
INTRODUCTION
Despite the high cure rates associated with the early diagnosis and removal of melanoma, patients with Stage IV metastatic disease have a 5-year survival expectancy of approximately 18% [1–3]. Melanoma thus accounts for the majority of deaths related to skin cancer. Davies et al. reported that approximately 37–59% of melanomas contain a mutation in the gene that encodes BRAF [4], which was associated with younger age at diagnosis and poorer survival [5, 6]. In recent years, drugs that target molecularly defined vulnerabilities (such as BRAF kinase) in human melanoma have been clinically validated as effective melanoma therapies [7]. Nearly all patients, however, experience a relapse of the disease due to the emergence of acquired drug resistance [8]. Resistance to therapy has now become a major obstacle for successful melanoma treatment. Efforts to overcome drug resistance with combination BRAF/MEK kinase inhibitors or monotherapy with immune checkpoint inhibitors have, so far, only prolonged time to progressive disease [9–14]. Here, we discuss the current views on the mechanisms of resistance, immunotherapy to overcome T cell dysfunction, and options to reinvigorate T cells, such as adoptive cell therapy.
Resistance to targeted therapy
Drug resistance is defined by either progression of disease or locoregional recurrence despite treatment, and further includes the appearance of new lesions [15, 16]. Resistance to BRAF inhibitor therapy is likely due to either a pre-existing intrinsic mechanism, or an acquired mechanism [14, 17–19], the latter being the case for most patients who initially respond but later progress [17]. Pre-existing intrinsic mechanisms of resistance primarily occur due to genetic alterations, mutations, loss of function, or overexpression of genes involved in either the PI3K/AKT signaling pathway or the MAPK/ERK pathway [8, 17, 18, 20–26]. The PI3K/AKT pathway involves the activation of mTOR, eventually resulting in reduced apoptosis, promoting growth, protein synthesis, and proliferation [17]. The MAPK/ERK pathway involves a cascade of activation and phosphorylation by kinases, promoting the proliferation, survival, and differentiation of cells [17]. The most common cause of acquired resistance is due to the reactivation of the MAPK/ERK pathway [8, 14, 17, 27–29], followed by the upregulation of the PI3K/AKT pathway [14]. Another potential mechanism of kinase inhibitor therapy is the activation of the noncanonical Hedgehog pathway observed in melanoma cell lines [30, 31]. The treatment of resistant cells to a specific Hedgehog pathway inhibitor, Gant61, observed in one study, restored sensitivity to the BRAF kinase inhibitor, vemurafenib [30]. Of note, however, tumor heterogeneity, i.e. the same patient tumor with more than one mechanism of resistance detected [14], may contribute to the persistence of disease progression despite combination kinase inhibitor therapy, and presents a new challenge against resistance.
Recently, we observed that acquired multidrug resistance is caused by a stress response induced by chemotherapy such as docetaxel, the BRAF kinase inhibitors vemurafenib and dabrafenib, the MEK inhibitor trametinib, hypoxia, or low nutrient environments [32]. The stress response involves chromatin remodeling and activation of signaling cascades and is characterized by an increase in expression of the nerve growth factor receptor CD271 [32], a proposed marker of melanoma stem cells [33], and one that may suppress CD8+ T cell function [34]. The stressed state is further signified by the loss of melanoma differentiation markers such as Melan-A and tyrosinase [32]. The loss of tumor-associated target antigens, which are normally expressed on the surface of melanoma cells, may prevent recognition by melanoma-specific cytotoxic T cells [34]. Tumor specific T cells are a key component of immune defense against melanoma, and impaired antigen recognition results in an inability of the immune system to control tumor growth.
Immunotherapy to overcome resistance
In an effort to address and combat resistance to monotherapy, current treatments for metastatic melanoma either combine the BRAF kinase inhibitor dabrafenib with the MEK inhibitor trametinib or recommend the monoclonal antibody, ipilimumab, against the immune checkpoint inhibitor cytotoxic T-lymphocyte antigen 4, CTLA-4, for first-line therapy [17, 35, 36]. Combined targeted therapy with two kinase inhibitors is indicated for unresectable or metastatic melanoma [17], showing improved overall survival in combination compared to the BRAF inhibitor alone [37]. However, resistance to combined BRAF/MEK kinase therapy is still described [13, 38]. Further studies and ongoing trials suggest that combination immunotherapy, pairing ipilimumab with the programmed cell death 1, PD-1, monoclonal antibody, nivolumab, prolongs progression-free survival in untreated metastatic melanoma patients compared to either nivolumab alone [39] or ipilimumab alone [39–41]. However, an increased incidence of severe drug-related adverse events was observed in over half of those patients undergoing combination therapy [39–41], including diarrhea, fatigue, pruritis, rash, nausea, colitis and fever. Many were reversible with immunosuppressants but some patients required additional systemic glucocorticoids, infliximab, or mycophenolate immunosuppressive therapy instead.
CTLA-4 blockade toxicity or therapeutic response may be related to gut-resident bacteria, according to recent studies [42]. One study found that T cell responses specific for Bacteroides species within the gut were associated with anti-CTLA-4 efficacy both in mice and in patients [43]. This effect was eliminated in mice treated with antibiotics to eradicate Bacteroides from the gut and reversed when Bacteroides was replaced by gavage. Furthermore, the study found that melanoma patients treated with CTLA-4 blockade grew gut-specific Bacteroides with antitumor characteristics. Another recent study evaluating the influence of the intestinal microbiome on immunotherapy-related colitis found that increased Bacteroides representation within the gut correlated with resistance to anti-CTLA-4-induced colitis [44]. Together, these studies may indicate an adjunct to combination immunotherapy with promoting, establishing, or replenishing favorable gut microbiota to both induce anti-tumor activity and prevent therapy-mediated toxicity.
