borresen-Dale AL and Botstein D. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A. 2003; 100(14):8418-8423.
5. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C and Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2(5):401-404.
6. Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF and den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011; 32(5):557-563.
7. Janku F, Wheler JJ, Naing A, Stepanek VM, Falchook GS, Fu S, Garrido-Laguna I, Tsimberidou AM, Piha-Paul SA, Moulder SL, Lee JJ, Luthra R, Hong DS and Kurzrock R. PIK3CA mutations in advanced cancers: characteristics and outcomes. Oncotarget. 2012; 3(12):1566-1575.
8. Tsimberidou AM, Iskander NG, Hong DS, Wheler JJ, Falchook GS, Fu S, Piha-Paul S, Naing A, Janku F, Luthra R, Ye Y, Wen S, Berry D and Kurzrock R. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clin Cancer Res. 2012; 18(22):6373-6383.
9. Munoz J, Swanton C and Kurzrock R. Molecular profiling and the reclassification of cancer: divide and conquer. Am Soc Clin Oncol Educ Book. 2013; 2013:127-134.
10. Cizkova M, Susini A, Vacher S, Cizeron-Clairac G, Andrieu C, Driouch K, Fourme E, Lidereau R and Bieche I. PIK3CA mutation impact on survival in breast cancer patients and in ERalpha, PR and ERBB2-based subgroups. Breast Cancer Res. 2012; 14(1):R28.
11. Janku F, Wheler JJ, Westin SN, Moulder SL, Naing A, Tsimberidou AM, Fu S, Falchook GS, Hong DS, Garrido-Laguna I, Luthra R, Lee JJ, Lu KH and Kurzrock R. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012; 30(8):777-782.
12. Wheler J, Tsimberidou AM, Moulder S, Cristofanilli M, Hong D, Naing A, Pathak R, Liu S, Feng L and Kurzrock R. Clinical outcomes of patients with breast cancer in a phase I clinic: the M. D. Anderson cancer center experience. Clin Breast Cancer. 2010; 10(1):46-51.
13. Hortobagyi GN, Piccart-Gebhart MJ, Rugo HS, Burris HA, Campone M, Noguchi S, Perez A, Deleu I, Shtivelband M, Provencher L, Masuda N, Dakhil SR, Anderson I, Chen D, Damasck A, Huang A, et al. Correlation of molecular alterations with efficacy of everolimus in hormone receptor-positive, HER2-negative advanced breast cancer: Results from BOLERO-2. J Clin Oncol. 2013; 31(18_suppl):LBA509.
14. Janku F, Wheler JJ, Naing A, Falchook GS, Hong DS, Stepanek VM, Fu S, Piha-Paul SA, Lee JJ, Luthra R, Tsimberidou AM and Kurzrock R. PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Cancer Res. 2013; 73(1):276-284.
15. Moroney JW, Schlumbrecht MP, Helgason T, Coleman RL, Moulder S, Naing A, Bodurka DC, Janku F, Hong DS and Kurzrock R. A phase I trial of liposomal doxorubicin, bevacizumab, and temsirolimus in patients with advanced gynecologic and breast malignancies. Clin Cancer Res. 2011; 17(21):6840-6846.
16. Gradishar WJ. Emerging approaches for treating HER2-positive metastatic breast cancer beyond trastuzumab. Ann Oncol. 2013.
17. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Janne PA, Costa DB, Varella-Garcia M, Kim WH, Lynch TJ, Fidias P, Stubbs H, Engelman JA, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010; 363(18):1693-1703.
18. Henary H, Hong DS, Falchook GS, Tsimberidou A, George GC, Wen S, Wheler J, Fu S, Naing A, Piha-Paul S, Janku F, Kim KB, Hwu P and Kurzrock R. Melanoma patients in a phase I clinic: molecular aberrations, targeted therapy and outcomes. Ann Oncol. 2013; 24(8):2158-2165.
