
INTRODUCTION
Neuroblastoma is the most common extracranial solid tumor in children. Half of all patients are classified as high-risk and demonstrate a poor prognosis. Although intensive multimodal treatment, including high-dose chemotherapy, improves clinical outcomes in patients with high-risk neuroblastoma, many patients still experience treatment failure [1] and have limited treatment options after relapse. Furthermore, high-risk neuroblastoma survivors suffer numerous long-term complications from previous intensive treatments [1, 2]. Novel treatment approaches are needed both to improve patient survival and to reduce treatment toxicities. Therefore, an emergent unmet clinical need in neuroblastoma is accurate prediction of outcome to facilitate the development of individualized treatment protocols.
Genomic analyses for pediatric neuroblastoma have identified recurrent somatic mutations in cancer-related genes such as ALK, PTPN11, ATRX, MYCN, and NRAS [3]. A recent analysis on structural variants using whole genome sequencing revealed that TERT promoter rearrangements characterize a subgroup of high-risk neuroblastoma with poor prognosis comparable to MYCN amplified tumors [4, 5]. ARID1 alterations also reportedly predict poor outcome in patients with neuroblastoma [6].
In this study, we analyzed 72 cases of pediatric neuroblastoma with CancerSCAN™ (Supplementary Table 1) to find potential biomarkers to predict prognosis and identify patients likely to benefit from molecularly targeted therapies. CancerSCAN™ is a targeted deep sequencing panel and was developed mainly to identify genetic alterations for targeted therapy and the driver mutations of cancers.
RESULTS
Genomic profiling of neuroblastoma
Tumor samples from 72 children with neuroblastoma were analyzed using targeted panel sequencing. At least one mutation in one of the 83 genes of the panel was found in 63 of 72 patients (87.5%). Across 83 genes in 72 tumor samples, we detected 180 single nucleotide variants (SNVs) and short insertions/deletions (indels) and 25 copy number variants (CNVs) (Supplementary Table 2). The prevalence of SNVs/indels and CNVs for each gene is shown in Figure 1. Alterations in ALK were detected in 12 of 72 patients (16.7%). Nonetheless, we did not detect any sign of ALK translocation. The second most common sequence alterations were in BRCA1 (13.9%). Because BRCA1 is located on chromosome 17q, copy number gain was also detected with other genes located in 17q in patients with 17q gain. In addition, six SNV/indels in BRCA1 were detected with a range of allele frequency between 2.6~34.0% (Supplementary Table 2) and predicted to be deleterious in function. The prevalence of somatic mutation in BRCA1 remains to be elucidated in a larger study. Copy number loss in ATM was also associated with chromosome 11q deletion. In the ATRX gene, three novel missense mutations (A1988S, V2189A, and R498G) were detected. The mutation rate did not vary based on risk group (Supplementary Figure 1).
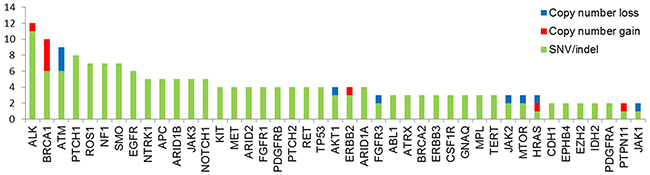
Figure 1: Mutation profiles of 72 patients with neuroblastoma. Data are included for nonsynonymous single nucleotide variants as well as small insertion and deletion (SNVs/indels), and copy number (CN) gain and loss. Genes with more than one genetic alteration were included.
Candidates for targeted therapies in neuroblastoma
Molecular target candidates for targeted therapy were detected in 16 of 72 patients (22.2%). Six cases with SNVs such as ALK R1275Q, F1174I, and R1192G, and one copy number gain in ALK could be potential candidates for ALK inhibitors [7–9]. Three R1275Q, an activating ALK mutation, were also confirmed with digital PCR method (Supplementary Figure 2) [10]. PARP inhibitors could be administered in 3 patients with BRCA1 truncating mutations and 3 patients with ATM copy number loss [11–13]. In addition, HRAS Q61R, MET exon14 skipping mutation, ERBB2 copy number gain, and STK11 copy number loss were each detected in one patient, respectively (Supplementary Table 3).
