
INTRODUCTION
Endometrial carcinoma (EC) is the most common malignancy of the female genital tract [1–3]. Estrogen stimulation is an important pathogenic factor contributing to endometrial carcinoma [4, 5]. Estrogen receptors (ERα and ERβ) are ligand-dependent transcription factors that mediate the effects of estrogen via gene regulation [6]. In addition, ERα can initiate several non-genomic signaling events in an estrogen-independent manner via post-translational modifications, such as phosphorylation. The cross-talk between ERα and signal transduction pathways leads to a series of cellular effects, including adhesion, migration, survival, and proliferation, which play a key role in tumorigenesis [7, 8].
The p21-activated kinases (Paks) are a family of serine/threonine kinases comprising six isoforms (group I includingPak1-3, and group II including Pak4-6) in humans, based on their structure and function [9, 10]. Interestingly, Paks have been found to mediate tamoxifen resistance in breast cancer [11, 12]. Pak1 signaling promotes trans-activation of ERα in breast cancer cells by phosphorylating ERα at serine 305 in the absence of its ligand estrogen, resulting in tamoxifen resistance [11]. Pak4 binds to ERα and promotes its transcriptional activity by phosphorylating ERα-Ser 305, and ERα in turn binds to the Pak4 promoter and induces Pak4 transcription. The Pak4-ERα interaction decreases sensitivity to tamoxifen in MCF-7 human breast cancer cells [12]. However, tamoxifen use increases the risk of endometrial cancer due to its estrogenic effects on the endometrium [13, 14], suggesting different regulatory mechanisms of estrogen signaling in breast and endometrial cancer.
Pak4 is one of the major downstream kinases in oncogenic signaling [15, 16]. Pak4 is upregulated and activated by various stimuli [17, 18]. For example, it promotes prostate cancer cell migration in response to hepatocyte growth factor (HGF) [19]. In gestational trophoblastic disease, Pak4 is activated by human chorionic gonadotropin (hCG) via PI3K/PKB signaling [20]. We have previously demonstrated that the Pak4 expression increases with the progression of EC [21]. Furthermore, we have observed a nuclear localization of Pak4, especially the activated, phosphorylated Pak4 form (p-Pak4ser474) in endometrial cancer tissues [21], suggesting that Pak4 might activate ERα and contribute to estrogen-induced EC pathogenesis.
To explore this possibility, we have investigated the relationship between Pak4 and estrogen signaling in endometrial cancer. We tested the hypothesis that a positive feedback loop exists in which estrogen stimulates Pak4 expression and activation, which in turn promotes ERα trans-activation, and endometrial cancer cell proliferation. This feedback loop also involves PI3K/AKT signaling, cyclin D1, and cell cycle progression. These studies define a novel mechanism underlying estrogen signaling regulation, and suggest that Pak4 might be an important therapeutic target in endometrial cancer.
RESULTS
Estrogen up-regulates Pak4 expression and activation
ER-positive human Ishikawa and RL95-2 endometrial cancer cells, as well as estrogen-responsive breast cancer MCF-7 cells were treated with a low-dose E2 (10 nM). We observed that E2 treatment led to a time-dependent increase in both Pak4 mRNA and protein levels (Figure 1A and 1B). In Ishikawa cells, the Pak4 protein levels started to rise after 2 days, and gradually peaked in 6 days. Similar trends were also found in RL95-2 cells. As for MCF-7 breast cancer cells, the levels of Pak4 mRNA and protein increased after 3 days of E2 stimulation.
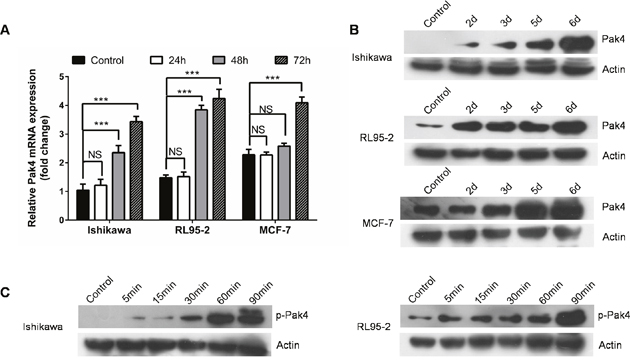
Figure 1: Estrogen increases Pak4 expression and activation. (A-B) E2 induces Pak4 mRNA and protein levels. Serum-starved Ishikawa, RL95-2 and MCF-7 cells were treated with 10 nM E2, and cells were harvested at the indicated time. (A) The levels of Pak4 mRNA were determined by qRT-PCR, using β-actin as an internal control. Values represent mean ± s.d. (n = 3). ***P<0.001 compared with control. (B) The protein levels of Pak4 were assessed by Western blot. (C) Ishikawa and RL95-2 cells were serum-starved for 24 h, and then treated with 10 nM E2 for indicated times. The levels of p-Pak4ser474 were measured by Western blot, using β-actin as a loading control. All experiments were carried out in triplicates.
Western blotting revealed a time-dependent increase in the levels of p-Pak4 Ser474 (the activated form) in Ishikawa and RL95-2 cells in the presence of E2. The level of p-Pak4 was increased after 5 min of E2 stimulation, and lasted for at least 90 min (Figure 1C), indicating that estrogen activates Pak4.
Estrogen activates Pak4 via PI3K/AKT signaling
We next investigated the estrogen downstream signaling involved in the Pak4 activation. We found that estrogen increased AKT phosphorylation within 15min in Ishikawa cells, and 5 min in RL95-2 cells, and lasted for at least 90 min (Figure 2A). In order to elucidate the role of PI3K/AKT in estrogen-induced Pak4 activation, we treated RL95-2 cells with LY 294002, a specific PI3K inhibitor, in the presence of estrogen. LY 294002 significantly blocked the E2 mediated AKT stimulation, and partially blocked Pak4 phosphorylation (Figure 2B and 2C), suggesting that PI3K/AKT signaling mediates the estrogen-induced Pak4 activation.