19. Wheler JJ, Yelensky R, Moulder S, Tsimberidou AM, Falchook GS, Janku F, Atkins JT, Zinner R, Karp DD, Hong DS, Stephens PJ, Wolff RA and Kurzrock R. Next-generation sequencing (NGS) in patients with advanced metastatic breast cancer: Identification of molecular alterations and analysis of associateions with treatment on phase I studies at MD Anderson Cancer Center. J Clin Oncol. 2013; 31:abstr 1051.
20. Reva B. Revealing selection in cancer using the predicted functional impact of cancer mutations. Application to nomination of cancer drivers. BMC Genomics. 2013; 14 Suppl 3:S8.
21. Augello MA, Burd CJ, Birbe R, McNair C, Ertel A, Magee MS, Frigo DE, Wilder-Romans K, Shilkrut M, Han S, Jernigan DL, Dean JL, Fatatis A, McDonnell DP, Visakorpi T, Feng FY, et al. Convergence of oncogenic and hormone receptor pathways promotes metastatic phenotypes. J Clin Invest. 2013; 123(1):493-508.
22. Axelrod M, Gordon VL, Conaway M, Tarcsafalvi A, Neitzke DJ, Gioeli D and Weber MJ. Combinatorial drug screening identifies compensatory pathway interactions and adaptive resistance mechanisms. Oncotarget. 2013; 4(4):622-635.
23. Saini KS, Loi S, de Azambuja E, Metzger-Filho O, Saini ML, Ignatiadis M, Dancey JE and Piccart-Gebhart MJ. Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer Treat Rev. 2013; 39(8):935-946.
24. Willyard C. ‘Basket studies’ will hold intricate data for cancer drug approvals. Nat Med. 2013; 19(6):655-655.
25. Bhargava R, Gerald WL, Li AR, Pan Q, Lal P, Ladanyi M and Chen B. EGFR gene amplification in breast cancer: correlation with epidermal growth factor receptor mRNA and protein expression and HER-2 status and absence of EGFR-activating mutations. Mod Pathol. 2005; 18(8):1027-1033.
26. Fabi A, Merola R, Ferretti G, Di Benedetto A, Antoniani B, Ercolani C, Nistico C, Papaldo P, Ciccarese M, Sperduti I, Vici P, Marino M, Gori S, Botti C, Malaguti P, Cognetti F, et al. Epidermal growth factor receptor gene copy number may predict lapatinib sensitivity in HER2-positive metastatic breast cancer. Expert Opin Pharmacother. 2013; 14(6):699-706.
27. Baselga J, Bradbury I, Eidtmann H, Di Cosimo S, de Azambuja E, Aura C, Gomez H, Dinh P, Fauria K, Van Dooren V, Aktan G, Goldhirsch A, Chang TW, Horvath Z, Coccia-Portugal M, Domont J, et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet. 2012; 379(9816):633-640.
28. Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, Hobday TJ, Okusaka T, Capdevila J, de Vries EG, Tomassetti P, Pavel ME, Hoosen S, Haas T, Lincy J, Lebwohl D, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011; 364(6):514-523.
29. Lodish MB and Stratakis CA. Endocrine tumours in neurofibromatosis type 1, tuberous sclerosis and related syndromes. Best Pract Res Clin Endocrinol Metab. 2010; 24(3):439-449.
30. Janku F, Hong DS, Fu S, Piha-Paul SA, Naing A, Falchook GS, Tsimberidou AM, Stepanek VM, Moulder SL, Lee JJ, Luthra R, Zinner RG, Broaddus RR, Wheler JJ and Kurzrock R. Assessing PIK3CA and PTEN in early-phase trials with PI3K/AKT/mTOR inhibitors. Cell Rep. 2014; 6(2):377-387.