ARID1B and neuroblastoma
Five patients showed sequence alterations in ARID1B, consisting of four missense mutations and one small deletion (Figure 2). Sanger sequencing confirmed all four missense mutations but the small deletion. Three of the four missense mutations were predicted to be deleterious and the remaining one to be neutral. Protein expression was maintained in tissues with ARID1B mutations. Four of five patients with the ARID1B mutation belonged to the high-risk group (Figure 3). Four SNVs were detected in ARID1A in three patients. In the present study, there was no patient who have both sequence alterations in ARID1 and MYCN amplification (Figure 3). Only ARID1B gene mutation was associated with differential relapse-free survival (RFS) between patients with mutation and wild-type gene among genes listed in Figure 1. RFS at 3 years in patients with ARID1B mutations was lower than in those without (Figure 4A, p = 0.01). In the analysis of only high-risk patients, 3-year RFS in patients with (n = 4) and without ARID1B mutations (n = 27) was 37.5 ± 28.6% and 76.7 ± 10.2%, respectively (p = 0.25). Survival of patients whose tumors harbored ARID1B mutations, which was similar to that of patients with MYCN-amplified tumors, was significantly worse than that of high-risk patients with neither MYCN amplification nor ARID1B mutations (Figure 4B, p = 0.05). Median follow-up duration was 37 months in patients with ARID1B mutations, 20 months in patients with MYCN amplification, and 29 months in patients with neither MYCN amplification nor ARID1B mutations, respectively. Whereas three high-risk patients with relapsed tumors with neither MYCN amplification nor ARID1B mutations were rescued with salvage treatment after relapse, two relapsed patients with ARID1B mutations died of tumor progression.
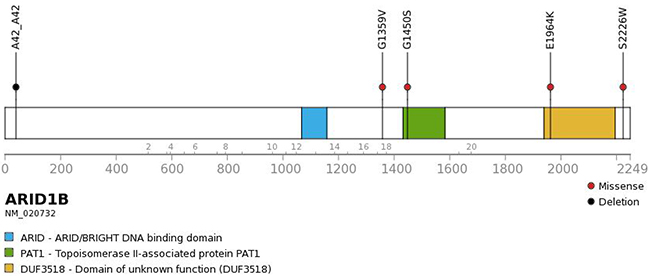
Figure 2: Genomic alterations affecting ARID1B. Four missense mutations and one deletion were found in ARID1B.
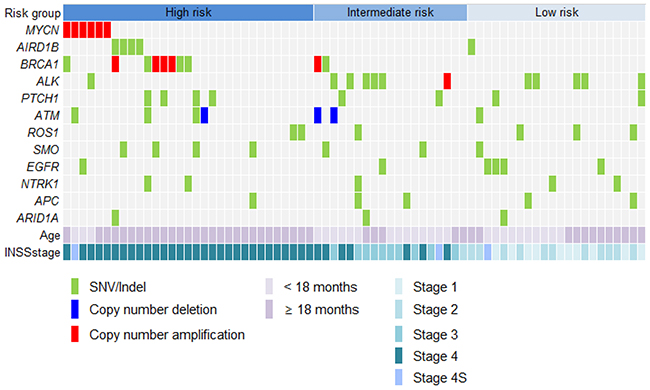
Figure 3: Targeted sequencing to identify genetic alterations. Four of five patients with ARID1B mutations were classified as high-risk. MYCN amplification was measured by fluorescence in-situ hybridization because MYCN was not included in the panel.
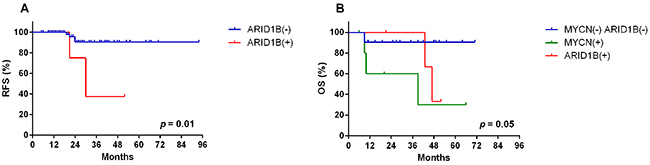
Figure 4: Relapse free survival and overall survival. (A) Three-year relapse-free survival (RFS) in patients with and without ARID1B mutations was 37.5 ± 28.6% and 90.5 ± 4.5%, respectively (p = 0.01) (n = 72). (B) In high-risk patients, the 3-year overall survival in patients with ARID1B mutations, with MYCN amplification, or with neither ARID1B mutation nor MYCN amplification were 33.3 ± 27.2%, 30.0 ± 23.9%, and 90.5 ± 6.4%, respectively (p = 0.05).