Furthermore, ipilimumab has shown improved overall survival in previously untreated or treated and refractory metastatic disease [35, 36]. The antibody target, CTLA-4 or CD152, is an immune inhibitory marker expressed on T cells after prolonged T cell activation and is constitutively expressed on memory CD4+ and CD8+ T cells as well as regulatory T cells [45, 46]. CTLA-4 competes with CD28 costimulation necessary for T cell activation, and thus suppresses the activation and further proliferation of T cells, resulting in a diminished immune response [45–48]. A study evaluating disease progression after ipilimumab therapy examined differences in the expression of immune antigens and candidate genes in patient melanoma tumor samples both pre and post-therapy [49]. The authors found that the expression of a tyrosine kinase cell cycle regulator, TTK, and the expression of a gene encoding the antiapoptotic protein survivin, BIRC5, both increased in tumors that progressed despite therapy. However, the expression of immune antigens, e.g. CD3, CD4, CD8, and CTLA-4, remained unchanged a year after ipilimumab therapy. While promising, the study was limited by a sample size of four patients. As part of the selection criteria to help elucidate mechanisms of ipilimumab resistance, the patient pool was narrowed down to those who developed some form of an autoimmune reaction to the therapy [49].
More recently, in an effort to increase the efficacy of ipilimumab monotherapy, a clinical trial combining the CTLA-4 inhibitor with an oncolytic virus derived from herpes simplex virus 1 (HSV-1) was developed, revealing a tolerable safety profile and greater efficacy in combination than with either alone [50]. An ongoing Phase III trial is currently evaluating the combination of pembrolizumab, the PD-1 inhibitor, with or without the same oncolytic HSV (NCT02263508). An oncolytic virus is one that selectively infects, replicates within, and kills tumor cells, allowing further dissemination of the virus throughout the tumor tissue [51]. Talimogene laherparepvec or T-VEC, the oncolytic HSV, recently underwent optimistic safety and efficacy trials in single-arm and comparable interventional studies of T-VEC monotherapy [52]. The oncolytic virus was modified by the deletion of two specific herpes simplex 1 viral genes, the neurovirulence factor ICP34.5 and the ICP47 gene, to evade the host’s anti-viral response thus allowing the virus to replicate within tumor cells, and to subsequently lyse them [51, 53–57]. The deletion of the ICP47 gene in particular is thought to induce a systemic tumor-specific immunity by deleting an inhibitor of antigen presentation, therefore rendering the tumor cells vulnerable to an immune response [58–61]. The virus was further modified by the insertion of a gene encoding the expression of human granulocyte macrophage colony-stimulating factor, GM-CSF, to promote anti-tumor immune responses by recruiting and activating antigen-presenting cells, APCs [62].
Phase I and II T-VEC trials revealed replication of the oncolytic virus within tumor cells, the expression of GM-CSF, and an overall response rate of 26% in the intralesional T-VEC group compared to 6% in the group given subcutaneous GM-CSF [63–65]. The Phase III trial revealed a higher durable response rate and longer overall survival in Stage IIIB, IIIC, or IV patients [52], likely due to the systemic tumor-specific immunity induced by the virus [58, 66]. Furthermore, T-VEC was determined to be safe and well tolerated with only minor viral prodromal adverse effects – fever, chills, myalgias, and mild skin site reactions [52, 63, 64]. While promising as monotherapy, the view that combination therapy to target different mechanisms of action – immune checkpoint inhibitors with oncolytic viruses enhancing the expression of pro-immune factors like GM-CSF as well as inducing systemic anti-tumor immunity–may provide more durable and efficacious response rates long term, as current trials suggest.
Immune exhaustion and dysfunction in the tumor microenvironment
Clinically, compared to ipilimumab, patients treated with monoclonal antibodies directed against PD-1, such as nivolumab and pembrolizumab, appear to have higher response rates, sustained tumor regression, and may be better tolerated [17, 67, 68]. Despite differences in clinical outcomes, both PD-1 and CTLA-4 are inhibitory markers expressed in T cell exhaustion–a dysfunctional state in response to persistent antigen stimulation and inflammation–along with other markers such as lymphocyte-activation gene 3 (Lag-3), and T cell immunoglobulin domain and mucin domain 3 (Tim-3) [69–71]. Both Lag-3 and Tim-3 are expressed on T cells, regulatory T cells, B cells, dendritic cells, NK cells, and NKT cells [72]. However, Lag-3 acts as a CD4 homolog by binding MHC class II [72–74], while Tim-3 is involved in T cell tolerance [72, 75]. Although much of the research on T cell exhaustion was done studying chronic viral infection, recent studies suggest that the tumor microenvironment in metastatic melanoma involves infiltrating lymphocytes (TILs) expressing characteristics of exhaustion – namely, PD-1 expression on T cells [69, 76, 77]. In examining the effector function of these PD-1+ TILs, Fourcade et al. showed that T cell function is impaired in those infiltrating CD8+ T cells or cytotoxic T lymphocytes (CTLs) that express PD-1 and more so in PD-1+ TIM-3+ infiltrating CTLs than single positive cells [78]. TILs that co-express many immune inhibitory markers are thus more dysfunctional [72, 79, 80]. This study, however, assessed CTL function by IFNγ, TNFα, or IL-2 cytokine release, but failed to show direct cytotoxic activity via tumor cell lysis.
Interestingly, in the exhausted state, CTLs may exhibit residual IFNγ production, albeit at low levels, but may express high levels of granzyme B with some residual cytotoxic capacity, although these studies were conducted by examining chronic viral infection, not the tumor microenvironment [69, 81, 82]. Spranger et al. showed that the PD-1 ligand, PD-L1, is expressed on melanoma cells with TILs, and indicates that IFNγ may increase PD-L1 expression; however, this study was performed in mice [83, 84]. These findings were validated in humans, where both melanoma tumor cells and infiltrating monocytes were shown to express PD-L1 in association with TILs in PD-L1+ melanomas [85]. This study also revealed the association of PD-L1 expression and TILs with IFNγ expression, which a prior human tumor cell line study revealed as an inducer of PD-L1 expression [86]. In a mouse model of tamoxifen-inducible liver cancer, tumor-specific CD8+ T cells, or TST cells, were determined to be dysfunctional early during tumorigenesis, suggesting that T cell dysfunction in later stages of cancer may be established during the initial tumor formation [87]. Furthermore, the study suggested that TST cell dysfunction was initially reversible but became fixed later on, was likely induced by persistent antigen exposure, and appears distinct although similar to exhausted T cells from chronic viral infection [87]. In another recent study evaluating the metabolic effects of T cell exhaustion in virus-infected mice, increased PD-1 signaling was associated with impaired glucose uptake and metabolism that occurred early during CD8+ T cell exhaustion [88]. This metabolic dysfunction was suggested to have been partially inhibited by PD-1 signaling [88]. While the mechanism of exhausted TILs within the tumor microenvironment appears to be variable and unclear, likely the functional impairment of exhausted TILs is reversible but requires further study [69].
