
INTRODUCTION
Adenosine triphosphate (ATP) is well known to provide the intracellular energy currency of cells, but is also present at low levels in the extracellular space under physiological conditions. Higher levels of extracellular ATP are perceived as a danger signal by cells in pathological processes [1]. The presence of high levels of extracellular ATP has been noted in the tumor microenvironment [2, 3]. Such heightened pericellular ATP levels have been demonstrated to increase tumor cell death through multiple mechanisms inclusive of: augmentation of antitumor immunity, direct induction of tumor cell death, and blockade of tumor angiogenesis [4, 5].
Although antitumor actions of extracellular ATP may occur through activation of several type 2-purinergic receptors (P2R), P2X7 is thus far the most important ATP-receptor [1, 5]. P2X7 is the only P2X receptor capable to form a membrane pore permeable to large molecules (up to 900 Da), resulting in tumor cell apoptosis [6].
Low levels of extracellular ATP that develop as a consequence of scavenging, or defective P2X7 activation per se, have been linked to immunosuppressive responses. These provoke cancer development by allowing tumor cells to escape from the P2X7 controlled pro-apoptotic mechanisms [7, 8]. Curiously, basal or low level activation of P2X7 receptor promotes tumor cell growth [9]. A therapeutic modality that increases P2X7 tumoricidal functions, thereby preventing cancer cell escape, would be an important advance.
We have recently highlighted the modulation of P2X7 receptor signaling in induction of cancer cell death and proposed the use of purine-related drugs as adjunctive agents in cancer therapy [4]. We have demonstrated that pulsed treatment of MCA38 colon cancer cells with high levels of ATP was able to induce cancer cell death, exclusively via P2X7 [4]. We have further delineated PI3K/AKT and AMPK-PRAS40-mTOR as the downstream pathways initiated by ATP-P2X7 signals. These two signaling axes act synergistically to elicit maximal levels of tumor cell death by disrupting homeostatic cell growth and the process of autophagy.
More recently, we have also shown that high levels of extracellular ATP promote human cervical cancer cell death through a joint mechanism linked by both P2X7 activation and associated adenosine formation [10]. Cells expressing high levels of P2X7 are susceptible to ATP-P2X7 cytotoxicity, whereas the death of cells with low P2X7 levels appeared to be caused by adenosine uptake and subsequent AMPK phosphorylation, dATP accumulation, p53 activation, as well as autophagy induction [10].
Interestingly, pore-forming activity of P2X7 has been linked to remodeling of membrane lipid rafts viz. the rate of agonist-evoked pore formation is enhanced by cholesterol depletion using methyl-β-cyclodextrin (MCD) whereas is inhibited by cholesterol loading [11]. Moreover, lipid raft remodeling can be induced by heat stress, non-proteotoxic membrane lipid-interacting chemicals, such as Bimoclomol, or the membrane fluidizer benzyl alcohol (BA) [12, 13]. These so-called “membrane-lipid therapy” promote changes in the nanostructure and physicochemical state of plasma membranes by interfering with protein and lipid interactions, thereby altering cell transmembrane signals [13]. However, whether such membrane-lipid therapies impact P2X7 functionality, and to what extent this is feasible, remain elusive.
Here, we demonstrate that mild heat stress (or hyperthermia; 40°C) augments ATP agonist efficacy thereby stimulating P2X7 pore opening and dilatation. Ultimately, this leads to dramatically enhanced MCA38 colon cancer cell death, as mediated through AKT/PRAS40/mTOR signaling. Hyperthermia-induced increases in membrane fluidity facilitate P2X7 pore formation and promote cell death. Furthermore, we have also shown that cisplatin or mitomycin C in combination with hyperthermia and extracellular ATP is more effective in inducing cancer cell death. Our results suggest the potential utility of P2X7 hyperactivation by hyperthermia as an adjunct therapy in the treatment of cancer.
RESULTS
Hyperthermia increases ATP tumoricidal activity via P2X7-AKT/PRAS40/mTOR signaling
We have previously shown that, at 37°C, 1 mM ATP will rapidly cause cytotoxicity in MCA38 colon cancer cells at 30 min [4]. Herein, we have investigated whether hyperthermia would sensitize cancer cells to ATP cytotoxicity. We first titrated growth responses of wild type (WT) MCA38 cells to increased ATP levels (ranging from 1, 2.5 and 5 mM) for 15 min at 37°C, 40°C or 42°C. As shown in Supplementary Figure 1, 42°C treatment was capable to promote cancer cell death per se. ATP cytotoxicity was selectively and dramatically enhanced by mild heat stress (hyperthermia; 40°C) in a dose-dependent manner (Figure 1A). In contrast, at 37°C, cytotoxicity was only noted with much higher concentration of ATP (5 mM). Therefore, 1 mM of ATP for 15 min and the temperature of 40°C were chosen for the hyperthermia experimentation, unless otherwise stated. We observed that elevation of cytotoxicity induced by combination of ATP and hyperthermia was associated with increased cell death (Figure 1B; upper panel) and cell shrinkage (Figure 1B; middle panel). We also found that ATP/hyperthermia-evoked cell death has both apoptotic and necrotic characteristics (Figure 1B; bottom panel). Moreover, such antitumor activity was near completely counteracted by co-treatment with APT102, a soluble ectonucleotidase, when cells were exposed to ATP (1 mM) for 30 min, either at 37°C or 40°C (Figure 1C), suggesting the exclusive involvement of ATP in antitumor actions under these experimental settings.
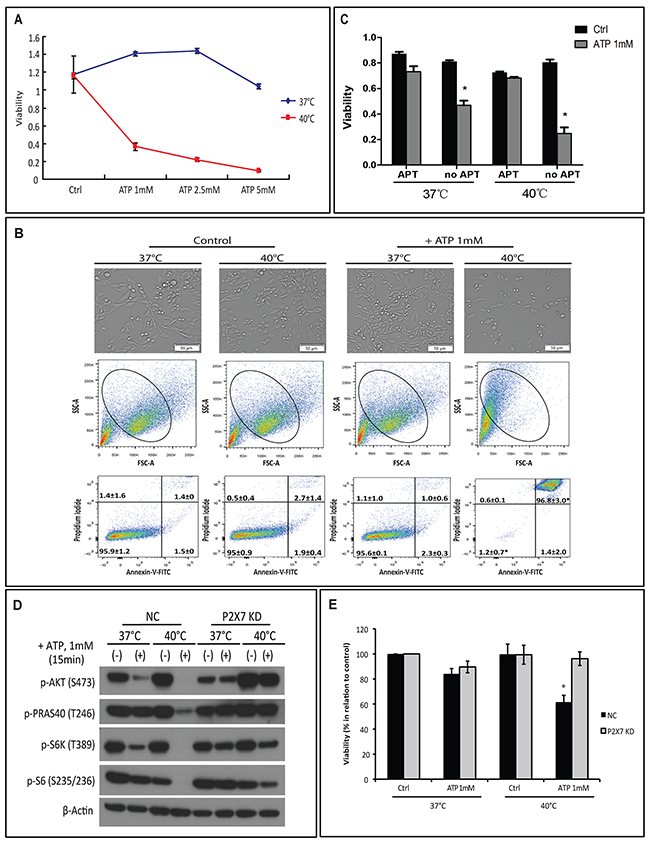
Figure 1: Hyperthermia potentiates ATP cytotoxicity in a P2X7-dependent manner. (A-B) MCA38 wild type (WT) cells were treated with exogenous ATP at indicated concentrations and temperatures for 15 min. Cells treated with media served as control (Ctrl). Cell viability was determined using CCK-8 (A) and images of live cells were captured by Celigo (upper) and cell apoptosis/necrosis was evaluated by FACS (middle and bottom) (B). (C) Cells were left untreated or pre-treated with 2.5 μg/ml of APT102 (soluble ectonucleotidase CD39) for 30 min before exposed to ATP 1 mM for 30 min at indicated temperatures. Cell viability was measured 24 hr later. (D-E) MCA38 P2X7 KD (P2X7-deficient) and NC (negative control) cells were treated with ATP (1 mM for 15 min) at 37°C or 40°C. AKT/PRAS40/mTOR signaling pathway was analyzed by Western blotting (D) and cell viability was evaluated after 24 hr (E). *p < 0.05 as compared to control (one-way ANOVA, followed by Tukey pos-test). Bars, 50 μM.