DISCUSSION
Although our approach is limited in its ability to detect alterations in non-coding regions or in genes outside our panel, the current panel covers genetic markers frequently involved in cancers and is sufficient to identify clinically relevant alterations in the context of targeted therapy in patients with neuroblastoma. ALK gene mutations were most frequent, as described in other studies [3, 14]. In addition, two thirds of ALK mutations were known to increase sensitivity to ALK inhibitors in the present study. In addition, we found druggable mutations in several other genes, including BRCA1, ATM, HRAS, MET, ERBB2, and STK11. Several targeted agents, including ALK inhibitors, are in clinical trials for treatment of neuroblastoma [15]. Recent studies for pediatric cancers demonstrate the promise of genome-guided therapy in children and adolescents although some challenges remain to be resolved such as rapid progression of disease and reemergence of resistance [16–18].
While mutation rates are higher in high-risk patients based on whole-genome sequencing, the mutation rate detected by panel sequencing was not different between risk groups [4]. The limited number of genes in the panel partly explains the results, however, involvement of major biologic pathways may also not vary between risk groups.
ARID1 genes are integral components of the SWI/SNF neural progenitor-specific chromatin-remodeling BAF complex essential for the self-renewal and differentiation of multipotent neural stem cells [19]. The chromatin remodeling function of the SWI/SNF complex is crucial for the gene expression program that converts precursor cells to their terminally differentiated counter parts [20]. AT-rich interacting domain (ARID) and domain of Unknown Function 3518 (DUF3518) in ARID1B bind to DNA and interact with the helicase subunits in the BAF complex, respectively [21–24]. Accordingly, missense mutations in these domains are likely to disrupt DNA-bind ability and interaction between ARID1B, BRG1, and BRM, which presumably compromise the function of BAF complexes and diminish their ability to regulate gene expression [25, 26]. Overall, the complex has been suggested to be an important tumor suppressor in human cancer. Tumor-specific deletions encompassing ARID1B have been reported in central nervous system tumors [27], and multiple members of this complex have been identified as tumor suppressor genes in ovarian cancer, pancreatic cancer, liver cancer, and neuroblastoma [6, 19, 28–30]. According to Sausen et al. [6], disrupted BAF complex signaling may preserve an undifferentiated progenitor state, leading them to suggest that alterations in ARID1 genes correlate with a more aggressive neuroblastoma phenotype. In the present study, four of five ARID1B mutations were found in high-risk patients, and two of them died due to progressive disease. Most studies about ARID1B-mediated disorders reported nonsense or frameshift mutations rather than missense mutation. However, this results were primarily from Coffin-Siris syndrome (CSS) cohorts and therefore might reflect ascertainment bias [26]. Most nonsense and frameshift mutations activate nonsense-mediated mRNA decay (NMD), therefore, these mutations are likely to cause NMD of the ARID1B transcript rather than the expression of the mutant ARID1B protein [31]. Truncating mutations that avoid NMD usually cause a distinct and more severe phenotype than that observed in NMD [32]. This finding suggests that there may be a different mutation pattern between CSS and cancers. TCGA data show that missense mutation is most frequent type of mutation in ARID1B in cancers. Although hemizygous deletion in ARID1B was the main type of mutation in neuroblastoma found in the previous study [6], all ARID1B changes were SNVs and small deletions in the present study. In recent study addressing the genomic landscape of schwannoma, 23% (23/99) of schwannoma harbored ARID1B mutations, in which only one mutation was frameshift deletion and others are missense mutations or non-frameshift indels [33]. The authors also proved maintenance of ARID1B expression in tissues with ARID1B mutation, which is probably due to remaining wild-type allele, therefore, suggest a haploinsufficient role of ARID1B in cancer. The effect of sequence alterations in the study need to be further validated. In addition, ARID1 mutations were not found in patients with MYCN amplification, which is different from the results of a previous study [6] in which samples harboring both MYCN amplification and ARID1 alteration were all of cell line origin. The further examination in a large population requires to validate the role of ARID1B mutation on prognosis and possible mutual exclusivity between MYCN and ARID1 in neuroblastoma patients.