Reinvigorate T cell function – adoptive T cell therapy
In addition to assessing the mechanism of exhausted or dysfunctional TILs, determining ways in which to reverse and reinvigorate the activity of TILs specific to the tumor, without promoting autoimmunity is necessary, especially if prognostic indicators for robust immunotherapy or targeted kinase therapy response cannot be clearly elucidated. The most recent trial involving adoptive cell therapy (ACT) revealed no benefit with the addition of total body irradiation given to those following the lymphodepleting chemotherapy regimen in preparation for ACT [89]. These ACT studies have been chronicled to summarily include lymphodepleting chemotherapy, administering ex vivo activated and expanded TILs, and high dose IL-2, resulting in a complete response rate of 24% in metastatic melanoma patients and an overall 3-year survival rate of 51% [90–99]. Interestingly, a pilot trial evaluating the co-administration of vemurafenib with ACT in metastatic melanoma patients revealed a relatively similar safety profile as either treatment alone, and favorable clinical responses [100]. Although the trial involved a small cohort of eleven patients, all of whom received both vemurafenib and ACT, the majority of the patients achieved a partial response, yet only two patients had complete regression. Further studies are needed to evaluate combination therapy involving different mechanisms of action, which may prove to be more synergistic than either alone.
To support the synergistic effect of combining ACT with other treatment modalities, in particular BRAF or MEK kinase inhibitors, prior studies have revealed that BRAF kinase inhibition may enhance T cell recognition by increasing expression of melanocyte differentiation antigens without impairing lymphocyte viability or function [101, 102]. Similarly, MEK inhibition has been shown to increase tumor antigen expression, but may impair lymphocyte proliferation and function without affecting viability [103]. While ACT remains a promising and evolving therapy that is personalized and patient-specific, predictive markers or factors that may predispose patient tumor cells to acquire resistance to the current therapy – BRAF and MEK kinase inhibitors–continues to demand investigation. Likely mechanisms to be explored include increased CD271 and PD-L1 expression in response to aberrant IFNγ release, thereby downregulating differentiation markers and impairing CTL activity, or alternatively, kinase signaling pathway reactivation–either through MAPK or PI3K pathways.
Potential biomarkers of resistance
Markers predictive of resistance, recurrence, and prevention of resistance would effectively redirect treatment towards combination therapy as first-line, ideally for patients in previously untreated but invasive disease, and not limited to those with advanced or unresectable melanoma. New biomarkers may even be used as new targets for combination therapy. A recent study evaluating potential biomarkers for response to treatment was conducted in metastatic melanoma patients treated with anti-CTLA-4 followed by anti-PD-1 therapy, using tumor biopsies at specific time points before and during treatment [104]. Response to therapy was defined as either with no radiographic evidence, stable disease, or decreased tumor volume, using the Response Evaluation Criteria in Solid Tumors (RECIST) criteria [16, 104]. The study demonstrated a higher density of CD8+ T cells present early during treatment in those tumors that responded to anti-CTLA-4, despite the lack of biomarkers clearly identified prior to treatment [104], although a prior study has shown a higher number of PD-1 and PD-L1 expressing cells too [105]. In those that responded to anti-PD-1 therapy, they found an increased expression of CD8, CD4, CD3, PD-1, PD-L1, and Lag3 early during treatment [104]. While it has been previously published that anti-CTLA-4 or PD-1 monotherapy induces infiltration by TILs [105–108], this study suggests that CD8+ T cells infiltrating the tumor before therapy are particularly correlated with tumor response during therapy. In conjunction with a prior study evaluating response to anti-PD-1 therapy [105], CD8+ T cell density may be useful in predicting response to either anti-PD-1 [104, 105, 108] or CTLA-4 therapy [107], and reduced expression of CD8, PD-1, and PD-L1 despite therapy may correlate with disease progression [105].
Ideally, a panel of biomarkers to indicate T cell dysfunction within the tumor microenvironment, or markers on melanoma cells specific to therapy resistance would be best for analyzing these states while accounting for variations in any one marker alone. These may include markers examining the T cell exhaustive state on TILs, such as CTLA-4, PD-1, TIM-3, and LAG-3. Markers on melanoma cells include PD-L1 and CD271 (Figure 1). The limitations to using a panel of biomarkers include the method by which to determine the expression of these markers efficiently. These markers are not ubiquitously expressed in tumor tissue or TILs, and may require larger samples to identify isolated areas of expression. Furthermore, while a panel of markers may be able to expand the number of possible markers expressed in a given area of tumor tissue, CD8+ T cells are likely to be in close proximity to the PD-1+ and corresponding PD-L1+ expressing cells in both the tumor and invasive margin [105]. This may likely extend to other proposed biomarkers involved in immune inhibition, exhaustion, or dysfunction as well.