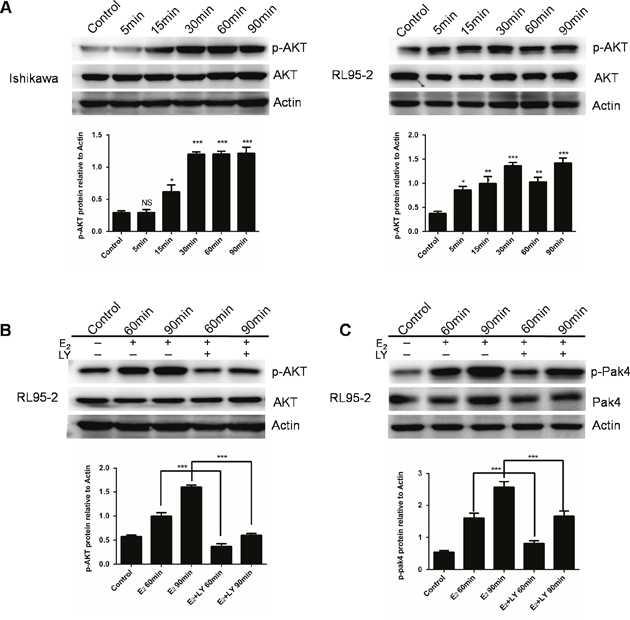
Figure 2: E2 activates Pak4 via PI3K/AKT pathway. (A) Ishikawa and RL95-2 cells were treated with 10 nM E2 for up to 90 min, and Western blot was used to detect p-AKT Ser473 and total AKT levels. (B-C) RL95-2 cells were treated with E2 for 60 or 90 min in the presence or absence of 20 μM LY 294002. Upper panel (B) p-AKT Ser473 and (C) p-Pak4ser474 levels were determined by western blotting, using β-actin as a loading control. Lower panel: Densitometric analysis of (B) p-AKT and (C) p-Pak4 in the immunoblots. Values represent mean ± s.d. (n = 3). *P< 0.05, ***P<0.001 compared with control, according to t-test. All experiments were carried out in triplicates.
Estrogen induces Pak4 and p-Pak4 nuclear levels in EC cells
We have previously observed mild nuclear and strong cytoplasmic Pak4 levels, and strong nuclear and moderate cytoplasmic p-Pak4 levels in EC tissues [21]. In this study, we further investigated the subcellular localization of Pak4 and p-Pak4 in EC cells by immunofluorescence staining. As shown in Figure 3A and 3B, Pak4 was found in the cytoplasm and in the nucleus, whereas mild cytoplasmic and strong nuclear p-Pak4 immunostaining was observed in human RL95-2 endometrial cancer cells. Moreover, we observed that estrogen treatment stimulated Pak4 and p-Pak4 nuclear accumulation (Figure 3A and 3B). Increased expression of Pak4 and p-Pak4 in cytoplasmic and nuclear fractions of RL95-2 in the presence of E2 was also confirmed by western analysis (Figure 3C).
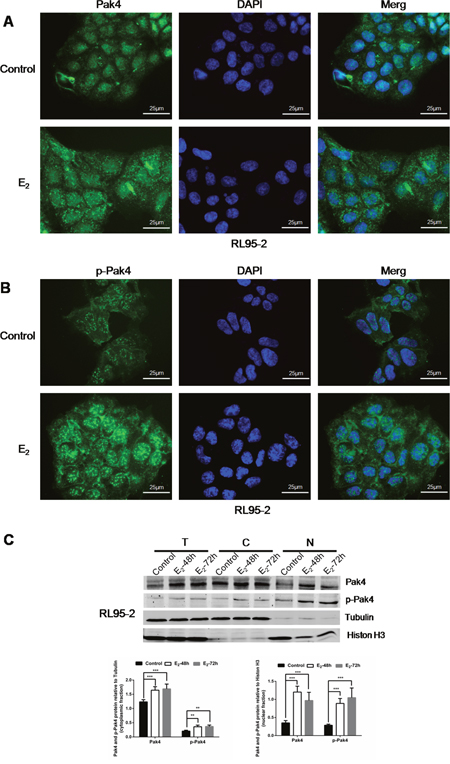
Figure 3: E2 promotes Pak4 and p-Pak4 nuclear accumulation. Immunofluorescence staining of (A) Pak4 and (B) p-Pak4 in RL95-2 cells treated with or without E2. Original magnification×400, bar=25μm. (C) Immunoblot analyses of Pak4 and p-Pak4 in subcellular protein fractions extracted from RL95-2 cells (T, total celllysate; C, cytoplasmic fraction; N, nuclear fraction). Cells were serum-starved for 24 h, and then treated with 10 nM E2, for indicated times.
Pak4 promotes ERα trans-activation
Following the nuclear accumulation of Pak4 and p-Pak4 after E2 stimulation, we then investigated the role of Pak4 in ERα trans-activation. Ishikawa cells that have relatively low Pak4 levels, were stably transfected with wild-type (wt) Pak4, constitutively active (ca) Pak4, or kinase-dead Pak4, whereas RL95-2 cells were stably transfected with two different shRNA constructs against human Pak4. The Pak4 mRNA and protein levels were substantially enhanced by wt Pak4 overexpression (Figure 4A) and reduced by Pak4 depletion (Figure 4B). Neither overexpression nor depletion of Pak4 affected the ERα mRNA levels (Figure 4A and 4B).