One limitation of this study is the lack of normal pair samples. The sequence alterations detected in this study do not exclude the possibility of low prevalence individual SNPs. We attempted to exclude the possibility by eliminating all sequence alterations known to be DNA polymorphisms in databases (dbSNP, Clinvar, SNPeffect 4.0, ESP5400, 1000 Genomes, Exac03, Korean SNP DB, and EVS) and publications. However, validation is necessary to further confirm the functions of each variant.
In conclusion, our results add to the current body of knowledge regarding the genomic characteristics of neuroblastoma. Around 20% of children with neuroblastoma might benefit from targeted therapy. In addition, we identified a subgroup of neuroblastoma with ARID1B mutation shows an aggressive behavior. These findings may provide a new biomarker to identify another subgroup of neuroblastoma with high-risk features. A neuroblastoma-specific panel using NGS could be developed to identify more comprehensive genomic information to molecularly treat and predict prognosis with sophistication based on genomic characteristics such as MYCN, TERT, ATRX, ALK, ARID1, and telomere length. In the near future, the risk stratification of patients with neuroblastoma can be changed based on genetic characteristics.
MATERIALS AND METHODS
Patients
Neuroblastoma tumor samples from 72 children diagnosed between 2008 and 2015 were included in this analysis (Supplementary Table 4). The proportion of formalin-fixed paraffin embedded (FFPE) tissue and fresh frozen tissue were 48.6% and 51.4%, respectively. Only specimens with > 30% tumor were included and the purity of tumor was more than 60% in 70% of samples. The study was approved by the Institutional Review Board of Samsung Medical Center (2014-08-060). All participants provided written informed consents.
Deep sequencing using CancerSCAN™
Genomics DNA (250 ng) from each tissue was sheared in a Covaris S220 ultrasonicator (Covaris, Woburn MA, USA) and used for the construction of a library with CancerSCAN™ probes and a SureSelect XT reagent kit, HSQ (Agilent Technologies) according to the manufacturer’s protocol. This panel was designed to enrich exons of 83 genes [34] covering 366.2 kb of the human genome. After enriched exome libraries were multiplexed, the libraries were sequenced using the 100-bp paired-end mode of the TruSeq Rapid PE Cluster Kit and TruSeq Rapid SBS kit on the Illumina HiSeq 2500 sequencing platform (Illumina Inc., San Diego, CA, USA). The DNA sequence data were aligned to the human genome reference (hg19) using the MEM algorithm in BWA 0.7.5 [35]. Duplicate read removal was performed using Picard v.193 and SAMTOOLS v0.1.18 [36]. Local alignment was optimized using the Genome Analysis Toolkit (GATK) v3.1-1 [37]. We also used BaseRecalibrator from GATK for base recalibration based on known single nucleotide polymorphisms (SNPs) and indels from Mills, dbSNP138, and 1000G gold standard, 1000G phase1 and Omni 2.5. Sequencing coverage is shown in Supplementary Table 5.
Bioinformatic analysis
Variant calling was done only in regions targeted in CancerSCAN™ v1. We detected SNVs using three tools: MuTect 1.1.4, LoFreq 0.6.1, and SNVer 0.5.3 [38–40]. We filtered out falsely detected variants from abnormally aligned strand biased and clustered reads by in-house developed scripts. ANNOVAR was used for annotating the detected variants by diverse resources including dbSNP138, COSMIC, TCGA, EPS5400, 1000 Genomes, Exac03, and the in-house Korean SNP DB. Indels were detected by Pindel 0.2.4 [41] and annotated by ANNOVAR. To filter out germline variants, we applied two algorithms: i) Except for hotspot mutations, variants with an allele frequency greater than or equal to 97% were filtered out; ii) Suspect germline variants were filtered out based on whether the allele frequency was ≥ 1% of previously mentioned database or ≥ 3% of 480 samples from healthy Korean subjects.