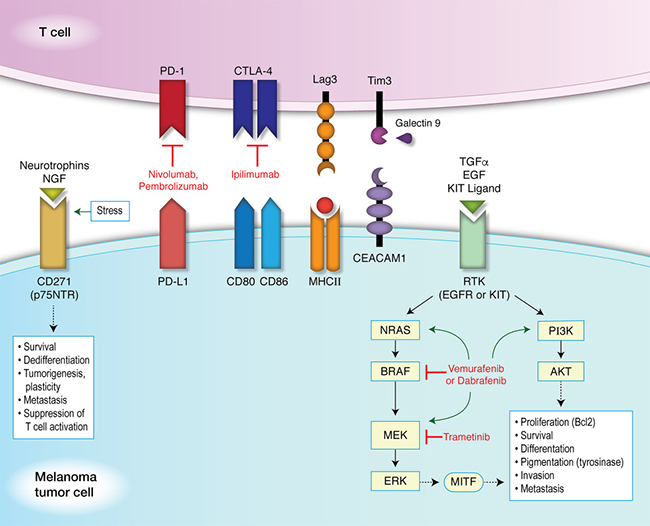
Figure 1: Targeted and immunotherapy against immune inhibitory receptors and resistance mechanisms. Immune inhibitory receptors and their ligands are paired as shown. Neurotrophins and nerve growth factors (NGF) bind to CD271 (p75NTR), whose expression can be induced under conditions of stress, resulting in downstream signaling pathways promoting survival, dedifferentiation, tumorigenesis, plasticity, metastasis, and the suppression of T cell activation. Transforming growth factor-α (TGFα), epidermal growth factor (EGF), and KIT ligand (stem cell factor or STEEL) can bind to a receptor tyrosine kinase (RTK), such as the EGF receptor (EGFR) or KIT receptor (c-KIT receptor or CD117). This promotes the activation of either the MAPK pathway or PI3K/AKT pathway. Kinase inhibitors can induce mutations in components of either pathway, as shown by green arrows. These induced mutations then promote the activation of downstream signaling, bypassing the targeted inhibition and resulting in the phosphorylation of microphthalmia-associated transcription factor (MITF). Expression of MITF leads to differentiation and pigmentation (via tyrosinase activity) as well as the proliferation and survival of melanocytes (through the upregulation of Bcl2). Activation of these pathways also promotes invasion and metastasis in melanoma tumor cells. Tim3 (HAVCR2) forms a heterodimer with CEACAM1 (carcinoembryonic antigen-related cell adhesion molecule) inducing T cell inhibition, and also binds its ligand, Galectin 9 to then suppress T helper cell type 1 (Th1) function and induce cell death.
Future immunotherapy combinations
To add to the current and ongoing research into potential biomarkers to predict responsiveness to therapy and mechanisms of response to therapy, mechanisms and predictors of resistance must also be considered. Treatment approaches that target different aspects of the “cancer-immunity cycle” [109] which may be dysfunctional are promising. These targets include factors within the tumor microenvironment that may modulate the T cell immune response against the tumor, or immune checkpoint inhibitors expressed on either T cells or tumor cells that may contribute to both resistance and T cell dysfunction in combination [110, 111]. New and effective therapy combinations are likely to be elucidated in the many ongoing clinical trials (Table 1). These studies either preemptively prevent resistance by combining a kinase inhibitor with immune inhibitor therapy or treat refractory patients with therapy tailored to the tumor’s resistance mechanism. A few trials evaluating combination immunotherapy and targeted kinase therapy include treating with ipilimumab, pembrolizumab, or an anti-PD-L1 antibody with BRAF and MEK kinase inhibitors. The BRAF kinase inhibitor vemurafenib is also being evaluated with adoptive cell therapy in one trial, while another is pairing vemurafenib with an anti-PD-L1 antibody. Two studies are currently recruiting those who relapse or progress. One study is examining progression after BRAF kinase therapy then specifically tailoring subsequent therapy to the tumor’s resistance mechanism, either with a cyclin dependent kinase inhibitor, fibroblast growth factor receptor (FGFR) inhibitor, PI3K inhibitor, or c-MET inhibitor (NCT02159066). The other study is comparing ipilimumab with or without nivolumab in those who relapsed after anti-PD-1 therapy. Another two studies are soon to begin recruiting patients to boost the costimulatory interaction between CD40 on APCs and CD40 ligand (CD40L) on activated T cells. One study will evaluate the effect of a CD40 agonist with pembrolizumab. The other study will use an adenovirus encoding a chimeric CD40L that is injected into the tumor directly. Interestingly, a Phase I trial is currently recruiting metastatic melanoma patients for treatment with patient T cells electroporated with RNA to express c-Met specific chimeric antigen receptors. More ongoing trials include pembrolizumab with or without T-VEC, ACT with or without pembrolizumab, indoximod (the indoleamine 2,3-dioxygenase inhibitor with a role in controlling inflammation and T cell tolerance [112]) with either pembrolizumab, ipilimumab, or nivolumab, and many others [17].
Table 1: Completed and active clinical trials for combination therapy in metastatic melanoma*
Regimen |
Phase |
Status |
Sponsor |
NCT identifier |
|---|---|---|---|---|
Ipilimumab and Dabrafenib +/− Trametinib |
I |
Completed |
GSK |
NCT01767454 |
Durvalumab (anti-PD-L1) and Trametinib +/− Dabrafenib |
I |
Active, not recruiting |
Med Immune |
NCT02027961 |
Pembrolizumab + Dabrafenib + Trametinib vs Pembrolizumab + Dabrafenib or Trametinib |
I/II |
Recruiting |
Merck |
NCT02130466 |
Nivolumab + Trametinib +/− Dabrafenib |
II |
Recruiting |
MD Anderson |
NCT02910700 |
Atezolizumab (anti-PD-L1) and Vemurafenib +/− Cobimetinib (MEK inhibitor) |
I |
Active, not recruiting |
Genentech |
NCT01656642 |
Atezolizumab (anti-PD-L1) +/− Vemurafenib and Cobimetinib (MEK inhibitor) |
III |
Recruiting |
Hoffmann-La Roche |
NCT02908672 |
Encorafenib (RAF inhibitor) and Binimetinib (MEK inhibitor) until progression then + Ribociclib (CDK4/6 inhibitor) or FGFR inhibitor or PI3K inhibitor or Capmatinib (c-MET inhibitor) |
II |
Active, not recruiting |
Array BioPharma |
NCT02159066 |
Ipilimumab +/− Nivolumab after progression or relapse post-anti-PD-1 |
II |
Recruiting |
Memorial Sloan Kettering |
NCT02731729 |
Vemurafenib + ACT |
I |
Terminated |
NCI |
NCT01585415 |
ACT +/− Pembrolizumab |
II |
Recruiting |
NCI |
NCT02621021 |
ACT using autologous T cells with retrovirally transduced TCR |
I |
Recruiting |
Loyola University |
NCT02870244 |
Pembrolizumab +/− T-VEC |
III |
Recruiting |
Amgen |
NCT02263508 |
IDO inhibitor and Ipilimumab or Pembrolizumab or Nivolumab |
I/II |
Recruiting |
NewLink Genetics |
NCT02073123 |
Pembrolizumab +/− Epacadostat (IDO inhibitor) |
III |
Recruiting |
Incyte |
NCT02752074 |
CD40 agonist with Pembrolizumab |
I/II |
Recruiting begins May 2017 |
MD Anderson |
NCT02706353 |
Pembrolizumab + Intratumoral adenovirus encoding chimeric CD40L |
I/II |
Not yet recruiting |
MD Anderson |
NCT02719015 |
Autologous T cells expressing c-MET chimeric antigen receptors |
I |
Recruiting |
University of Pennsylvania |
NCT03060356 |
*NCT = ClinicalTrials.gov number; Refs = references associated with study; GSK = GlaxoSmithKline; ACT = adoptive cell therapy; NCI = National Cancer Institute; TCR = T cell receptor; T-VEC = Talimogene laherparepvec; IDO = indoleamine 2,3-dioxygenase; CDK4/6 = cyclin-dependent kinases 4 and 6; FGFR = fibroblast growth factor receptor; PI3K = phosphatidyl-inositol-3 kinase; c-MET = tyrosine protein kinase Met or hepatocyte growth factor receptor encoded by MET gene [119].