We established previously that ATP induces MCA38 cancer cell death through two P2X7-mediated intracellular signaling networks: PI3K/AKT and AMPK-PRAS40-mTOR [4]. We next examined whether there was an exclusive link between hyperthermia and P2X7 signaling by employing the P2X7-deficient MCA38 cell line (P2X7 KD) [4].
We first confirmed resistance of P2X7 KD cells to cytotoxicity induced by ATP and hyperthermia. In the negative control cell line (NC), as linked to cell death, we noted additive effects of hyperthermia on the two ATP-P2X7 signaling axes: 40°C treatment decreased phosphorylation of both AKT and mTOR pathway components including PRAS40, S6K and S6 (Figure 1D and 1E). These effects were not seen in the P2X7 KD cell line (Figure 1D and 1E).
Hyperthermia facilitates ATP cytotoxicity by increasing P2X7 functionality
Next, we evaluated the impact of hyperthermia on P2X7 functionality including pore formation capacity, Ca2+ influx, and associations with pannexin or connexin channels. First, ethidium bromide uptake assay was performed to measure the membrane pore-opening activity of P2X7 in a non-selective manner. We observed that ATP markedly stimulates ethidium bromide uptake when cells were incubated at 40°C (Figure 2A).
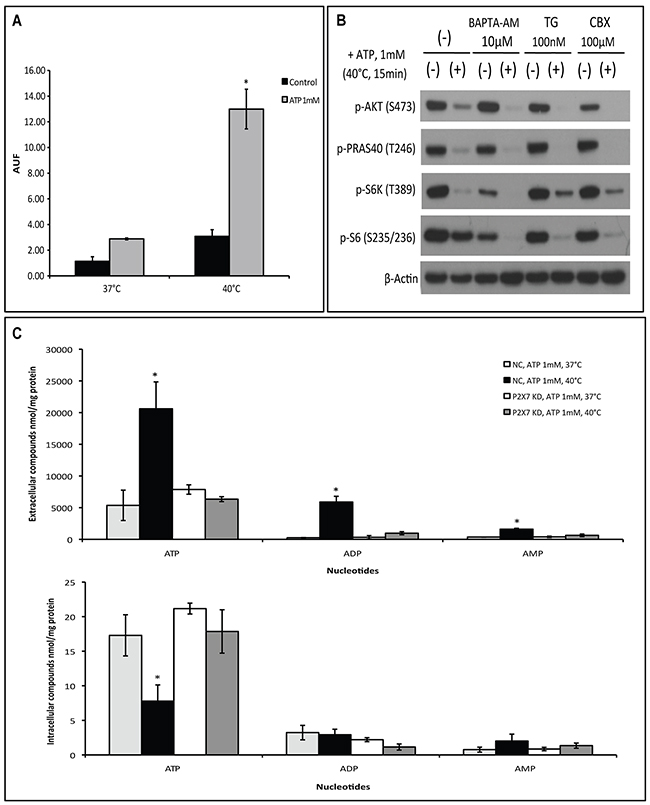
Figure 2: Hyperthermia increases ATP-tumor killing activity by enhancing P2X7 pore formation independently of Ca2+ influx and pannexin/connexin interaction. (A) P2X7 functionality upon ATP/hyperthermia treatment measured by etidium bromide (EtBr) uptake. Cells were left untreated or treated with ATP (1 mM) for 15 min at 37°C or 40°C, followed by whole cell fluorescence measurement (AUF), as described in Material and Methods. (B) Cells were pre-incubated with BAPTA-AM, thapsigargin (TG) or Carbenoxolone (CBX) prior to heat-ATP pulse treatment, followed by western blot analysis of AKT/PRAS40/mTOR signaling pathway. Cells treated with media served as control. (C) NC (negative control) and P2X7 KD (P2X7-deficient) cells were exposed to ATP for 15 min at 37°C or 40°C and extracellular and intracellular adenine nucleotide levels were determined by HPLC. *p < 0.05 in contrast to control (one-way ANOVA, followed by Tukey pos-test, mean ± SD).
Second, two chemical compounds that differently alter cytosolic Ca2+ levels: BAPTA-AM, a selective cell permeable calcium chelator that depletes Ca2+ intracellular stores, and thapsigargin (TG), a potent inhibitor of endoplasmic reticulum Ca2+-ATPase causing an immediate increase in cytoplasmic Ca2+ levels, were employed to examine changes in the ATP-P2X7 intracellular signaling pathway upon hyperthermia treatment. As shown in Figure 2B, neither BAPTA-AM nor TG was able to block hyperthermia-induced ATP-P2X7-AKT/PRAS40/mTOR signaling transduction. Of note, prevention of extracellular Ca2+ influx with EDTA also fails to counteract ATP cytotoxicity at 40°C (Supplementary Figure 2). Moreover, carbenoxolone (CBX), a pharmacological inhibitor of pannexin/connexin channels that are often linked to P2X7 activation, also failed to block the hyperthermia-associated changes to the ATP-P2X7 signaling cascade (Figure 2B).
It is known that non-selective pore opening upon P2X7 activation facilitates cytosolic adenine nucleotide efflux into the extracellular space [4]. We then assessed levels of both extracellular and intracellular adenine nucleotides in our experimental settings by HPLC. We found that co-treatment of ATP with hyperthermia significantly increased levels of extracellular adenine nucleotides (inclusive of ATP, ADP and AMP), accompanied with concomitant decreases in intracellular levels of ATP (Figure 2C). Such phenomena were not seen in cells deficient of P2X7 (Figure 2C). Of note, basal levels of extracellular nucleotides were not altered by the changes in temperatures (data not shown).
These data suggest that hyperthermia/ATP-evoked pore formation is an intrinsic property of P2X7 receptor that is independent of calcium signaling and pannexin/connexin channels.
Hyperthermia-induced P2X7 sensitivity to ATP is not associated with lipid raft cholesterol content
It has been demonstrated that membrane interaction of P2X7 with lipid rafts regulates the receptor channel properties [11], and heat stress perturbs lipid raft integrity [12, 13]. As such, we next evaluated the role of cholesterol within lipid rafts in hyperthermia and ATP-mediated hyperactivation of P2X7.