We used CancerSCAN™ software (unpublished) to detect CNVs. In CancerSCAN™, the software ‘Depth of Coverage’ in GATK v3.1-1 was used to calculate sequencing coverage in each exon. The median of mean coverage for total exons was calculated and divided by the gained average for normalization of mean coverage of exons. The median value of normalized exons of pattern matched normal reference datasets (HapMap cell lines, FFPE tissues from normal patients) was used as a reference value, and then the normalized median value of exons of each patient was divided by the reference value and transformed to the binary logarithm, respectively. Tumor purity for adjusting CNVs was calculated using normalized coverage and B allele frequencies at all exons. Using normal samples, coverage of sample was fitted and after that major ploidies and copy number in the neutral region were decided. Based on the inferred copy neutral region and major ploidies, tumor purity was calculated using the relationship between B allele frequencies and tumor ploidy data. Finally, we defined ‘Copy number loss’ as when the copy number was less than one and ‘Copy number gain’ as when the copy number was more than four using the above method.
MYCN amplification
Paraffin sections of each tumor were hybridized for interphase FISH studies, using commercial probes: Vysis LSI MYCN (2p24)/ CEP 2 (2p11.1-q11.1) (Abbott Molecular, Abbott Park, IL, USA). DAPI was used to counterstain nuclei. The stained sections were examined by a microscope equipped with fluorescence filters (BX51, Olympus, Tokyo, Japan). The fluorescent signals of MYCN (green) and CEP2 (orange) were counted in fifty non-overlapping nuclei of the tumor cells. MYCN amplification was defined as a 10-fold increase of the MYCN signal number compared to the reference probe located on chromosome centromere 2.
Statistical analysis
For two group comparisons, Student’s t-test was used to determine differences between mean values for normal distribution. Survival analysis was performed using the Kaplan-Meier method and survival differences between groups were examined by log-rank test. All data were analyzed for significance using GraphPadPrism 6 software (San Diego, CA, USA).
URLs
dbSNP, www.ncbi.nim.nih.gov/SNP/; EVS, http://evs.gs.washington.edu/EVS/; ClinVar, www.ncbi.nih.gov/clinvar/; SNPeffct 4.0, http://snpeffect.switchlab.org/
ACKNOWLEDGMENTS
The authors would like to thank their patients for participating and providing samples for this research. The authors also would like to thank So Yeon Lee and Soo Jin Park for assistance with sample acquisition and informed consent.
CONFLICTS OF INTEREST
There are no potential conflicts of interest to disclose.
GRANT SUPPORT
This work was supported by a grant from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI13C2096) and the National R&D Program for Cancer Control, Ministry for Health and Welfare, Republic of Korea (1520210).
REFERENCES
1. Sung KW, Son MH, Lee SH, Yoo KH, Koo HH, Kim JY, Cho EJ, Lee SK, Choi YS, Lim DH, Kim JS, Kim DW. Tandem high-dose chemotherapy and autologous stem cell transplantation in patients with high-risk neuroblastoma: results of SMC NB-2004 study. Bone Marrow Transplant. 2013; 48: 68-73. doi: 10.1038/bmt.2012.86.
2. Perwein T, Lackner H, Sovinz P, Benesch M, Schmidt S, Schwinger W, Urban C. Survival and late effects in children with stage 4 neuroblastoma. Pediatr Blood Cancer. 2011; 57: 629-35. doi: 10.1002/pbc.23036.
3. Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, Kim J, Lawrence MS, Lichenstein L, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013; 45: 279-84. doi: 10.1038/ng.2529.
4. Peifer M, Hertwig F, Roels F, Dreidax D, Gartlgruber M, Menon R, Kramer A, Roncaioli JL, Sand F, Heuckmann JM, Ikram F, Schmidt R, Ackermann S, et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature. 2015; 526: 700-4. doi: 10.1038/nature14980.
5. Valentijn LJ, Koster J, Zwijnenburg DA, Hasselt NE, van Sluis P, Volckmann R, van Noesel MM, George RE, Tytgat GA, Molenaar JJ, Versteeg R. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat Genet. 2015; 47: 1411-4. doi: 10.1038/ng.3438.
6. Sausen M, Leary RJ, Jones S, Wu J, Reynolds CP, Liu X, Blackford A, Parmigiani G, Diaz LA Jr, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE, et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet. 2013; 45: 12-7. doi: 10.1038/ng.2493.
7. Bresler SC, Wood AC, Haglund EA, Courtright J, Belcastro LT, Plegaria JS, Cole K, Toporovskaya Y, Zhao H, Carpenter EL, Christensen JG, Maris JM, Lemmon MA, et al. Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma. Sci Transl Med. 2011; 3: 108ra14. doi: 10.1126/scitranslmed.3002950.