Concluding remarks
The treatment of patients with metastatic melanoma has thus experienced huge progress with the use of molecular targeted inhibitors and immunotherapy. Recently published results of the Sunbelt Melanoma Trial revealed no survival benefit for adjuvant high dose interferon in those with a single positive sentinel lymph node (Stage III), whether they underwent complete lymph node dissection alone or with interferon therapy [113]. Despite the mixed and controversial results of studies evaluating the efficacy of high dose interferon therapy as a method to induce an effective immune response [114–118], the current trend towards combination immunotherapy appears much more promising. While targeted inhibitors can be effective initially, many patients relapse within one year. Immunotherapy, in the form of antibodies against specific tumor antigens to promote immune recognition and response, shows sustained progression-free survival and even complete recovery in many patients. Mechanisms of resistance in those patients who do not respond to drug therapy, or who respond but later progress, are largely unknown but are of utmost importance to reveal in order to define better treatment strategies for optimal patient-specific precision medicine.
CONFLICTS OF INTEREST
None.
FUNDING
This study was supported by an International Scholarship and a Full Tuition Fee Award from the University of Queensland (Dr Hilary Keller), as well as by a Perpetual Trustees Fellowship (Dr James Wells).
REFERENCES
1. Balch CM, Gershenwald JE, Soong SJ, Thompson JF. Update on the melanoma staging system: the importance of sentinel node staging and primary tumor mitotic rate. Journal of Surgical Oncology. 2011; 104:379–85.
2. Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, Buzaid AC, Cochran AJ, Coit DG, Ding S, Eggermont AM, Flaherty KT, Gimotty PA, et al. Final version of 2009 AJCC melanoma staging and classification. Journal of Clinical Oncology. 2009; 27:6199–206.
3. Howlader N, Noone A, Krapcho M, Miller D, Bishop K, Kosary C, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis D, Chen H, Feuer E, et al. SEER Cancer Statistics Review, 1975-2014. National Cancer Institute. https://seer.cancer.gov/csr/1975_2014/, based on November 2016 SEER data submission, posted to the SEER web site, April 2017.
4. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W. Mutations of the BRAF gene in human cancer. Nature. 2002; 417:949–54.
5. Long GV, Menzies AM, Nagrial AM, Haydu LE, Hamilton AL, Mann GJ, Hughes TM, Thompson JF, Scolyer RA, Kefford RF. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. Journal of Clinical Oncology. 2011; 29:1239–46.
6. Flaherty KT. Is it good or bad to find a BRAF mutation? Journal of Clinical Oncology. 2011; 29:1229–30.
7. Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K. Inhibition of mutated, activated BRAF in metastatic melanoma. New England Journal of Medicine. 2010; 363:809–19.
8. Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010; 468:973–7.
9. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L. Mutations associated with acquired resistance to PD-1 blockade in melanoma. New England Journal of Medicine. 2016; 375:819–29.
10. Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, Joshua AM, Patnaik A, Hwu WJ, Weber JS. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. Journal of the American Medical Association. 2016; 315:1600–9.
11. Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, Hodi FS, Schachter J, Pavlick AC, Lewis KD, Cranmer LD, Blank CU, O’Day SJ, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncology. 2015; 16:908–18.
12. Johnson DB, Flaherty KT, Weber JS, Infante JR, Kim KB, Kefford RF, Hamid O, Schuchter L, Cebon J, Sharfman WH, McWilliams RR, Sznol M, Lawrence DP, et al. Combined BRAF (Dabrafenib) and MEK inhibition (Trametinib) in patients with BRAFV600-mutant melanoma experiencing progression with single-agent BRAF inhibitor. Journal of Clinical Oncology. 2014; 32:3697–704.
13. Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. New England Journal of Medicine. 2012; 367:1694–703.
14. Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer discovery. 2014; 4:80–93.
15. Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC. New guidelines to evaluate the response to treatment in solid tumors. Journal of the National Cancer Institute. 2000; 92:205–16.
16. Eisenhauer E, Therasse P, Bogaerts J, Schwartz L, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). European journal of cancer. 2009; 45:228–47.
17. Spagnolo F, Ghiorzo P, Orgiano L, Pastorino L, Picasso V, Tornari E, Ottaviano V, Queirolo P. BRAF-mutant melanoma: treatment approaches, resistance mechanisms, and diagnostic strategies. OncoTargets and Therapy. 2015; 8:157–68.