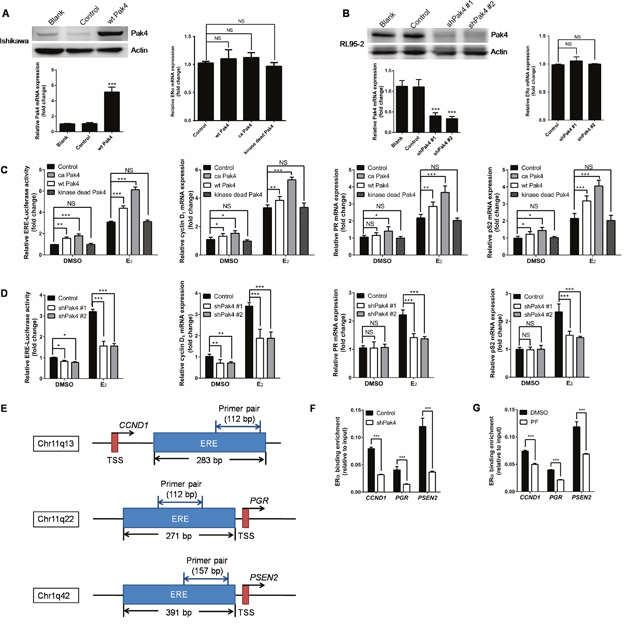
Figure 4: Pak4 enhances ERα transcription and ERα target gene expression. (A) Left: Protein and mRNA levels of Pak4 were measured in wt Pak4-overexpressing Ishikawa cells by Western blot and qRT-PCR analysis, respectively; Right: ERα mRNA levels detected by qRT-PCR. Values are the mean ± SD from at least three independent experiments. (B) Left: Western blot and qRT-PCR of Pak4 levels in two different shPak4-transfected RL95-2 cells. Right: ERα mRNA levels detected by qRT-PCR. Values are the mean ± SD from at least three independent experiments. (C) Ishikawa cells were stably transfected with wt Pak4, ca Pak4, kinase-dead Pak4, or the control vector. (D) RL95-2 cells were stably transfected with two different shPak4 or the control vector. The ERE-Luc reporter plasmids were transfected into Ishikawa and RL95-2 cells 24 h before E2 treatment, and luciferase assay was performed 48 h after E2 addition. The mRNA levels of ERα target genes were determined by qRT-PCR. Cells were treated with 10 nM E2 or vehicle for 48 h before RNA extraction. Values represent mean ± s.d. (n = 3), from three independent experiments. *P< 0.05, **P<0.01, ***P<0.001 compared with control, according to t-test. (E) Schematic representation of the estrogen response element and the primers used for ChIP–qPCR. ERE: estrogen response element. TSS: transcription start sites. (F-G) A summary of ChIP-qPCR results for ERα binding in RL95-2 cells with the primer pairs shown in (E). (F) shPak4 and control vector transfected RL95-2 cellswere treated with 10 nM E2 for 48 h before DNA extraction. (G) RL95-2 cells were treated with 10 nM E2 in the presence or absence of 1μM PF 3758309.
In order to elucidate the effect of abnormal Pak4 expression on ERα transcription, we performed an estrogen response element (ERE) luciferase assay. We found that wt Pak4 and ca Pak4 significantly increased ERE-dependent trans-activation of ERα in estrogen-treated Ishikawa cells (Figure 4C). Similarly, ca Pak4 also increased ERα target gene expression, including cyclin D1, PR, and pS2, both with and without E2 treatment. Wild-type Pak4 also induced mRNA expression of cyclin D1, PR, and pS2 in the presence of E2, but had no effect on PR expression in the absence of E2 (Figure 4C). Conversely, a stable knockdown of Pak4 decreased ERE luciferase activity in RL95-2 cells, both in the presence and absence of E2 (Figure 4D). Significantly reduced cyclin D1, PR, and pS2 mRNA levels were also observed in RL95-2 cells after shPak4 knockdown in the presence of E2. Interestingly, without E2 treatment, Pak4 inhibition only decreased cyclin D1 mRNA expression, but had no effect on PR and pS2 (Figure 4D).
To investigate whether Pak4 regulates the recruitment of ERα to ERE of target genes, we performed ChIP–qPCR using primers for ERE of CCND1, PGR, and PSEN2 genes (Figure 4E). Consistent with the results above, knockdown of Pak4 with shRNA (Figure 4F) or inhibition of Pak4 with Pak4-inhibitor PF 3758309 (Figure 4G) impaired the recruitment of ERα to ERE of the target genes.
Pak4 inhibition decreases E2-induced cell proliferation via alteration of G0/G1-phase cell cycle progression
We next investigated whether estrogen increases EC cell proliferation by activating Pak4. As shown in Figure 5A and 5C, E2 treatment promoted RL95-2 cell colony formation in soft agar, whereas depletion or functional inhibition of Pak4 (with Pak4 inhibitor PF 3758309) almost completely abrogated this effect. In addition, using MTT proliferation assay, we found that E2 induced RL95-2 cell proliferation, and that the E2-induced proliferation was reversed by shPak4 or PF 3758309 (Figure 5D). We next inhibited the estrogen pathway with a selective ERα inhibitor ICI 182,780, and then rescued the phenotype with overexpression of wt Pak4, ca Pak4, or kinase-dead Pak4. We observed that ICI 182,780 treatment inhibited the E2-induced colony formation, and overexpression of wt Pak4 or ca Pak4 partly rescued the number of colonies (Figure 5A and 5B). MTT proliferation assay provided similar results (Figure 5E).