First, we examined the involvement of cholesterol, an essential component of plasma membrane lipid rafts, in hyperthermia/ATP-induced cytotoxicity. Cells were pre-treated with two chemical cholesterol disruptors: methyl β-cyclodextrin (MCD), which depletes lipid rafts cholesterol or filipin, which is highly fluorescent and binds specifically to cholesterol, before cells were exposed to ATP and/or hyperthermia. In Supplementary Figure 3A, we show that filipin forms a multimeric globular complex with membrane cholesterol, whereas MCD (10 mM) effectively depletes rafts of cholesterol. We observed that MCD or filipin had no effects on ATP cytotoxicity at 40°C (Figure 3A).
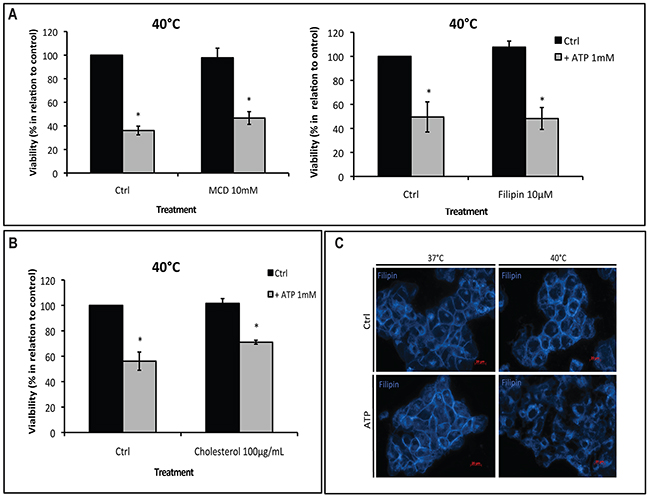
Figure 3: Hyperthermia-increased P2X7 functionality is independent of lipid rafts. (A-B) MCA38 cells were treated with methyl β-cyclodextrin (MCD) (left) or filipin (right) (A) or cholesterol (B), before being pulse treated with ATP (1 mM) for 15 min at 40°C. 24 hr later, cell viability was evaluated. Cells treated with media served as control (Ctrl). (C) Cholesterol staining with filipin showing cholesterol rearrangement at the plasma membrane after treatment with hyperthermia and ATP. *p < 0.05 in contrast with control (two-way ANOVA, followed by Bonferroni pos-test, mean ± SD). Bars, 20 μM.
Alternatively, stabilization of lipid rafts by loading cells with soluble cholesterol also failed to alter ATP tumor-killing activity (Figure 3B), indicating that cholesterol content within lipid rafts is not responsible for controlling P2X7 functionality. In addition, immunoprecipitation assays suggested there were no physical interactions between P2X7 and the two lipid raft markers caveolin-1 or flotillin-2 (Supplementary Figure 3B).
However, interestingly, combination of hyperthermia and ATP indeed led to reorganization of membrane cholesterol (Figure 3C), indicating that changes in the plasma membrane structure per se may modulate P2X7 function.
Hyperthermia-induced membrane fluidity is responsible for enhanced P2X7 sensitivity to ATP
It is known that, besides lipid raft reorganization, alterations in the nanostructure and physicochemical state of plasma membrane e.g. membrane fluidity may also interfere with protein and receptor functions [12, 13]. We thus replaced hyperthermia with a membrane fluidizer benzyl alcohol (BA) at 37°C in our experimental settings to test if BA treatment could mimic these hyperthermia effects on P2X7 functionality initiated by ATP, as described above. As shown in Figure 4A, in NC cells, BA increased ATP cytotoxicity at 37°C in a dose-dependent manner. This is concurrent with increased cell shrinkage (Figure 4B, upper and middle) and cell apoptosis/necrosis (indicated by percentages of Annexin-V+/PI+ cells) (Figure 4B, bottom right) as well as blockade of AKT/PRAS40/mTOR signaling (Figure 4C). And such effects were near completely blocked in P2X7 KD cells. Moreover, co-treatment of BA with ATP at 37°C resulted in cholesterol rearrangement in the plasma membrane that is similar to ATP effects at 40°C (Figure 4D). In parallel, neither cholesterol disruption by filipin nor cholesterol loading was able to counteract BA-induced ATP cytotoxicity at 37°C (Supplementary Figure 4).
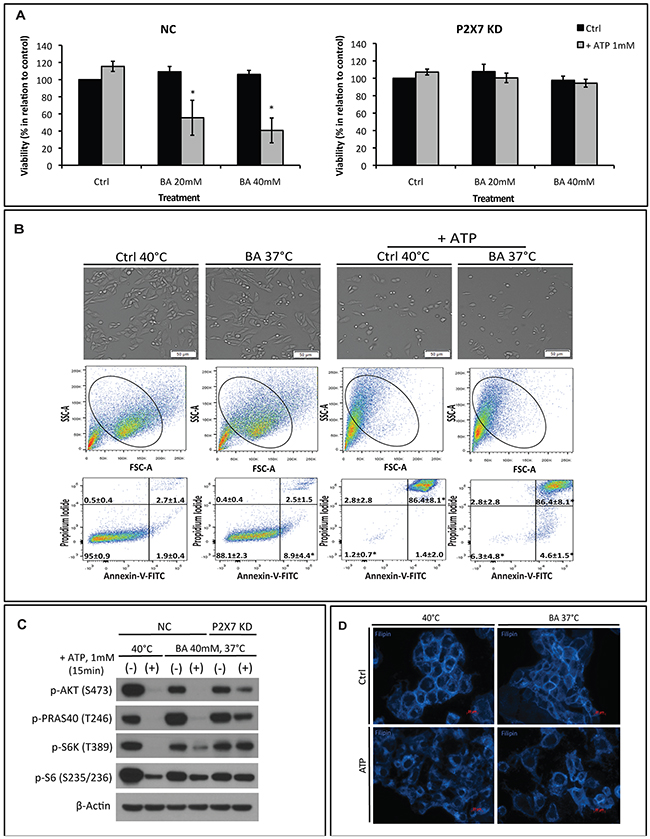
Figure 4: The membrane fluidizer benzyl alcohol (BA) acts similarly as hyperthermia leading to P2X7 hyperactivation at 37°C. NC (negative control) or P2X7 KD (P2X7-deficient) cells were exposed to BA alone or together with ATP for 15 min at 37°C (in order to mimic the heat effect per se) or 40°C. Cells treated with media served as control (Ctrl). (A) Cell viability and (B) images of live cells (upper panel) and FACS analyses of NC cells (middle and bottom panels) were determined. (C) Western blot analysis of AKT/PRAS40/mTOR pathway components was performed immediately after treatment. (D) Re-organization of cholesterol-rich microdomains in co-treated NC cells was visualized using filipin. *p < 0.05 when compared to control (two-way ANOVA, followed by Bonferroni pos-test, mean ± SD). Bars, 50 μM (B) and 20 μM (D).
Taken together, these results imply that enhancements in membrane fluidity caused by hyperthermia augment P2X7 receptor function.
Hyperthermia and extracellular ATP potentiate chemotherapy cytotoxicity, eliciting maximal cell death
Next, we investigated the therapeutic potential of combining hyperthermia and ATP with conventional chemotherapeutic drugs (i.e. cisplatin or mitomycin C). Cells were exposed to cisplatin or mitomycin C prior to ATP and/or hyperthermia treatment. We noted that cisplatin induced cancer cell death in a dose-dependent manner, while mitomycin C alone produced a slight cytotoxic effect only at the higher concentration (Figure 5A). Combination of both drugs with hyperthermia and extracellular ATP exhibited additive effects on their cytotoxicity (Figure 5A and 5B). Indeed, this cumulative effect was more pronounced when cells were exposed to lower concentrations of cisplatin and all concentrations of mitomycin C (Figure 5B), suggestive of the potential utility of hyperthermia and ATP in increasing therapeutic efficacy of both drugs while decreasing side effects caused by higher chemotherapy dosage.