8. Schonherr C, Ruuth K, Yamazaki Y, Eriksson T, Christensen J, Palmer RH, Hallberg B. Activating ALK mutations found in neuroblastoma are inhibited by Crizotinib and NVP-TAE684. Biochem J. 2011; 440: 405-13. doi: 10.1042/BJ20101796.
9. Togashi Y, Mizuuchi H, Kobayashi Y, Hayashi H, Terashima M, Sakai K, Banno E, Mizukami T, Nakamura Y, de Velasco MA, Fujita Y, Tomida S, Mitsudomi T, et al. An activating ALK gene mutation in ALK IHC-positive/FISH-negative nonsmall-cell lung cancer. Ann Oncol. 2015; 26: 1800-1. doi: 10.1093/annonc/mdv240.
10. Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, Kitano TK, Hodel MR, Petersen JF, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011; 83: 8604-10. doi: 10.1021/ac202028g.
11. Dedes KJ, Wilkerson PM, Wetterskog D, Weigelt B, Ashworth A, Reis-Filho JS. Synthetic lethality of PARP inhibition in cancers lacking BRCA1 and BRCA2 mutations. Cell Cycle. 2011; 10: 1192-9. doi: 10.4161/cc.10.8.15273.
12. Bouwman P, Jonkers J. Molecular pathways: how can BRCA-mutated tumors become resistant to PARP inhibitors? Clin Cancer Res. 2014; 20: 540-7. doi: 10.1158/1078-0432.CCR-13-0225.
13. Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, Mitchell G, Fried G, Stemmer SM, Hubert A, Rosengarten O, Steiner M, Loman N, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015; 33: 244-50. doi: 10.1200/JCO.2014.56.2728.
14. Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, Nakagawara A, Hayashi Y, Mano H, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008; 455: 971-4. doi: 10.1038/nature07399.
15. Schulte JH, Schulte S, Heukamp LC, Astrahantseff K, Stephan H, Fischer M, Schramm A, Eggert A. Targeted therapy for neuroblastoma: ALK inhibitors. Klin Padiatr. 2013; 225: 303-8. doi: 10.1055/s-0033-1357132.
16. Parsons DW, Roy A, Yang Y, Wang T, Scollon S, Bergstrom K, Kerstein RA, Gutierrez S, Petersen AK, Bavle A, Lin FY, Lopez-Terrada DH, Monzon FA, et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol. 2016 Jan 28. doi: 10.1001/jamaoncol.2015.5699. [Epub ahead of print].
17. Harris MH, DuBois SG, Glade Bender JL, Kim A, Crompton BD, Parker E, Dumont IP, Hong AL, Guo D, Church A, Stegmaier K, Roberts CW, Shusterman S, et al. Multicenter feasibility study of tumor molecular profiling to inform therapeutic decisions in advanced pediatric solid tumors: the Individualized Cancer Therapy (iCat) Study. JAMA Oncol. 2016 Jan 28. doi: 10.1001/jamaoncol.2015.5689. [Epub ahead of print].
18. Mody RJ, Wu YM, Lonigro RJ, Cao X, Roychowdhury S, Vats P, Frank KM, Prensner JR, Asangani I, Palanisamy N, Dillman JR, Rabah RM, Kunju LP, et al. Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA. 2015; 314: 913-25. doi: 10.1001/jama.2015.10080.
19. Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010; 463: 474-84. doi: 10.1038/nature08911.
20. Xu F, Flowers S, Moran E. Essential role of ARID2 protein-containing SWI/SNF complex in tissue-specific gene expression. J Biol Chem. 2012; 287: 5033-41. doi: 10.1074/jbc.M111.279968.
21. Patsialou A, Wilsker D, Moran E. DNA-binding properties of ARID family proteins. Nucleic Acids Res. 2005; 33: 66-80. doi: 10.1093/nar/gki145.
22. Wang X, Nagl NG, Wilsker D, Van Scoy M, Pacchione S, Yaciuk P, Dallas PB, Moran E. Two related ARID family proteins are alternative subunits of human SWI/SNF complexes. Biochem J. 2004; 383: 319-25. doi: 10.1042/BJ20040524.