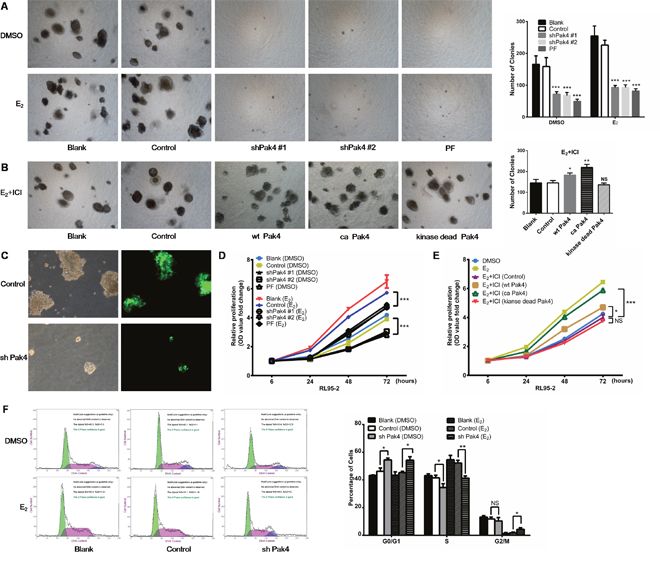
Figure 5: Pak4 inhibition suppresses E2-induced cell proliferation and cell cycle progression. (A) Soft agar colony assays of Pak4 knockdown, Pak4 inhibitor PF 3758309 treated RL95-2 cells and control cells. Cells were cultured in the medium with or without E2 for 2 weeks. (B) RL95-2 cells were transfected with wt Pak4, ca Pak4, kinase-dead Pak4, or the control vector. Cells were cultured in the medium with 10 nM E2 and 100 nM ICI 182,780 for 2 weeks. Representative images (left) were captured with an inverted phase contrast microscope (magnification, ×200). Columns (right), represent the number of colonies from three independent experiments, each in triplicates; values represent mean ± s.d.; *P< 0.05, **P<0.01, ***P<0.001. (C) RL95-2 cells were stably transfected with shPak4 or control vector with GFP. The fluorescence images showing the transfection efficiency, as well as the decreased size of colonies in Pak4 knockdown cells compared with control cells. Original magnification, ×400. (D) MTT assay of Pak4 knockdown, Pak4 inhibitor PF 3758309 treated RL95-2 cells and control cells. Cells were either treated with E2, vehicle or left untreated as indicated. (E) RL95-2 cells were transfected with wt Pak4, ca Pak4, kinase-dead Pak4, or the control vector. Cells were treated with 10 nM E2, 10 nM E2+ 100 nM ICI 182,780, or vehicle as indicated. All experiments were carried out in triplicates. (F) Cell-cycle profiles of shPak4 RL95-2 cells were assessed by FACS using DNA content profiles (left). Cells were either treated with E2, vehicle, or left untreated for 96 h before measurement. The percentages of cells in each compartment were calculated (right). Values represent mean ± s.d. (n = 3). *P< 0.05, **P<0.01 compared with control, according to t-test. All experiments were carried out in triplicates.
To assess the mechanism of Pak4-induced cellular proliferation, the cell-cycle profiles of RL95-2 cells were determined using propidium iodide staining and fluorescence-activated cell sorter analysis. Knockdown of Pak4 in RL95-2 cells resulted in accumulation of cells in G0/G1 phase and a decrease in S phase compared with controls, both in the presence and absence of E2 (Figure 5F).
Pak4 inhibition suppresses estrogen-induced tumor growth in nude mice
To investigate the role of Pak4 in vivo, RL95-2 cells with stably knocked-down Pak4 (shPak4) or control cells were injected subcutaneously into the flanks of nude mice. Both groups were administered subcutaneously 17β-estradiol 90-day-release pellets. The growth rate of tumors in mice inoculated with shPak4 RL95-2 cells was significantly lower than in tumors formed by control cells (P=0.0198; Figure 6A). The Pak4 suppression in shPak4 tumors was confirmed by immunohistochemistry staining (Figure 6B).
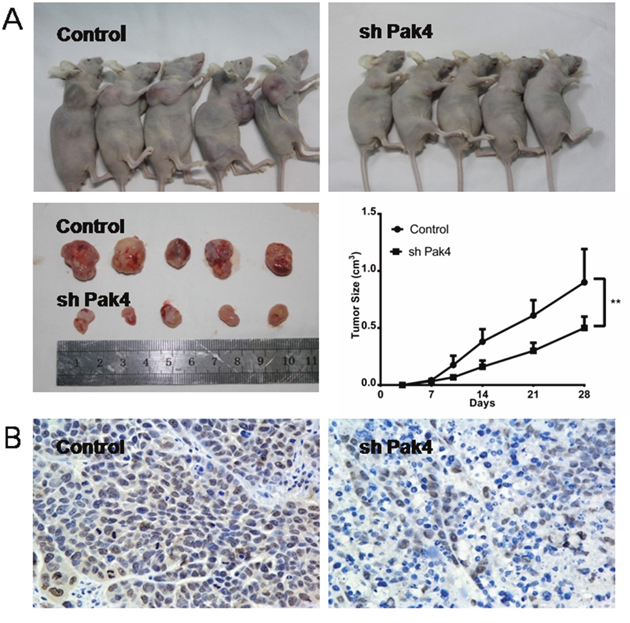
Figure 6: Pak4 depletion inhibits tumor growth in vivo. (A) Growth rates of tumors in nude mice inoculated with shPak4 RL95-2 cells or control cells. Values represent mean ± s.d. (n = 5). **P<0.01 compared with control, according to t-test. (B) Immunohistochemical staining of Pak4 in control and shPak4 tumors.
DISCUSSION
Cross-talk between ERα and signaling pathways contributes to the pathogenesis of endometrial carcinoma [22], but the underlying mechanisms are not clear. In the present study, we identified a positive feedback loop between Pak4 kinase and ERα signaling that promotes EC cell proliferation, and determined the role of PI3K/AKT signaling pathway and cyclin D1 in the process (Figure 7).
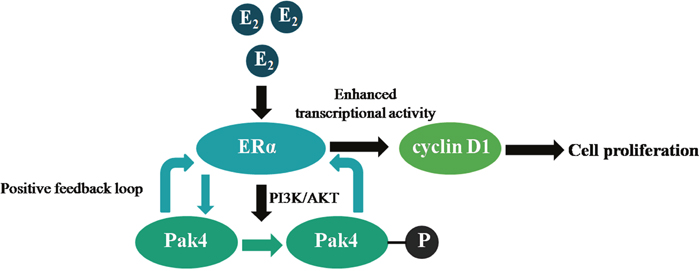
Figure 7: Illustration of a positive feedback loop between Pak4 and ERα signaling in endometrial cancer. Estrogen increases Pak4 expression and activation via PI3K/AKT pathway, the increased and activated Pak4 in turn enhances ERα transcriptional activity and cyclin D1 expression, which facilitates EC cell proliferation.