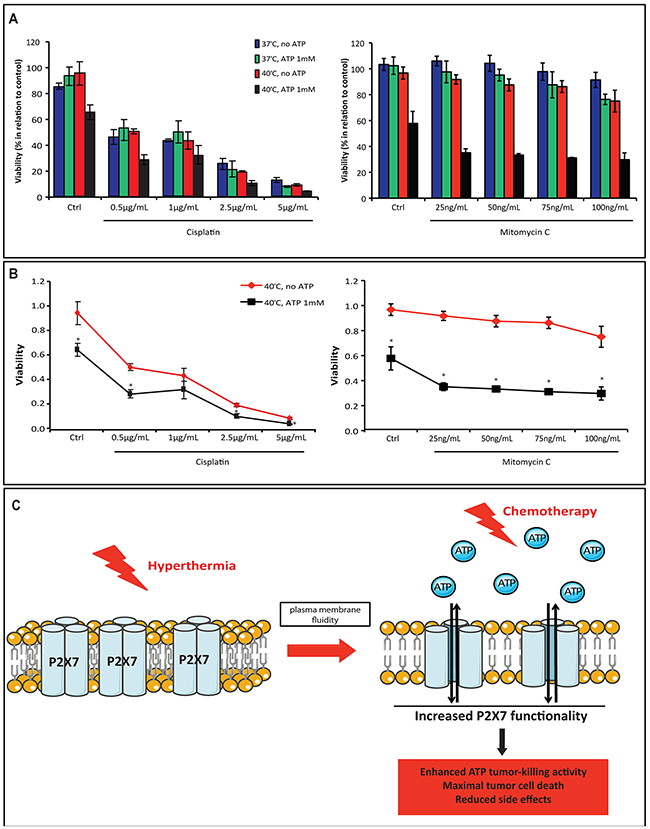
Figure 5: Combination of hyperthermia and ATP with chemotherapy drugs enhances the therapeutic efficacy. (A-B) MCA38 WT cells were left untreated or treated with cisplatin or mitomycin C at the indicated concentrations overnight prior to ATP pulse treatment (ATP 1 mM, 15 min). Cell viability was evaluated after 24 hr. Overall response (A) and drug dose response curve at 40°C (B) were analyzed. (C) Schematic illustration showing the use of hyperthermia with ATP as a new approach to elicit maximal tumor cell death in association with traditional chemotherapy. Hyperthermia, by promoting plasma membrane fluidity, increases P2X7 sensitivity to ATP and therefore enhances ATP tumor-killing activity. The combination of hyperthermia, ATP and conventional chemotherapy would consequently elicit maximal tumor cell death while circumventing side effects of high dose of cytotoxic drugs. *p < 0.05 when compared cells treated with ATP to cells without ATP treatment (two-way ANOVA, followed by Bonferroni pos-test, mean ± SD).
Collectively, our data bring a new insight into cancer therapeutics: combination of hyperthermia with ATP or a purine-related drug as an adjunctive therapy to elicit maximal tumor cell death while circum-venting side effects of chemotherapy (as illustrated in Figure 5C).
DISCUSSION
It has been proposed that high levels of extracellular ATP accumulate within tumor sites, post radiation and chemotherapy [3, 14]. These high levels of extracellular ATP in the tumor microenvironment inhibit tumor growth through the P2X7 receptor, i.e. stimulating antitumor immune responses via P2X7-NLRP3 inflammasome activation [14–16] as well as exerting P2X7-directed cytotoxicity in tumor cells and tumor vascular endothelial cells (thereby limiting angiogenesis) [4, 7, 8, 10, 17–25]. Therefore, maintaining extracellular ATP at high levels in tumor tissues and/or increasing P2X7 functionality might be effective approaches in enhancing the efficacy of traditional cancer therapies.
Local (or superficial) hyperthermia has been used as adjuvant to treat melanoma [26–28]. The clinical use of hyperthermia for cancer either alone or in combination with radiotherapy or chemotherapy has been exploited with mixed effects [28–31]. Improved clinical outcomes have been noted for many types of cancer including prostate [32, 33], breast [34, 35], cervix [36, 37] as well as head and neck cancer [38, 39]. Extracorporeal shock wave therapy (ESWT) is another therapeutic tool utilizing concurrent hyperthermia to treat breast cancer-related lymphedema [40, 41].
Although still controversial, combinations of hyper-thermia with conventional cancer therapies appear to contribute to improved control, cure and/or palliation of certain cancers. Synergistic effects of hyperthermia with chemotherapy are still considered as options in isolated limb perfusion and intraperitoneal chemotherapy for sarcoma and melanoma [28, 42, 43].
Herein, we have identified a novel strategy utilizing hyperthermia that we show to augment ATP-elicited P2X7 functionality thereby maximizing induction of cancer cell death. We have further delineated the underlying molecular mechanisms that could boost effectiveness of chemotherapies such as cisplatin and mitomycin C.
Specifically, we found that hyperthermia effectively sensitizes P2X7 receptor functionality by altering membrane fluidity in response to tumoricidal ATP in MCA38 colon cancer cells. This effect is mediated through the two P2X7 downstream signaling: PI3K/AKT and mTOR that we recently identified [4]. There is growing evidence linking the cellular response to heat stress to changes in lipid composition and architecture of plasma membranes [12]. Nagy et al. [12] showed that either thermal stress or the membrane fluidizer, benzyl alcohol (BA), was able to produce profound alterations in the plasma membrane microdomains and thereby generate stress signal to activate Hsp genes. In this study, we demonstrated that, at 37°C, BA was able to reproduce the hyperthermia effect in potentiating ATP-P2X7 induced cancer cell death, linking increases in membrane fluidity to enhanced P2X7 functionality. Despite hyperthermia and BA being able to activate Hsp gene and increase HSP expression [12, 13, 44], we were unable to observe any HSP modification or interaction with P2X7 in our experimental settings (not shown). This effect was not seen with ethyl alcohol (not shown), which is not considered as a membrane fluidizer. Ethyl alcohol does however have a role in inhibiting NLRP3 inflammasome activation, attenuating IL-1β and caspase-1 cleavage and secretion – pathways related to P2X7 receptor that controls immune system activation [45].
Changes in temperature can also alter solubility, redistribution and activity of lipid raft proteins [46, 47]. Evidences for the involvement of P2X receptors in lipid raft biology are emerging [48, 49] and the presence of P2X7 receptor in this lipid structure has also been described [11, 50, 51]. Accordingly, P2X7 association with lipid rafts varies by cell type. When present in non-raft compartments, P2X7 seems to have increased ion channel activity. In contrast, P2X7 is conferred with a more resistant mode of gating when localized in lipid rafts [11, 50, 51]. In MCA38 cells, P2X7 receptor appears to be present at the non-raft compartments of plasma membrane and its activation occurs independently of cholesterol lipid rafts.
Furthermore, we have demonstrated that hype-rthermia and extracellular ATP have the potential to synergistically increase cytotoxicity of cisplatin and mitomycin C, maximizing tumor cell death. Our data not only reinforce the possible justification of the use of hyperthermia as an adjunctive modality to treat cancer, but also provide a new insight into such treatment option with detailed molecular underpinnings.