31. Finn RS, Crown JP, Boer K, Lang I, Parikh RJ, Breazna A, Ho SN, Kim ST, Randolph S and Slamon DJ. Results of a Randomized Phase 2 Study of Pd 0332991, a Cyclin-Dependent Kinase (Cdk) 4/6 Inhibitor, in Combination with Letrozole Vs Letrozole Alone for First-Line Treatment of Er +/Her2-Advanced Breast Cancer (Bc). Annals of Oncology. 2012; 23:43-43.
32. Ho HK, Yeo AH, Kang TS and Chua BT. Current strategies for inhibiting FGFR activities in clinical applications: opportunities, challenges and toxicological considerations. Drug Discov Today. 2014; 19(1):51-62.
33. Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, Kalyana-Sundaram S, Wang R, Ning Y, Hodges L, Gursky A, Siddiqui J, Tomlins SA, Roychowdhury S, Pienta KJ, Kim SY, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013; 45(12):1446-1451.
34. Said R, Hong DS, Warneke CL, Lee JJ, Wheler JJ, Janku F, Naing A, Falchook GS, Fu S, Piha-Paul S, Tsimberidou AM and Kurzrock R. P53 mutations in advanced cancers: clinical characteristics, outcomes, and correlation between progression-free survival and bevacizumab-containing therapy. Oncotarget. 2013; 4(5):705-714.
35. Abdulghani J, Allen JE, Dicker DT, Liu YY, Goldenberg D, Smith CD, Humphreys R and El-Deiry WS. Sorafenib sensitizes solid tumors to Apo2L/TRAIL and Apo2L/TRAIL receptor agonist antibodies by the Jak2-Stat3-Mcl1 axis. PLoS One. 2013; 8(9):e75414.
36. Schroeder RD, Angelo LS and Kurzrock R. NF2/merlin in hereditary neurofibromatosis 2 versus cancer: biologic mechanisms and clinical associations. Oncotarget. 2014; 5(1):67-77.
37. Kurzrock R, Sherman SI, Ball DW, Forastiere AA, Cohen RB, Mehra R, Pfister DG, Cohen EE, Janisch L, Nauling F, Hong DS, Ng CS, Ye L, Gagel RF, Frye J, Muller T, et al. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol. 2011; 29(19):2660-2666.
38. Morandi A, Plaza-Menacho I and Isacke CM. RET in breast cancer: functional and therapeutic implications. Trends Mol Med. 2011; 17(3):149-157.
39. Tutt A, Robson M, Garber JE, Domchek S, Audeh MW, Weitzel JN, Friedlander M and Carmichael J. Phase II trial of the oral PARP inhibitor olaparib in BRCA-deficient advanced breast cancer. Journal of Clinical Oncology. 2009; 27(18).
40. Harry BL, Eckhardt SG and Jimeno A. JAK2 inhibition for the treatment of hematologic and solid malignancies. Expert Opin Investig Drugs. 2012; 21(5):637-655.
41. Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C, DeMarini DJ, Cox DS, Xu Y, Morris SR, Peddareddigari VG, Le NT, Hart L, Bendell JC, Eckhardt G, Kurzrock R, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 2012; 13(8):773-781.
42. Villarroel MC, Rajeshkumar NV, Garrido-Laguna I, De Jesus-Acosta A, Jones S, Maitra A, Hruban RH, Eshleman JR, Klein A, Laheru D, Donehower R and Hidalgo M. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol Cancer Ther. 2011; 10(1):3-8.
43. Cecchi F, Rabe DC and Bottaro DP. Targeting the HGF/Met signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012; 16(6):553-572.
44. Metzelder S, Wang Y, Wollmer E, Wanzel M, Teichler S, Chaturvedi A, Eilers M, Enghofer E, Neubauer A and Burchert A. Compassionate use of sorafenib in FLT3-ITD-positive acute myeloid leukemia: sustained regression before and after allogeneic stem cell transplantation. Blood. 2009; 113(26):6567-6571.
45. Baselga J, Campone M, Piccart M, Burris HA, 3rd, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, Beck JT, Ito Y, Yardley D, Deleu I, Perez A, Bachelot T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012; 366(6):520-529.