Pak4 is overexpressed and/or activated in various human cancer cells [16, 23, 24]. It is upregulated in cancers of breast, stomach, ovary, pancreas, and prostate [12, 25–30]. Although in our previous study, we observed overexpression and activation of Pak4 in estrogen-induced EC cells, it was not clear whether it was related to estrogen signaling [21]. In this study, we found that the expression and activation of Pak4 was increased in response to estrogen stimulation in EC cells, and that the activation occurred via PI3K/AKT pathway. In HGF signaling, Pak4 is activated through PI3K in MDCK cells [17]. However, LY294002, the inhibitor of PI3K, had no effect on HGF-induced Pak4 activation in prostate cancer cell lines [29]. In this study, we observed that LY294002 treatment partially blocked E2-induced Pak4 phosphorylation. These findings suggest that the relationship between Pak4 and PI3K/AKT pathway may be cell specific.
In the present study, E2 treatment also induced Pak4 phosphorylation, suggesting that in addition to the increased expression, the activation of Pak4 may play an important role in EC progression. In our previous study, we found a positive correlation between nuclear p-Pak4 expression and EC progression [21]. Here, we observed Pak4 and p-Pak4 nuclear accumulation after E2 stimulation. We also demonstrated that wild-type Pak4 increased ERα trans-activation in the presence or absence of estrogen. A more pronounced effect was observed when cells were transfected with constitutively active Pak4, suggesting that Pak4 may activate ERα in an estrogen-independent manner, and may contribute to the development of hormone independence. Therefore, the protein level and activation of Pak4 may potentially affect the effectiveness of anti-estrogen therapies; this is consistent with previous reports of Pak1 signaling-dependent activation of ER-S305 leading to enhanced S118 phosphorylation, and development of tamoxifen resistance in breast cancer [11]. Another study reported that GNE-2861, a small molecule selectively inhibiting group II PAKs, overcomes tamoxifen resistance in breast cancer [12]. Therefore, the emergence of Pak4 inhibitors [31, 32] suggests that targeting Pak4 expression or activity may represent a novel strategy to increase the response to hormonal treatment in endometrial cancer.
Furthermore, we demonstrated that depletion or functional inhibition of Pak4 abrogated cell proliferation induced by E2, whereas increased Pak4 expression and activation rescued the impaired cell proliferation caused by inhibiting the estrogen pathway. We also demonstrated that Pak4 knockdown decreased cyclin D1 expression with and without E2 treatment. Cyclin D1, a D-type cyclin regulating G1-phase cell-cycle progression, has been identified as a critical downstream effector of estrogen signaling and is frequently overexpressed in endometrial cancer [33–36]. Pak4 has been shown to control cell cycle progression by regulating the cyclin-dependent kinase inhibitor p21 [37]. Downregulation of Pak4 decreases proliferation and increases apoptosis and S phase arrest in Hep-2 cells via activation of the ATM/Chk1/2/p53 pathway [38]. Our results show that knockdown of Pak4 induces G1-S arrest, which is consistent with the effect of cyclin D1 downregulation [39].
In summary, our results demonstrate the presence of a positive feedback loop between Pak4 and ERα signaling in endometrial cancer. Estrogen stimulation leads to increased Pak4 expression, as well as hyper-activation via PI3K/AKT pathway, and the increased and activated Pak4 in turn enhances ERα trans-activation. This positive feedback loop promotes endometrial cancer cell proliferation by increasing cyclin D1 expression and altering cell cycle progression. The correlation between Pak4 and ERα signaling not only reveals an underlying mechanism of estrogen-related tumor progression, but also provides a rationale for multi-targeted therapies against endometrial cancer.
MATERIALS AND METHODS
Cell culture
Ishikawa, RL95-2, and MCF-7 cells were purchased from American Type Culture Collection (Manassas, VA). Cells were maintained in DMEM/F12 (Gibco, Auckland, NZ) supplemented with 10% FBS (Gibco, Carlsbad, CA) in a 37°C, 5% CO2 incubator.
Plasmids and transfection
The Pak4 mutants including ca Pak4 (E474) and kinase-dead Pak4 (M350) were generated by site-specific mutagenesis from the Pak4 wild-type using the Quick Change Site Directed Mutagenesis Kit (Stratagene, LaJolla, CA, USA). The ca Pak4 mutant was generated by mutating Ser474 to glutamic acid. The resulting Pak4 (E474) mutant showed enhanced auto-phosphorylation activity [40]. The kinase-dead Pak4 (M350) mutant carries a mutation in which the conserved lysine in subdomain II is converted to a non-phosphorylated methionine, resulting in a completely inactive kinase [40].
To stably express Pak4 in Ishikawa cells, wt Pak4, ca Pak4, kinase-dead Pak4, or the control vector pEGFP-N1 (Clontech Laboratories, Palo Alto, CA) were transfected into cells at 70% confluency in 12-well culture plates, using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) and then selected with G418 (800 μg/mL, SIGMA Chemical, St Louis, MO, USA). To stably silence Pak4 in RL95-2 cells, cells were transfected with two different shRNA constructs against human Pak4 or the control vector psiHIV-U6 (GeneCopoeia, Germantown, MD), and then selected with puromycin (0.5μg/mL, SIGMA Chemical, St Louis, MO, USA).
Estrogen treatment, PI3K, Pak4 and ERα inhibitor treatment
For western blot analysis, cells were seeded in 6-well plates at 70% confluency and cultured for 24 h in serum and phenolred-free medium, then treated with 10 nM 17β-estradiol (SIGMA Chemical, St Louis, MO, USA) or vehicle (DMSO, 0.1%) for the indicated times. For long-term treatment, cell were seeded in 6-well plates, at 70% confluency at different time points, cultured in a medium containing 10% charcoal-stripped FBS (HyClone, Logan, UT). Cells were plated for 24 h before treatment with the PI3K inhibitor LY 294002 (20 μM, SIGMA Chemical, St Louis, MO, USA), Pak4 inhibitor PF3758309 (1μM, Selleck, Shanghai, CHINA), ERα inhibitor ICI 182,780, (100 nM, SIGMA Chemical, St Louis, MO, USA), with DMSO as control.