On an important note, this strategy would only be applicable for tumor cells expressing the P2X7A receptor splice variant, which is the well-characterized full-length receptor capable of inducing pore formation and apoptosis [52]. Tumors cells expressing truncated/defective P2X7 variant might fail to undergo cell death even with agonist stimulus [53–56].
Taken together, our work highlights the importance of in vivo studies to validate the therapeutic potential of hyperthermia, in combination with high levels of ATP or purine-related drugs, to achieve maximal efficacy of more standard cancer treatments.
MATERIALS AND METHODS
Reagents and antibodies
Carbenoxolone (CBX), BAPTA-AM and thapsigargin (TG) were purchased from Tocris Bioscience (Ellisville, MO). APT102, the recombinant form of soluble CD39, was kindly provided by Dr. Ridong Chen at APT Therapeutics Inc. (St. Louis, MO). All other chemicals and cell culture media were from Sigma-Aldrich (St. Louis, MO) and other culture reagents from Life Technologies (Carlsbad, CA), unless otherwise stated.
Rabbit anti-mouse P2X7 antibody (#APR-004) was obtained from Alomone labs (Jerusalem, Israel); beta-actin (AC-15, #ab6276) from Abcam (Cambridge, MA); Flotillin-2 (B6, #sc-28320) from Santa Cruz Biotechnology, Inc. (Dallas, TX); Caveolin-1 (7C8, #NB100-615) from Novus Biologicals (Littleton, CO); phospho-AKT (S473) (#9271), phospho-PRAS40 (T246) (#2997), phospho-S6K (T389) (#9205), phospho-S6 (S235/236) (#2211) from Cell Signaling Technology (Danvers, MA); HRP-conjugated goat anti-mouse (#31438), donkey anti-rabbit (#31458) and mouse anti-goat IgG (#31400) and the SuperSignal West Femto Maximum Sensitivity Substrate reagents (#PI-34096) were from Thermo Scientific (Rockford, IL).
Tumor cell lines
Syngeneic C57BL/6 murine MCA38 colon cancer cells (a gift of Dr. Nicholas P. Restifo, National Cancer Institute) were kindly provided by Dr. Alan B. Frey at New York University School of Medicine [4, 24, 57]. Cells were also tested for Mycoplasma and other infections by mouse IMPACT III PCR Profile via RADIL (Columbia, MO). Cells were maintained in culture flask in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin at 37°C in a 5% CO2 atmosphere at 100% humidity. Generation of P2X7 knockdown cell line has been previously established [4]. Selection medium contains 3 μg/ml of puromycin. P2X7-defiecient (P2X7 KD) cells were infected with mouse P2X7 shRNA and negative control (NC) cells were infected with an empty shRNA vector control.
Hyperthermia treatment
MCA38 cells were pulse-treated with medium or ATP for 15-30 min at 37°C (incubator) or 40°C (water bath) with 5 min pre-incubation. This mild heat temperature was chosen considering the increased cytotoxicity caused by higher temperatures (Supplementary Figure 1). Also, according to Gombos et al [13], at 39.5°C, heat-induced lipid raft destabilization was seen after 10 min of thermal stress and a spontaneous recovery of raft integrity occurred in 45 min. As such, the time course of 15-30 min is adequate to study thermal stress-induced plasma membrane destabilization.
Antagonist/drugs/plasma membrane disturbance-treatments
Cells were pre-incubated with antagonist carbenoxolone (100 μM), BAPTA-AM (10 μM), thapsigargin (100 nM), and EDTA (0.6 mM) for 30 min, and with soluble CD39 APT102 (2.5 μg/ml) for 15-30 min, before being exposed to pulse treatment with ATP or control medium. Concentration and incubation time for APT102 was based on our previous work [58], which is sufficient to promote total ATP degradation. The cholesterol depleting agents, Methyl-β-cyclodextrin (MCD; 10 mM) or Filipin (10 μM), were incubated for 20 min and removed before ATP treatment. Cholesterol loading at the plasma membrane was obtained by adding water-soluble cholesterol at 100 μg/ml for 30 min before and during ATP exposure. The membrane fluidizer benzyl alcohol (BA), a documented nondenaturant, was added together with ATP for 15 min in order to mimic the hyperthermia effect per se. The chemotherapy drugs Cisplatin and Mitomycin C were added at different concentrations overnight prior to ATP pulse treatment. All compounds were pre-mixed in complete culture medium (RPMI + 10% FBS) at their final concentrations tested before incubation with the cells.
Cell viability measurements
Cells (7.5 × 103) were seeded into 96-well plates and cultured for 24 hr. Cells were then treated as described above, replaced with fresh culture media, and grown for additional 24 hr. Cells viability was evaluated using Cell Counting Kit-8 (CCK-8, Dojindo Molecular Tech. Inc., Rockville, MD) that measures the activity of cellular dehydrogenases (correlating with cell proliferation), as previously established [24, 59].
Annexin V and propidium iodide (PI) staining
Phosphatidylserine externalization was determined by the annexin fluorescence signal of an annexin V-fluorescein isothiocyanate conjugate (BioLegend Inc, San Diego, CA) according to the manufacturer’s protocol. Immediately after treatment, cells were trypsinized and centrifuged for 5 min at 1200 rpm, and the supernatant was discarded. Cells were resuspended in Annexin V Binding Buffer at a concentration of 0.25-1.0 × 107 cells/ml and then an aliquot of 100 μl was taken and incubated with 5 μl of FITC-Annexin V and 10 μl of PI for 15 min at room temperature in the dark. Samples were analyzed on BD FACS LSR II cytometer (BD Bioscience, San Diego, CA) using FlowJo V.10 software for analysis (Tree Star Inc, Ashland, OR).
Analysis of intracellular and extracellular nucleotide levels by high-performance liquid chromatography
Intracellular and extracellular levels of ATP, ADP, and AMP were determined by high performance liquid chromatography (HPLC) as previously described [59–61] with slight modifications. Cells were seeded into 35 mm × 10 mm dishes in culture media and grown for 24 hr until reach 70% of confluence. Cells were then treated with ATP (1 mM) or medium in a final volume of 2 ml for 15 min, at 37°C or 40°C. The supernatant was removed, transferred to a new tube and centrifuged at 2,000 rpm for 10 min at 0°C. A new supernatant aliquot was taken, centrifuged at 5,000 rpm for 5 min at 0°C, precipitated by 5% of 8 M perchloric acid (PCA) and stored at -80°C for subsequent HPLC analyses. The remaining cells were washed with ice-cold HBSS five times to remove excess extracellular ATP. Some wells were lysed with 130 μl of protein lysis buffer for protein concentration measurement; and some wells were harvested with 600 μl of HBSS containing 5% of 8 M PCA and then subjected to three frozen-thaw cycles. Cells were then scraped, transferred to a 1.5 ml Eppendorf tube, pulse sonicated on ice, and then stored at -80°C for subsequent HPLC analysis. 40 μl of each sample were analyzed using a Waters 484 system (Waters Corporation, Milford, MA) as previously described [59–61].
Ethidium bromide uptake assay
This assay was performed as established previously [4, 62] with slight modifications. Briefly, cells (1.5× 104) were seeded into black 96-well plates. 24 hr later, cells were washed once with HBSS containing Ca2+/Mg2+ and incubated with 1.27 μM of ethidium bromide (a cell impermeable organic dye) in the absence or presence of ATP (1 mM) for 15 min at 37°C or 40°C, followed by whole cell fluorescence measurement (in arbitrary units of fluorescence, AUF) at 544/610 nm excitation/emission using the SoftMax Pro software on a SpectraMax M5 Microplate Reader (Molecular Devices, Sunnyvale, CA).