23. Inoue H, Furukawa T, Giannakopoulos S, Zhou S, King DS, Tanese N. Largest subunits of the human SWI/SNF chromatin-remodeling complex promote transcriptional activation by steroid hormone receptors. J Biol Chem. 2002; 277: 41674-85. doi: 10.1074/jbc.M205961200.
24. Hurlstone AF, Olave IA, Barker N, van Noort M, Clevers H. Cloning and characterization of hELD/OSA1, a novel BRG1 interacting protein. Biochem J. 2002; 364: 255-64. doi: 10.1042/bj3640255.
25. Cajuso T, Hanninen UA, Kondelin J, Gylfe AE, Tanskanen T, Katainen R, Pitkanen E, Ristolainen H, Kaasinen E, Taipale M, Taipale J, Bohm J, Renkonen-Sinisalo L, et al. Exome sequencing reveals frequent inactivating mutations in ARID1A, ARID1B, ARID2 and ARID4A in microsatellite unstable colorectal cancer. Int J Cancer. 2014; 135: 611-23. doi: 10.1002/ijc.28705.
26. Sim JC, White SM, Lockhart PJ. ARID1B-mediated disorders: mutations and possible mechanisms. Intractable Rare Dis Res. 2015; 4: 17-23. doi: 10.5582/irdr.2014.01021.
27. Ichimura K, Mungall AJ, Fiegler H, Pearson DM, Dunham I, Carter NP, Collins VP. Small regions of overlapping deletions on 6q26 in human astrocytic tumours identified using chromosome 6 tile path array-CGH. Oncogene. 2006; 25: 1261-71. doi: 10.1038/sj.onc.1209156.
28. Jones S, Wang TL, Shih Ie M, Mao TL, Nakayama K, Roden R, Glas R, Slamon D, Diaz LA Jr, Vogelstein B, Kinzler KW, Velculescu VE, Papadopoulos N. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010; 330: 228-31. doi: 10.1126/science.1196333.
29. Shain AH, Pollack JR. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS One. 2013; 8: e55119. doi: 10.1371/journal.pone.0055119.
30. Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, Arai Y, Takahashi H, Shirakihara T, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012; 44: 760-4. doi: 10.1038/ng.2291.
31. Khajavi M, Inoue K, Lupski JR. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur J Hum Genet. 2006; 14: 1074-81. doi: 10.1038/sj.ejhg.5201649.
32. Inoue K, Khajavi M, Ohyama T, Hirabayashi S, Wilson J, Reggin JD, Mancias P, Butler IJ, Wilkinson MF, Wegner M, Lupski JR. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat Genet. 2004; 36: 361-9. doi: 10.1038/ng1322.
33. Agnihotri S, Jalali S, Wilson MR, Danesh A, Li M, Klironomos G, Krieger JR, Mansouri A, Khan O, Mamatjan Y, Landon-Brace N, Tung T, Dowar M, et al. The genomic landscape of schwannoma. Nat Genet. 2016; 48: 1339-48. doi: 10.1038/ng.3688.
34. Park YH, Shin HT, Jung HH, Choi YL, Ahn T, Park K, Lee A, Do IG, Kim JY, Ahn JS, Park WY, Im YH. Role of HER2 mutations in refractory metastatic breast cancers: targeted sequencing results in patients with refractory breast cancer. Oncotarget. 2015; 6: 32027-38. doi: 10.18632/oncotarget.5184.
35. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010; 26: 589-95. doi: 10.1093/bioinformatics/btp698.
36. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25: 2078-9. doi: 10.1093/bioinformatics/btp352.
37. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20: 1297-303. doi: 10.1101/gr.107524.110.
38. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013; 31: 213-9. doi: 10.1038/nbt.2514.
39. Wilm A, Aw PP, Bertrand D, Yeo GH, Ong SH, Wong CH, Khor CC, Petric R, Hibberd ML, Nagarajan N. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012; 40: 11189-201. doi: 10.1093/nar/gks918.
40. Wei Z, Wang W, Hu P, Lyon GJ, Hakonarson H. SNVer: a statistical tool for variant calling in analysis of pooled or individual next-generation sequencing data. Nucleic Acids Res. 2011; 39: e132. doi: 10.1093/nar/gkr599.
41. Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009; 25: 2865-71. doi: 10.1093/bioinformatics/btp394.