Biochemical fractionation
Cytoplasmic and nuclear extracts from RL95-2 cells were isolated using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce; Thermo Scientific, Rockford, IL, USA). Primary antibodies were the following: rabbit anti-Pak4 (1:1000, Abcam, Cambridge, UK), rabbit anti-p-Pak4 Ser474 (1:1000, Cell Signaling Technology), mouse anti-β-actin (1:2000, ProteinTech Group, Chicago, IL), rabbit anti-tubulin (1:1000, Cell Signaling Technology), and rabbit anti-histone H3 (1:1000, Cell Signaling Technology).
Luciferase reporter assays
The reporter gene ERE-Luc was constructed using the enhanced luciferase reporter gene pGL3-promotor. Three tandem repeats of the consensus ERE oligo (GGTCACTGTGACC) were inserted into the Mlu I-Bgl II site of the multiple cloning site of pGL3-promotor, upstream of SV40 promoter. The ERE-Luc reporter and phRL/CMV (Renilla luciferase) plasmids were co-transfected into Pak4 overexpressing Ishikawa cells and Pak4 knockdown RL95-2 cells. After 24 h, cells were treated with or without E2. Luciferase assay was performed using Dual-Luciferase Reporter Assay System (E1531, Promega, Mannheim, Germany) after 48 h treatment with E2.
Chromatin immunoprecipitation (ChIP)–PCR
The ChIP assay was performed in RL95-2 cells using the Pierce Agarose ChIP kit (Pierce; Thermo Scientific, Rockford, IL, USA) following the manufacturer’s instructions. The antibody used in ChIP analysis was anti-ERα (1:50, Epigentek Group Inc, Brooklyn, NY, USA). Specific primers for qRT-PCR were: CCND1: 5’-CGCTTCCCAGCACCAACA-3’, 5’-CAAAGAGGCAGGCACCAC-3’; PSEN2: 5’-ATGT GAGAACAACCGGGAGGA-3’, 5’-TCGGAACTAAGC GACGACCTT-3’; PGR: 5’-GAGAAAGTGGGTGTTG AATGTG-3’, 5’-TGACGACAGGATGGAGGC-3’. Enrichment was calculated using the comparative Ctmethod. IgG was used as a negative control.
Soft agar colony assay
Cells were seeded on 0.3% top agar in a growth medium over a layer of 0.6% agar in a 6-well plate, at a density of 1×104 cells/mL (100 μL/well). Growth medium with or without E2 or E2+ICI 182,780 or PF 3758309 was added to the wells every 3 to 4 days. After 2 weeks of incubation, colonies of more than 50 cells were produced. Colonies were photographed and counted with an inverted microscope.
RNA extraction and qRT-PCR
Total RNA was extracted from cell lines using TRIzol reagent (Invitrogen, Life Technologies; Shanghai, PR China). cDNA was reverse-transcribed from total RNA using Prime Script RT reagent Kit (Takara, Dalian, PR China). Real-time PCR was performed using SYBR Premix Ex Taq (Takara, Dalian, PR China) and analyzed with an ABI Prism 7000 Sequence Detection System. The oligonucleotide primers used were: Pak4: 5’-ATGTGGTGGAGATGTACAACAGCTA-3’, 5’-GTTCATCC TGGTGTGGGTGAC-3’. The primers for ERα, cyclin D1, PR, and pS2 were described previously [41]. Gel electrophoresis was used to confirm PCR purity. All data were obtained in triplicates in three independent experiments.
Western blot
Cells were lysed using ProteoJET Mammalian Cell Lysis Reagent (MBI Fermentas, ON, Canada) with a protease inhibitor cocktail (Roche Diagnostics, Basel, Switzerland). Total protein concentration was estimated using the BCA method (Pierce, Rockford, IL, USA). A total of 60 μg of protein was separated on an 8% sodium dodecyl sulfate–polyacrylamide gel and transferred to a polyvinylidene fluoride (PVDF) membrane. Membranes were incubated with primary antibodies. Signal was detected using the BeyoECL Plus (Beyotime, Shanghai, PR China).
Primary antibodies were: rabbit anti-Pak4 (1:1000, Abcam, Cambridge, UK), rabbit anti-p-Pak4 Ser474 (1:1000, Cell Signaling Technology), mouse anti-β-actin (1:2000, ProteinTech Group, Chicago, IL), rabbit anti-AKT1 (1:1000, Epitomics, Burlingame, CA, USA), and rabbit anti-p-AKT Ser473 (1:1000, Epitomics, Burlingame, CA, USA).
Cell cycle analysis
Cells were fixed in 70% ice-cold ethanol. The cells were stained with 25μg/mL propidium iodide (KeyGen Biotech, Shanghai, PR China) in fluorescence-activated cell sorting buffer (PBS containing 0.1% bovine serum albumin, 0.05% of Triton X-100, and 50 μg/mL of RNaseA). After incubation for 30 min at room temperature, the cells were analyzed by flow cytometry (Becton Dickinson FACScan). Tests were performed in triplicates.
Proliferation assay
Cells (2 ×103cells/well) were seeded in 96-well culture plates in a growth medium. Cell viability was measured every 24 h by MTT assay following the manufacturer’s instructions (Beyotime, Shanghai, PR China). Medium was changed every other day. Each experiment was repeated in triplicates.
Cell immunofluorescence
Cells treated with or without E2 were seeded on coverslips and cultured in DMEM/10% FBS overnight. Cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 and blocked with goat serum before incubation with antibodies against Pak4 (1:100, Abcam), or p-Pak4 Ser474 (1:100, Cell Signaling Technology) at 4°C overnight. The cells were incubated with Alexa 594-labeled secondary antibodies (1:200; Invitrogen, Burlington, ON, Canada), and counterstained with 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI; Beyotime) before analysis using a microscope (Leica TCS SP8). The control slides were treated with PBS instead of the primary antibody.
Studies in vivo
The 5 × 106 shPak4 or vector-transfected RL95-2 cells were inoculated s.c. (five mice per group) into four-week-old BALB/c female nude mice. Estrogen was administered to the animals subcutaneously as 17-beta-estradiol 90-day-release pellets (0.72 mg/pellet; IRA, Toledo, OH) as described previously [42]. The diameters of s.c. tumors were measured perpendicularly weekly, and volumes were calculated using the following standard formula: tumor volumes (cm3) = (the longest diameter) × (the shortest diameter)2× 0.5. Mice were sacrificed at 28 days post-injection. Tumors were excised and measured. All experimental protocols were approved by the Ethics Committee for Animal Experimentation at Tongji University.