18. Johannessen CM, Johnson LA, Piccioni F, Townes A, Frederick DT, Donahue MK, Narayan R, Flaherty KT, Wargo JA, Root DE. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature. 2013; 504:138–42.
19. Bagrodia S, Smeal T, Abraham RT. Mechanisms of intrinsic and acquired resistance to kinase-targeted therapies. Pigment cell & melanoma research. 2012; 25:819–31.
20. Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012; 487:505–9.
21. Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012; 487:500–4.
22. Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu X, Gimotty PA, Kee D. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer cell. 2010; 18:683–95.
23. Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010; 468:968–72.
24. Trunzer K, Pavlick AC, Schuchter L, Gonzalez R, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT, Kim KB, Weber JS. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. Journal of Clinical Oncology. 2013; 31:1767–74.
25. Xing F, Persaud Y, Pratilas C, Taylor B, Janakiraman M, She Q, Gallardo H, Liu C, Merghoub T, Hefter B. Concurrent loss of the PTEN and RB1 tumor suppressors attenuates RAF dependence in melanomas harboring V600EBRAF. Oncogene. 2012; 31:446–57.
26. Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, Kong X, Bosenberg MC, Wiesner T, Rosen N. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature. 2015; 520:368–72.
27. Poulikakos PI, Rosen N. Mutant BRAF melanomas—dependence and resistance. Cancer cell. 2011; 19:11–5.
28. Shi H, Kong X, Ribas A, Lo RS. Combinatorial treatments that overcome PDGFRβ-driven resistance of melanoma cells to V600EB-RAF inhibition. Cancer research. 2011; 71:5067–74.
29. Sun C, Wang L, Huang S, Heynen GJ, Prahallad A, Robert C, Haanen J, Blank C, Wesseling J, Willems SM. Reversible and adaptive resistance to BRAF (V600E) inhibition in melanoma. Nature. 2014; 508:118–22.
30. Faião-Flores F, Alves-Fernandes DK, Pennacchi PC, Sandri S, Vicente AL, Scapulatempo-Neto C, Vazquez VL, Reis RM, Chauhan J, Goding CR, Smalley KS, Maria-Engler SS. Targeting the hedgehog transcription factors GLI1 and GLI2 restores sensitivity to vemurafenib-resistant human melanoma cells. Oncogene. 2016; 36:1849–61.
31. Sabbatino F, Wang Y, Wang X, Flaherty KT, Yu L, Pepin D, Scognamiglio G, Pepe S, Kirkwood JM, Cooper ZA. PDGFRα up-regulation mediated by sonic hedgehog pathway activation leads to BRAF inhibitor resistance in melanoma cells with BRAF mutation. Oncotarget. 2014; 5:1926–41. doi: 10.18632/oncotarget.1878.
32. Menon DR, Das S, Krepler C, Vultur A, Rinner B, Schauer S, Kashofer K, Wagner K, Zhang G, Rad EB. A stress-induced early innate response causes multidrug tolerance in melanoma. Oncogene. 2014; 34:4448–59.
33. Shakhova O, Sommer L. Testing the cancer stem cell hypothesis in melanoma: the clinics will tell. Cancer letters. 2013; 338:74–81.
34. Furuta J, Inozume T, Harada K, Shimada S. CD271 on melanoma cell is an IFN-γ-inducible immunosuppressive factor that mediates downregulation of melanoma antigens. Journal of Investigative Dermatology. 2014; 134:1369–77.
35. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC. Improved survival with ipilimumab in patients with metastatic melanoma. New England Journal of Medicine. 2010; 363:711–23.
36. Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. New England Journal of Medicine. 2011; 364:2517–26.
37. Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L. Improved overall survival in melanoma with combined dabrafenib and trametinib. New England Journal of Medicine. 2015; 372:30–9.
38. Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, Rosenberg M, Goetz EM, Sullivan RJ, Farlow DN. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer discovery. 2014; 4:61–8.
39. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. New England Journal of Medicine. 2015; 373:23–34.
40. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, Shaheen M, Ernstoff MS, Minor D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. New England Journal of Medicine. 2015; 372:2006–17.
41. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K. Nivolumab plus ipilimumab in advanced melanoma. New England Journal of Medicine. 2013; 369:122–33.
42. Pitt JM, Vétizou M, Waldschmitt N, Kroemer G, Chamaillard M, Boneca IG, Zitvogel L. Fine-tuning cancer immunotherapy: optimizing the gut microbiome. Cancer Research. 2016; 76:4602–7.
43. Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CP. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015; 350:1079–84.
44. Dubin K, Callahan MK, Ren B, Khanin R, Viale A, Ling L, No D, Gobourne A, Littmann E, Huttenhower C. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nature communications. 2016; 7.
45. Baksh K, Weber J. Immune checkpoint protein inhibition for cancer: preclinical justification for CTLA-4 and PD-1 blockade and new combinations. Seminars in Oncology. 2015; 42:363–77.
46. Levings MK, Sangregorio R, Roncarolo MG. Human CD25+ CD4+ T regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. The Journal of experimental medicine. 2001; 193:1295–302.
47. Walker LS, Sansom DM. Confusing signals: Recent progress in CTLA-4 biology. Trends in immunology. 2015; 36:63–70.
48. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994; 1:405–13.
49. Bresler SC, Min L, Rodig SJ, Walls AC, Xu S, Geng S, Hodi FS, Murphy GF, Lian CG. Gene expression profiling of anti-CTLA4-treated metastatic melanoma in patients with treatment-induced autoimmunity. Laboratory Investigation. 2017; 97:207–16.
50. Puzanov I, Milhem MM, Minor D, Hamid O, Li A, Chen L, Chastain M, Gorski KS, Anderson A, Chou J. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. Journal of Clinical Oncology. 2016; 34:2619–26.
51. Liu B, Robinson M, Han Z, Branston R, English C, Reay P, McGrath Y, Thomas S, Thornton M, Bullock P. ICP34. 5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene therapy. 2003; 10:292–303.
52. Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. Journal of Clinical Oncology. 2015; 33:2780–8.