Western blotting
Cells were maintained in 35 mm × 10 mm dishes in culture media for 24 hr until reach 70% of confluence. Cells were treated with freshly made compounds and the reaction was stopped immediately by washing the cells with ice-cold PBS three times. Cells were then lysed in ice-cold modified-RIPA buffer (50 mM Tris-HCl, pH 7.4; 1% NP-40; 0.25% sodium deoxycholate; 150 mM NaCl) supplemented with Protease and Phosphatase Inhibitor Cocktails (Thermo Scientific). The lysates were kept on ice for at least 30 min and then centrifuged at 14,000 rpm for 20 minutes at 4°C. The measurement of protein concentrations and detailed procedures of immunoblotting were described previously [59, 63].
Immunoprecipitation
Cells were seeded into 100 mm × 20 mm dishes and kept in culture media until reach 80% of confluence. Cells were then treated and the reaction was stopped immediately by washing the cells with ice-cold PBS three times. Cell lysate was obtained as described in Western Blotting section. 150 μg of total cellular protein was incubated with 2 μl of P2X7 or Caveolin-1 antibody or 10 μl of Flotillin-2 antibody with an end-over-end rotation at 4°C for 4 hr. Then, 20 μl of Protein G-Sepharose fast flow (Sigma Aldrich) were added into each mixture and incubated at 4°C on a rotating device overnight. The next day, the mixture were centrifuged at 2500 rpm, for 5 min at 4°C, washed three times with lysis buffer, eluted with 2X SDS reducing sample buffer (Bio-Rad, Hercules, CA) containing 2% of 2-mercaptoethanol and resolved on a 4-12% gradient SDS-page gel (Bio-Rad, Hercules, CA) according to western blot methods.
Filipin staining
Cells were seeded into 24-well plates at a low confluence and cultured over a cover slip for 3 days until reach 50% of confluence. Then, cells were treated with medium or ATP (1 mM) for 15 min at 37°C or 40°C. The supernatant was removed and cells were fixed with 2% paraformaldehyde (PFA) (Electron Microscopy Sciences, Hatfield, PA). Fixed cells were incubated with filipin (0.5 mg/ml) in PBS for 60 min [64], followed by two washes with PBS and two washes with distilled water. Cell-containing coverslips were then dried, mounted in a microscope glass slide and visualized/analyzed using a Zeiss AxioVert 200M Inverted Fluorescent Microscope (Peabody, MA). Pictures were taken at 40X magnification.
Statistical analyses
Statistical analyses were performed using Prism 5 (GraphPad, La Jolla, CA). Data are expressed as percentage of control and presented as mean ± SD of at least three independent experiments. Statistical analyses for comparison among multiple groups were performed by one-way analysis of variance (ANOVA), followed by a Tukey post-hoc test. When more than one molecule was mixed to the same well at the same time, a two-way ANOVA was performed, followed by a Bonferroni post-test. Values were considered significant at p < 0.05.
Abbreviations
ADP, adenosine diphosphate; AMP, adenosine monophosphate; ATP, adenosine triphosphate; APT102, soluble ectonucleotidase CD39; BAPTA-AM, 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetra acetic acid tetrakis(acetoxymethyl ester); BA, benzyl alcohol; CBX, carbenoxolone; eATP, extracellular ATP; EDTA, ethylenediamine tetraacetic acid; HPLC, high performance liquid chromatography; MCD, methyl-β-cyclodextrin; NC, negative control cell line infected with an empty shRNA vector control; P2R, type 2-purinergic receptors; P2X7 KD, P2X7-deficient MCA38 cell line; PCA, perchloric acid; PFA, paraformaldehyde; PI, propidium iodide; TG, thapsigargin; TME, tumor microenvironment; WT, wild type.
Author contributions
Conceptualization, P.A.M., Y.W. and S.C.R.; Methodology, P.A.M., S.B., L.E.B.S., H.Z., J.Z., W.J., Y.W., and S.C.R.; Investigation, P.A.M., S.B., L.E.B.S., Y.W., and S.C.R.; Formal analysis, P.A.M. and S.B.; Writing – Original Draft, P.A.M.; Writing – Review & Editing, P.A.M., M.R.W., G.L., A.B., Y.W., and S.C.R.; Funding Acquisition, Y.W. and S.C.R.; Resources, Y.W. and S.C.R.; Supervision, Y.W. and S.C.R.
ACKNOWLEDGMENTS
We sincerely thank Dr. Ridong Chen from APT Therapeutics, Inc. for providing APT102.
CONFLICTS OF INTEREST
The authors declare no competing or financial interests.
FUNDING
This work has been supported by the National Institute of Health awards P01HL107152 and R21 CA164970; grants from the Ben and Rose Cole Charitable PRIA foundation, generosity of the family of Jane O. Siegel and the Leona M. and Harry B. Helmsley Charitable Trust to S.C.R., as well as the Charlotte F. & Irving W. Rabb Research Award to Y.W. together with the Doctoral scholarship from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Fundação Lemann/Brazil to the first author P.A.M.
REFERENCES
1. Roger S, Jelassi B, Couillin I, Pelegrin P, Besson P, Jiang LH. Understanding the roles of the P2X7 receptor in solid tumour progression and therapeutic perspectives. Biochim Biophys Acta. 2015; 1848:2584-602.
2. Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007; 7:961-7.
3. Pellegatti P, Raffaghello L, Bianchi G, Piccardi F, Pistoia V, Di Virgilio F. Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase. PLoS One. 2008; 3:e2599.
4. Bian S, Sun X, Bai A, Zhang C, Li L, Enjyoji K, Junger WG, Robson SC, Wu Y. P2X7 integrates PI3K/AKT and AMPK-PRAS40-mTOR signaling pathways to mediate tumor cell death. PLoS One. 2013; 8:e60184.
5. Feng L, Tapper EB, Sun X, Gehring M, Robson SC, Wu Y. (2014). Purinegic Modulation and CD39/ENTPD1 in Cancer. In: Atta-ur-Rahman, Iqbal Choudhary M (eds) Frontiers in Anti-Cancer Drug Discovery, Bentham Science Publishers, Beijing, China, pp 229-292.
6. Adinolfi E, Callegari MG, Ferrari D, Bolognesi C, Minelli M, Wieckowski MR, Pinton P, Rizzuto R, Di Virgilio F. Basal activation of the P2X7 ATP receptor elevates mitochondrial calcium and potential, increases cellular ATP levels, and promotes serum-independent growth. Mol Biol Cell. 2005; 16:3260-72.
7. Huang S, Chen Y, Wu W, Ouyang N, Chen J, Li H, Liu X, Su F, Lin L, Yao Y. miR-150 promotes human breast cancer growth and malignant behavior by targeting the pro-apoptotic purinergic P2X7 receptor. PLoS One. 2013; 8:e80707.
8. Gehring MP, Kipper F, Nicoletti NF, Sperotto ND, Zanin R, Tamajusuku AS, Flores DG, Meurer L, Roesler R, Filho AB, Lenz G, Campos MM, Morrone FB. P2X7 receptor as predictor gene for glioma radiosensitivity and median survival. Int J Biochem Cell Biol. 2015; 68:92-100.