Statistical analysis
All statistical analyses were performed using SPSS 16.0 (Microsoft, Redmond, WA, USA) or Prism (GraphPad, San Diego, CA, USA). Each experiment was performed as least three times, and data were expressed as the means ± SD where applicable. A P value < 0.05 was considered to be significant.
Author contributions
WL and XPW conceived and designed the experiments. WL, TS, JJQ, KW and DZ performed the experiments. WL, YPZ and LY analyzed the data. WL wrote the manuscript. All authors read and approved thefinal manuscript.
ACKNOWLEDGMENTS
We thank Professor Zhi-Yong Mao for kindly providing experimental facilities in Tongji University.
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
GRANT SUPPORT
The study was supported by the National Natural Science Foundation of China (No. 81402144, No. 81472427, No. 81672574, and No. 81471436), the Promising Youth Training Plan of Tongji University (No.1400804), the Promising Young Doctors Training Plan of Shanghai, and The leading edge technology joint research projects of Shanghai Shenkang hospital development center (No. SHDC12015110).
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017; 67: 7-30.
2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65: 87-108.
3. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016; 66: 115-32.
4. Wright JD, Barrena Medel NI, Sehouli J, Fujiwara K, Herzog TJ. Contemporary management of endometrial cancer. Lancet. 2012; 379: 1352-60.
5. Morice P, Leary A, Creutzberg C, Abu-Rustum N, Darai E. Endometrial cancer. Lancet. 2016; 387: 1094-108.
6. Edwards DP. Regulation of signal transduction pathways by estrogen and progesterone. Annu Rev Physiol. 2005; 67: 335-76.
7. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005; 307: 1625-30.
8. Kavarthapu R, Tsai Morris CH, Dufau ML. Prolactin induces up-regulation of its cognate receptor in breast cancer cells via transcriptional activation of its generic promoter by cross-talk between ERalpha and STAT5. Oncotarget. 2014; 5: 9079-91. doi: 10.18632/oncotarget.2376.
9. Eswaran J, Soundararajan M, Kumar R, Knapp S. UnPAKing the class differences among p21-activated kinases. Trends Biochem Sci. 2008; 33: 394-403.
10. Ha BH, Morse EM, Turk BE, Boggon TJ. Signaling, regulation, and specificity of the type II p21-activated kinases. J Biol Chem. 2015; 290: 12975-83.
11. Rayala SK, Talukder AH, Balasenthil S, Tharakan R, Barnes CJ, Wang RA, Aldaz CM, Khan S, Kumar R. P21-activated kinase 1 regulation of estrogen receptor-alpha activation involves serine 305 activation linked with serine 118 phosphorylation. Cancer Res. 2006; 66: 1694-701.
12. Zhuang T, Zhu J, Li Z, Lorent J, Zhao C, Dahlman-Wright K, Stromblad S. p21-activated kinase group II small compound inhibitor GNE-2861 perturbs estrogen receptor alpha signaling and restores tamoxifen-sensitivity in breast cancer cells. Oncotarget. 2015; 6: 43853-68. doi: 10.18632/oncotarget.6081.
13. van Leeuwen FE, Benraadt J, Coebergh JW, Kiemeney LA, Gimbrere CH, Otter R, Schouten LJ, Damhuis RA, Bontenbal M, Diepenhorst FW, van Tinteren H, Coebergh JW, Kiemeney LA, et al. Risk of endometrial cancer after tamoxifen treatment of breast cancer. Lancet. 1994; 343: 448-52.
14. Droog M, Nevedomskaya E, Kim Y, Severson T, Flach KD, Opdam M, Schuurman K, Gradowska P, Hauptmann M, Dackus G, Hollema H, Mourits M, Nederlof P, et al. Comparative cistromics reveals genomic cross-talk between FOXA1 and ERalpha in tamoxifen-associated endometrial carcinomas. Cancer Res. 2016; 76: 3773-84.
15. Radu M, Semenova G, Kosoff R, Chernoff J. PAK signalling during the development and progression of cancer. Nat Rev Cancer. 2014; 14: 13-25.
16. Fulciniti M, Martinez-Lopez J, Senapedis W, Oliva S, Bandi RL, Amodio N, Xu Y, Szalat RL, Gulla A, Samur MK, Roccaro A, Linares M, Cea M, et al. Functional role and therapeutic targeting of p21-associated kinase 4 (PAK4) in multiple myeloma. Blood. 2017; 129:2233-45.
17. Wells CM, Abo A, Ridley AJ. PAK4 is activated via PI3K in HGF-stimulated epithelial cells. J Cell Sci. 2002; 115: 3947-56.
18. Cammarano MS, Nekrasova T, Noel B, Minden A. Pak4 induces premature senescence via a pathway requiring p16INK4/p19ARF and mitogen-activated protein kinase signaling. Mol Cell Biol. 2005; 25: 9532-42.
19. Ahmed T, Shea K, Masters JR, Jones GE, Wells CM. A PAK4-LIMK1 pathway drives prostate cancer cell migration downstream of HGF. Cell Signal. 2008; 20: 1320-8.
20. Zhang HJ, Siu MK, Yeung MC, Jiang LL, Mak VC, Ngan HY, Wong OG, Zhang HQ, Cheung AN. Overexpressed PAK4 promotes proliferation, migration and invasion of choriocarcinoma. Carcinogenesis. 2011; 32: 765-71.
21. Lu W, Xia YH, Qu JJ, He YY, Li BL, Lu C, Luo X, Wan XP. p21-activated kinase 4 regulation of endometrial cancer cell migration and invasion involves the ERK1/2 pathway mediated MMP-2 secretion. Neoplasma. 2013; 60: 493-503.
22. Hecht JL, Mutter GL. Molecular and pathologic aspects of endometrial carcinogenesis. J Clin Oncol. 2006; 24: 4783-91.