53. Chou J, Roizman B. The gamma 1 (34.5) gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programed cell death in neuronal cells. Proceedings of the National Academy of Sciences. 1992; 89:3266–70.
54. He B, Chou J, Brandimarti R, Mohr I, Gluzman Y, Roizman B. Suppression of the phenotype of gamma (1) 34.5-herpes simplex virus 1: failure of activated RNA-dependent protein kinase to shut off protein synthesis is associated with a deletion in the domain of the alpha47 gene. Journal of virology. 1997; 71:6049–54.
55. Brown SM, MacLean AR, McKie EA, Harland J. The herpes simplex virus virulence factor ICP34. 5 and the cellular protein MyD116 complex with proliferating cell nuclear antigen through the 63-amino-acid domain conserved in ICP34. 5, MyD116, and GADD34. Journal of virology. 1997; 71:9442–9.
56. Goldsmith K, Chen W, Johnson DC, Hendricks RL. Infected cell protein (ICP) 47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. The Journal of experimental medicine. 1998; 187:341–8.
57. Poppers J, Mulvey M, Khoo D, Mohr I. Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. Journal of virology. 2000; 74:11215–21.
58. Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Science. 2016; 107:1373–9.
59. Goldsmith K, Chen W, Johnson DC, Hendricks RL. Infected cell protein (ICP) 47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. Journal of Experimental Medicine. 1998; 187:341–8.
60. Fruh K, Ahn K, Djaballah H, Sempé P. A viral inhibitor of peptide transporters for antigen presentation. Nature. 1995; 375:415–8.
61. York IA, Roop C, Andrews DW, Riddell SR, Graham FL, Johnson DC. A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell. 1994; 77:525–35.
62. Kaufman HL, Ruby CE, Hughes T, Slingluff CL. Current status of granulocyte–macrophage colony-stimulating factor in the immunotherapy of melanoma. Journal for immunotherapy of cancer. 2014; 2:1.
63. Hu JC, Coffin RS, Davis CJ, Graham NJ, Groves N, Guest PJ, Harrington KJ, James ND, Love CA, McNeish I. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clinical Cancer Research. 2006; 12:6737–47.
64. Senzer NN, Kaufman HL, Amatruda T, Nemunaitis M, Reid T, Daniels G, Gonzalez R, Glaspy J, Whitman E, Harrington K. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor–encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. Journal of Clinical Oncology. 2009; 27:5763–71.
65. Kaufman HL, Kim DW, DeRaffele G, Mitcham J, Coffin RS, Kim-Schulze S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Annals of surgical oncology. 2010; 17:718–30.
66. Goldufsky J, Sivendran S, Harcharik S, Pan M, Bernardo S, Stern RH, Friedlander P, Ruby CE, Saenger Y, Kaufman HL. Oncolytic virus therapy for cancer. Oncolytic Virotherapy. 2013; 2:31.
67. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS. Safety and tumor responses with lambrolizumab (anti–PD-1) in melanoma. New England Journal of Medicine. 2013; 369:134–44.
68. Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, Brahmer JR, Lawrence DP, Atkins MB, Powderly JD. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. Journal of Clinical Oncology. 2014; 32:1020–30.
69. Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends in immunology. 2015; 36:265–76.
70. Wherry EJ. T cell exhaustion. Nature immunology. 2011; 12:492–9.
71. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nature Reviews Immunology. 2015; 15:486–99.
72. Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory pathways in immunotherapy for cancer. Annual review of immunology. 2016; 34:539–73.
73. Huard B, Gaulard P, Faure F, Hercend T, Triebel F. Cellular expression and tissue distribution of the human LAG-3-encoded protein, an MHC class II ligand. Immunogenetics. 1994; 39:213–7.
74. Baixeras E, Huard B, Miossec C, Jitsukawa S, Martin M, Hercend T, Auffray C, Triebel F, Piatier-Tonneau D. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. Journal of Experimental Medicine. 1992; 176:327–37.
75. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, Manning S, Greenfield EA, Coyle AJ, Sobel RA. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002; 415:536–41.
76. Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, Johnson D, Swetter S, Thompson J, Greenberg PD. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nature medicine. 1999; 5:677–85.
77. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, Rosenberg SA. Tumor antigen–specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009; 114:1537–44.
78. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, Kirkwood JM, Kuchroo V, Zarour HM. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. The Journal of experimental medicine. 2010; 207:2175–86.
79. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, Wherry EJ. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nature immunology. 2009; 10:29–37.
80. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. The Journal of experimental medicine. 2010; 207:2187–94.
81. Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. The Lancet. 2015; 386:444–51.
82. Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, Bikoff EK, Robertson EJ, Lauer GM, Reiner SL. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science. 2012; 338:1220–5.
83. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015; 523:231–5.
84. Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF. Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Science translational medicine. 2013; 5: 200ra116-200ra116.
85. Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Science translational medicine. 2012; 4:127ra37–ra37.
86. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nature medicine. 2002; 8:793–800.
87. Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, Lauer P, Brockstedt DG, Knoblaugh SE, Hämmerling GJ. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity. 2016; 45:389–401.
88. Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, Stelekati E, McLane LM, Paley MA, Delgoffe GM. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8+ T cell exhaustion. Immunity. 2016; 45:358–73.
89. Goff SL, Dudley ME, Citrin DE, Somerville RP, Wunderlich JR, Danforth DN, Zlott DA, Yang JC, Sherry RM, Kammula US. Randomized, Prospective Evaluation Comparing Intensity of Lymphodepletion Before Adoptive Transfer of Tumor-Infiltrating Lymphocytes for Patients With Metastatic Melanoma. Journal of Clinical Oncology. 2016; 34:2389–97.
90. Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE, Matory YL, Skibber JM, Shiloni E, Vetto JT. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. New England Journal of Medicine. 1985; 313:1485–92.
91. Rosenberg SA, Yannelli JR, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, Seipp CA, Einhorn JH, White DE. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. Journal of the National Cancer Institute. 1994; 86:1159–66.
92. Dudley ME, Wunderlich JR, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry RM, Marincola FM, Leitman SF, Seipp CA. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. Journal of immunotherapy (Hagerstown, Md: 1997). 2002; 25:243.
93. Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, Levy D, Kubi A, Shoshani N, Zikich D, Ohayon Y. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clinical Cancer Research. 2013; 19:4792–800.
94. Pilon-Thomas S, Kuhn L, Ellwanger S, Janssen W, Royster E, Marzban S, Kudchadkar R, Zager J, Gibney G, Sondak VK. Brief Communication: Efficacy of Adoptive Cell Transfer of Tumor Infiltrating Lymphocytes after Lymphopenia Induction for Metastatic Melanoma. Journal of immunotherapy (Hagerstown, Md: 1997). 2012; 35:615.
95. Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, Wu R, Lizee G, Mahoney S, Alvarado G. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clinical Cancer Research. 2012; 18:6758–70.
96. Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE, Mavroukakis SA. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. Journal of Clinical Oncology. 2005; 23:2346–57.
97. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clinical Cancer Research. 2011; 17:4550–7.
98. Wrzesinski C, Paulos CM, Kaiser A, Muranski P, Palmer DC, Gattinoni L, Yu Z, Rosenberg SA, Restifo NP. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. Journal of immunotherapy (Hagerstown, Md: 1997). 2010; 33:1.
99. Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang J, Citrin DE, Leitman SF. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. Journal of Clinical Oncology. 2008; 26:5233–9.
100. Deniger DC, Kwong ML, Pasetto A, Dudley ME, Wunderlich JR, Langhan MM, Lee CR, Rosenberg SA. A Pilot Trial of the Combination of Vemurafenib with Adoptive Cell Therapy in Patients with Metastatic Melanoma. Clinical Cancer Research. 2016; 23:351–62.
101. Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, Ferrone CR, Flaherty KT, Lawrence DP, Fisher DE, Tsao H, Wargo JA. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer research. 2010; 70:5213–9.
102. Comin-Anduix B, Chodon T, Sazegar H, Matsunaga D, Mock S, Jalil J, Escuin-Ordinas H, Chmielowski B, Koya RC, Ribas A. The oncogenic BRAF kinase inhibitor PLX4032/RG7204 does not affect the viability or function of human lymphocytes across a wide range of concentrations. Clinical Cancer Research. 2010; 16:6040–8.
103. Vella LJ, Pasam A, Dimopoulos N, Andrews M, Knights A, Puaux AL, Louahed J, Chen W, Woods K, Cebon JS. MEK inhibition, alone or in combination with BRAF inhibition, affects multiple functions of isolated normal human lymphocytes and dendritic cells. Cancer immunology research. 2014; 2:351–60.
104. Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, Miller JP, Bassett RL, Gopalakrishnan V, Wani K, De Macedo MP, Austin-Breneman JL, Jiang H, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer discovery. 2016; 6:827–37.
105. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014; 515:568–71.
106. Huang RR, Jalil J, Economou JS, Chmielowski B, Koya RC, Mok S, Sazegar H, Seja E, Villanueva A, Gomez-Navarro J. CTLA4 blockade induces frequent tumor infiltration by activated lymphocytes regardless of clinical responses in humans. Clinical Cancer Research. 2011; 17:4101–9.
107. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS. Genetic basis for clinical response to CTLA-4 blockade in melanoma. New England Journal of Medicine. 2014; 371:2189–99.
108. Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, Chen L, Pardoll DM, Topalian SL, Anders RA. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti–PD-1 therapy. Clinical Cancer Research. 2014; 20:5064–74.
109. Ji RR, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, Alaparthy S, Berman D, Jure-Kunkel M, Siemers NO, Jackson JR, Shahabi V. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother. 2012; 61:1019–31.
110. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013; 39:1–10.
111. Schumacher TN, Kesmir C, van Buuren MM. Biomarkers in cancer immunotherapy. Cancer Cell. 2015; 27:12–4.
112. Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends in immunology. 2016; 37:193–207.
113. McMasters KM, Egger ME, Edwards MJ, Ross MI, Reintgen DS, Noyes RD, Martin RC, Goydos JS, Beitsch PD, Urist MM. Final results of the Sunbelt Melanoma Trial: a multi-institutional prospective randomized phase III study evaluating the role of adjuvant high-dose interferon alfa-2b and completion lymph node dissection for patients staged by sentinel lymph node biopsy. Journal of Clinical Oncology. 2016; 34:1079–86.
114. Kirkwood JM, Strawderman MH, Ernstoff MS, Smith TJ, Borden EC, Blum RH. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. Journal of clinical oncology. 1996; 14:7–17.
115. Kirkwood JM, Ibrahim JG, Sondak VK, Richards J, Flaherty LE, Ernstoff MS, Smith TJ, Rao U, Steele M, Blum RH. High-and low-dose interferon alfa-2b in high-risk melanoma: first analysis of intergroup trial E1690/S9111/C9190. Journal of clinical oncology. 2000; 18:2444–58.
116. Kirkwood JM, Ibrahim JG, Sosman JA, Sondak VK, Agarwala SS, Ernstoff MS, Rao U. High-dose interferon alfa-2b significantly prolongs relapse-free and overall survival compared with the GM2-KLH/QS-21 vaccine in patients with resected stage IIB-III melanoma: results of intergroup trial E1694/S9512/C509801. Journal of Clinical Oncology. 2001; 19:2370–80.
117. Mocellin S, Pasquali S, Rossi CR, Nitti D. Interferon alpha adjuvant therapy in patients with high-risk melanoma: a systematic review and meta-analysis. Journal of the National Cancer Institute. 2010; 102:493–501.
118. Eggermont AM, Suciu S, Testori A, Kruit WH, Marsden J, Punt CJ, Santinami M, Salès F, Schadendorf D, Patel P. Ulceration and stage are predictive of interferon efficacy in melanoma: results of the phase III adjuvant trials EORTC 18952 and EORTC 18991. European journal of cancer. 2012; 48:218–25.
119. National Library of Medicine (NLM) at the National Institutes of Health (NIH). ClinicalTrials.gov. 2017. https://clinicaltrials.gov/ct2/home (accessed June 1 2017).