9. Adinolfi E, Raffaghello L, Giuliani AL, Cavazzini L, Capece M, Chiozzi P, Bianchi G, Kroemer G, Pistoia V, Di Virgilio F. Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res. 2012; 72:2957-69.
10. de Andrade Mello P, Filippi-Chiela EC, Nascimento J, Beckenkamp A, Santana DB, Kipper F, Casali EA, Nejar Bruno A, Paccez JD, Zerbini LF, Wink MR, Lenz G, Buffon A. Adenosine uptake is the major effector of extracellular ATP toxicity in human cervical cancer cells. Mol Biol Cell. 2014; 25:2905-18.
11. Robinson LE, Shridar M, Smith P, Murrell-Lagnado RD. Plasma membrane cholesterol as a regulator of human and rodent P2X7 receptor activation and sensitization. J Biol Chem. 2014; 289:31983-94.
12. Nagy E, Balogi Z, Gombos I, Akerfelt M, Björkbom A, Balogh G, Török Z, Maslyanko A, Fiszer-Kierzkowska A, Lisowska K, Slotte PJ, Sistonen L, Horváth I, et al. Hyperfluidization-coupled membrane microdomain reorganization is linked to activation of the heat shock response in a murine melanoma cell line. Proc Natl Acad Sci U S A. 2007; 104:7945-50.
13. Gombos I, Crul T, Piotto S, Güngör B, Török Z, Balogh G, Péter M, Slotte JP, Campana F, Pilbat AM, Hunya A, Tóth N, Literati-Nagy Z, et al. Membrane-lipid therapy in operation: the HSP co-inducer BGP-15 activates stress signal transduction pathways by remodeling plasma membrane rafts. PLoS One. 2011; 6:e28818.
14. Aymeric L, Apetoh L, Ghiringhelli F, Tesniere A, Martins I, Kroemer G, Smyth MJ, Zitvogel L. Tumor cell death and ATP release prime dendritic cells and efficient anticancer immunity. Cancer Res. 2010; 70:855-8.
15. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, Perfettini JL, Schlemmer F, Tasdemir E, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009; 15:1170-8.
16. Adinolfi E, Capece M, Franceschini A, Falzoni S, Giuliani AL, Rotondo A, Sarti AC, Bonora M, Syberg S, Corigliano D, Pinton P, Jorgensen NR, Abelli L, et al. Accelerated tumor progression in mice lacking the ATP receptor P2X7. Cancer Res. 2015; 75:635-44.
17. Fang WG, Pirnia F, Bang YJ, Myers CE, Trepel JB. P2-purinergic receptor agonists inhibit the growth of androgen-independent prostate carcinoma cells. J Clin Invest. 1992; 89:191-6.
18. Wang Q, Wang L, Feng YH, Li X, Zeng R, Gorodeski GI. P2X7 receptor-mediated apoptosis of human cervical epithelial cells. Am J Physiol Cell Physiol. 2004; 287:C1349-58.
19. Li X, Zhou L, Feng YH, Abdul-Karim FW, Gorodeski GI. The P2X7 receptor: a novel biomarker of uterine epithelial cancers. Cancer Epidemiol Biomarkers Prev. 2006; 15:1906-13.
20. Shabbir M, Ryten M, Thompson C, Mikhailidis D, Burnstock G. Purinergic receptor-mediated effects of ATP in high-grade bladder cancer. BJU Int. 2008; 101:106-12.
21. Deli T, Csernoch L. Extracellular ATP and cancer: an overview with special reference to P2 purinergic receptors. Pathol Oncol Res. 2008; 14:219-31.
22. Shabbir M, Burnstock G. Purinergic receptor-mediated effects of adenosine 5’-triphosphate in urological malignant diseases. Int J Urol. 2009; 16:143-50.
23. Tamajusuku AS, Villodore ES, Paulus R, Coutinho-Silva R, Battasstini AM, Wink MR, Lenz G. Characterization of ATP-induced cell death in the GL261 mouse glioma. J Cell Biochem. 2010; 109:983-91.
24. Feng L, Sun X, Csizmadia E, Han L, Bian S, Murakami T, Wang X, Robson SC, Wu Y. Vascular CD39/ENTPD1 directly promotes tumor cell growth by scavenging extracellular adenosine triphosphate. Neoplasia. 2011; 13:206-16.
25. Gehring MP, Pereira TC, Zanin RF, Borges MC, Braga Filho A, Battastini AM, Bogo MR, Lenz G, Campos MM, Morrone FB. P2X7 receptor activation leads to increased cell death in a radiosensitive human glioma cell line. Purinergic Signal. 2012; 8:729-39.
26. Engin K, Leeper DB, Tupchong L, Waterman FM, Mansfield CM. Thermoradiation therapy for superficial malignant tumors. Cancer. 1993; 72:287-96.
27. Jones EL, Oleson JR, Prosnitz LR, Samulski TV, Vujaskovic Z, Yu D, Sanders LL, Dewhirst MW. Randomized trial of hyperthermia and radiation for superficial tumors. J Clin Oncol. 2005; 23:3079-85.
28. Cihoric N, Tsikkinis A, van Rhoon G, Crezee H, Aebersold DM, Bodis S, Beck M, Nadobny J, Budach V, Wust P, Ghadjar P. Hyperthermia-related clinical trials on cancer treatment within the ClinicalTrials.gov registry. Int J Hyperthermia. 2015; 31:609-14.
29. Luk K, Francis M, Perez CA, Johnson RJ. Combined radiation and hyperthermia: comparison of two treatment schedules based on data from a registry established by the Radiation Therapy Oncology Group (RTOG). Int J Radiat Oncol Biol Phys. 1984; 10:801-9.
30. Irish CE, Brown J, Galen WP, Gallucci JJ, Hyman MD, Horowitz IJ, Snedecor PA, Baker HW. Thermoradiotherapy for persistent cancer in previously irradiated fields. Cancer. 1986; 57:2275-9.
31. Seegenschmiedt MH, Sauer R, Miyamoto C, Chalal JA, Brady LW. Clinical experience with interstitial thermoradiotherapy for localized implantable pelvic tumors. Am J Clin Oncol. 1993; 16:210-22.
32. Van den Berg CA, Van de Kamer JB, De Leeuw AA, Jeukens CR, Raaymakers BW, van Vulpen M, Lagendijk JJ. Towards patient specific thermal modelling of the prostate. Phys Med Biol. 2006; 51:809-25.
33. Hurwitz MD, Hansen JL, Prokopios-Davos S, Manola J, Wang Q, Bornstein BA, Hynynen K, Kaplan ID. Hyperthermia combined with radiation for the treatment of locally advanced prostate cancer: long-term results from Dana-Farber Cancer Institute study 94–153. Cancer. 2011; 117:510-6.
34. Moros EG, Peñagaricano J, Novàk P, Straube WL, Myerson RJ. Present and future technology for simultaneous superficial thermoradiotherapy of breast cancer. Int J Hyperthermia. 2010; 26:699-709.
35. Zagar TM, Oleson JR, Vujaskovic Z, Dewhirst MW, Craciunescu OI, Blackwell KL, Prosnitz LR, Jones EL. Hyperthermia combined with radiation therapy for superficial breast cancer and chest wall recurrence: a review of the randomised data. Int J Hyperthermia. 2010; 26:612-7.