23. Callow MG, Clairvoyant F, Zhu S, Schryver B, Whyte DB, Bischoff JR, Jallal B, Smeal T. Requirement for PAK4 in the anchorage-independent growth of human cancer cell lines. J Biol Chem. 2002; 277: 550-8.
24. Kim H, Woo DJ, Kim SY, Yang EG. p21-activated kinase 4 regulates HIF-1α translation in cancer cells. Biochem Biophys Res Commun. 2017; 486:270-76.
25. Dart AE, Box GM, Court W, Gale ME, Brown JP, Pinder SE, Eccles SA, Wells CM. PAK4 promotes kinase-independent stabilization of RhoU to modulate cell adhesion. J Cell Biol. 2015; 211: 863-79.
26. Guo Q, Su N, Zhang J, Li X, Miao Z, Wang G, Cheng M, Xu H, Cao L, Li F. PAK4 kinase-mediated SCG10 phosphorylation involved in gastric cancer metastasis. Oncogene. 2014; 33: 3277-87.
27. Siu MK, Chan HY, Kong DS, Wong ES, Wong OG, Ngan HY, Tam KF, Zhang H, Li Z, Chan QK, Tsao SW, Stromblad S, Cheung AN. p21-activated kinase 4 regulates ovarian cancer cell proliferation, migration, and invasion and contributes to poor prognosis in patients. Proc Natl Acad Sci U S A. 2010; 107: 18622-7.
28. Tyagi N, Marimuthu S, Bhardwaj A, Deshmukh SK, Srivastava SK, Singh AP, McClellan S, Carter JE, Singh S. p-21 activated kinase 4 (PAK4) maintains stem cell-like phenotypes in pancreatic cancer cells through activation of STAT3 signaling. Cancer Lett. 2016; 370: 260-7.
29. Park MH, Lee HS, Lee CS, You ST, Kim DJ, Park BH, Kang MJ, Heo WD, Shin EY, Schwartz MA, Kim EG. p21-Activated kinase 4 promotes prostate cancer progression through CREB. Oncogene. 2013; 32: 2475-82.
30. He LF, Xu HW, Chen M, Xian ZR, Wen XF, Chen MN, Du CW, Huang WH, Wu JD, Zhang GJ. Activated-PAK4 predicts worse prognosis in breast cancer and promotes tumorigenesis through activation of PI3K/AKT signaling. Oncotarget. 2017; 8:17573-85. doi: 10.18632/oncotarget.7466.
31. Baskaran Y, Ang KC, Anekal PV, Chan WL, Grimes JM, Manser E, Robinson RC. An in cellulo-derived structure of PAK4 in complex with its inhibitor Inka1. Nat Commun. 2015; 6: 8681.
32. Murray BW, Guo C, Piraino J, Westwick JK, Zhang C, Lamerdin J, Dagostino E, Knighton D, Loi CM, Zager M, Kraynov E, Popoff I, Christensen JG, et al. Small-molecule p21-activated kinase inhibitor PF-3758309 is a potent inhibitor of oncogenic signaling and tumor growth. Proc Natl Acad Sci U S A. 2010; 107: 9446-51.
33. Prall OW, Sarcevic B, Musgrove EA, Watts CK, Sutherland RL. Estrogen-induced activation of Cdk4 and Cdk2 during G1-S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2. J Biol Chem. 1997; 272: 10882-94.
34. Cao QJ, Einstein MH, Anderson PS, Runowicz CD, Balan R, Jones JG. Expression of COX-2, Ki-67, cyclin D1, and P21 in endometrial endometrioid carcinomas. Int J Gynecol Pathol. 2002; 21: 147-54.
35. Moreno-Bueno G, Rodriguez-Perales S, Sanchez-Estevez C, Marcos R, Hardisson D, Cigudosa JC, Palacios J. Molecular alterations associated with cyclin D1 overexpression in endometrial cancer. Int J Cancer. 2004; 110: 194-200.
36. Wu W, Slomovitz BM, Soliman PT, Schmeler KM, Celestino J, Milam MR, Lu KH. Correlation of cyclin D1 and cyclin D3 overexpression with the loss of PTEN expression in endometrial carcinoma. Int J Gynecol Cancer. 2006; 16: 1668-72.
37. Nekrasova T, Minden A. PAK4 is required for regulation of the cell-cycle regulatory protein p21, and for control of cell-cycle progression. J Cell Biochem. 2011; 112: 1795-806.
38. Sun X, Liu B, Wang J, Li J, Ji WY. Inhibition of p21-activated kinase 4 expression suppresses the proliferation of Hep-2 laryngeal carcinoma cells via activation of the ATM/Chk1/2/p53 pathway. Int J Oncol. 2013; 42: 683-9.
39. Carlson B, Lahusen T, Singh S, Loaiza-Perez A, Worland PJ, Pestell R, Albanese C, Sausville EA, Senderowicz AM. Down-regulation of cyclin D1 by transcriptional repression in MCF-7 human breast carcinoma cells induced by flavopiridol. Cancer Res. 1999; 59: 4634-41.
40. Abo A, Qu J, Cammarano MS, Dan C, Fritsch A, Baud V, Belisle B, Minden A. PAK4, a novel effector for Cdc42Hs, is implicated in the reorganization of the actin cytoskeleton and in the formation of filopodia. EMBO J. 1998; 17: 6527-40.
41. Chen X, Yan Q, Li S, Zhou L, Yang H, Yang Y, Liu X, Wan X. Expression of the tumor suppressor miR-206 is associated with cellular proliferative inhibition and impairs invasion in ERalpha-positive endometrioid adenocarcinoma. Cancer Lett. 2012; 314:41-53.
42. Che Q, Liu BY, Liao Y, Zhang HJ, Yang TT, He YY, Xia YH, Lu W, He XY, Chen Z, Wang FY, Wan XP. Activation of a positive feedback loop involving IL-6 and aromatase promotes intratumoral 17beta-estradiol biosynthesis in endometrial carcinoma microenvironment. Int J Cancer. 2014; 135: 282-94.