36. Vasanthan A, Mitsumori M, Park JH, Zhi-Fan Z, Yu-Bin Z, Oliynychenko P, Tatsuzaki H, Tanaka Y, Hiraoka M. Regional hyperthermia combined with radiotherapy for uterine cervical cancers: a multi-institutional prospective randomized trial of the international atomic energy agency. Int J Radiat Oncol Biol Phys. 2005; 61:145-53.
37. Franckena M, van der Zee J. Use of combined radiation and hyperthermia for gynecological cancer. Curr Opin Obstet Gynecol. 2010; 22:9-14.
38. Huilgol NG, Gupta S, Dixit R. Chemoradiation with hyperthermia in the treatment of head and neck cancer. Int J Hyperthermia. 2010; 26:21-5.
39. Huilgol NG, Gupta S, Sridhar CR. Hyperthermia with radiation in the treatment of locally advanced head and neck cancer: a report of randomized trial. J Cancer Res Ther. 2010; 6:492-6.
40. Bae H, Kim HJ. Clinical outcomes of extracorporeal shock wave therapy in patients with secondary lymphedema: a pilot study. Ann Rehabil Med. 2013; 37:229-34.
41. Cebicci MA, Sutbeyaz ST, Goksu SS, Hocaoglu S, Oguz A, Atilabey A. Extracorporeal shock wave therapy for breast cancer-related lymphedema: a pilot study. Arch Phys Med Rehabil. 2016; 97:1520-5.
42. Pennacchioli E, Fiore M, Gronchi A. Hyperthermia as an adjunctive treatment for soft-tissue sarcoma. Expert Rev Anticancer Ther. 2009; 9:199-210.
43. Rastrelli M, Campana LG, Valpione S, Tropea S, Zanon A, Rossi CR. Hyperthermic isolated limb perfusion in locally advanced limb soft tissue sarcoma: a 24-year single-centre experience. Int J Hyperthermia. 2016; 32:165-72.
44. Yamada K, Fukao Y, Hayashi M, Fukazawa M, Suzuki I, Nishimura M. Cytosolic HSP90 regulates the heat shock response that is responsible for heat acclimation in Arabidopsis thaliana. J Biol Chem. 2007; 282:37794-804.
45. Hoyt LR, Ather JL, Randall MJ, DePuccio DP, Landry CC, Wewers MD, Gavrilin MA, Poynter ME. Ethanol and other short-chain alcohols inhibit NLRP3 inflammasome activation through protein tyrosine phosphatase stimulation. J Immunol. 2016; 197:1322-34.
46. Vigh L, Escribá PV, Sonnleitner A, Sonnleitner M, Piotto S, Maresca B, Horváth I, Harwood LJ. The significance of lipid composition for membrane activity: new concepts and ways of assessing function. Prog Lipid Res. 2005; 44:303-44.
47. Vigh L, Torok Z, Balogh G, Glatz A, Piotto S, Horvatah I. (2007). Membrane-Regulated Stress Response. In: Csermely P, Vigh L (eds) Molecular Aspects of the Stress Response: Chaperones, Membranes and Networks, Springer, New York, USA, pp 114-131.
48. Vacca F, Amadio S, Sancesario G, Bernardi G, Volonté C. P2X3 receptor localizes into lipid rafts in neuronal cells. J Neurosci Res. 2004; 76:653-61.
49. Vial C, Evans RJ. Disruption of lipid rafts inhibits P2X1 receptor-mediated currents and arterial vasoconstriction. J Biol Chem. 2005; 280:30705-11.
50. Garcia-Marcos M, Pochet S, Marino A, Dehaye JP. P2X7 and phospholipid signalling: the search of the “missing link” in epithelial cells. Cell Signal. 2006; 18:2098-104.
51. Barth K, Weinhold K, Guenther A, Young MT, Schnittler H, Kasper M. Caveolin-1 influences P2X7 receptor expression and localization in mouse lung alveolar epithelial cells. FEBS J. 2007; 274:3021-33.
52. Rassendren F, Buell GN, Virginio C, Collo G, North RA, Surprenant A. The permeabilizing ATP receptor, P2X7. Cloning and expression of a human cDNA. J Biol Chem. 1997; 272:5482-6.
53. Skarratt KK, Fuller SJ, Sluyter R, Dao-Ung LP, Gu BJ, Wiley JS. A 5’ intronic splice site polymorphism leads to a null allele of the P2X7 gene in 1-2% of the Caucasian population. FEBS Lett. 2005; 579:2675-8.
54. Cheewatrakoolpong B, Gilchrest H, Anthes JC, Greenfeder S. Identification and characterization of splice variants of the human P2X7 ATP channel. Biochem Biophys Res Commun. 2005; 332:17-27.
55. Feng YH, Li X, Wang L, Zhou L, Gorodeski GI. A truncated P2X7 receptor variant (P2X7-j) endogenously expressed in cervical cancer cells antagonizes the full-length P2X7 receptor through hetero-oligomerization. J Biol Chem. 2006; 281:17228-37.
56. Feng YH, Li X, Zeng R, Gorodeski GI. Endogenously expressed truncated P2X7 receptor lacking the C-terminus is preferentially upregulated in epithelial cancer cells and fails to mediate ligand-induced pore formation and apoptosis. Nucleosides Nucleotides Nucleic Acids. 2006; 25:1271-6.
57. Sun X, Wu Y, Gao W, Enjyoji K, Csizmadia E, Müller CE, Murakami T, Robson SC. CD39/ENTPD1 expression by CD4+Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology. 2010; 139:1030-40.
58. Moeckel D, Jeong SS, Sun X, Broekman MJ, Nguyen A, Drosopoulos JH, Marcus AJ, Robson SC, Chen R, Abendschein D. Optimizing human apyrase to treat arterial thrombosis and limit reperfusion injury without increasing bleeding risk. Sci Transl Med. 2014; 6:248ra105.
59. Sun X, Han L, Seth P, Bian S, Li L, Csizmadia E, Junger WG, Schmelzle M, Usheva A, Tapper EB, Baffy G, Sukhatme VP, Wu Y, et al. Disordered purinergic signaling and abnormal cellular metabolism are associated with development of liver cancer in Cd39/Entpd1 null mice. Hepatology. 2013; 57:205-16.
60. Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, Nizet V, Insel PA, Junger WG. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science. 2006; 314:1792-95.
61. Corriden R, Chen Y, Inoue Y, Beldi G, Robson SC, Insel PA, Junger WG. Ectonucleoside triphosphate diphosphohydrolase 1 (E-NTPDase1/CD39) regulates neutrophil chemotaxis by hydrolyzing released ATP to adenosine. J Biol Chem. 2008; 283:28480-6.
62. Casas-Pruneda G, Reyes JP, Pérez-Flores G, Pérez-Cornejo P, Arreola J. Functional interactions between P2X4 and P2X7 receptors from mouse salivary epithelia. J Physiol. 2009; 587:2887-901.
63. Wu Y, Sun X, Kaczmarek E, Dwyer KM, Bianchi E, Usheva A, Robson SC. RanBPM associates with CD39 and modulates ecto-nucleotidase activity. Biochem J. 2006; 396:23-30.
64. Eskelinen EL, Schmidt CK, Neu S, Willenborg M, Fuertes G, Salvador N, Tanaka Y, Lüllmann-Rauch R, Hartmann D, Heeren J, von Figura K, Knecht E, Saftig P. Disturbed cholesterol traffic but normal proteolytic function in LAMP-1/LAMP-2 double-deficient fibroblasts. Mol Biol Cell. 2004; 15:3132-45.