
INTRODUCTION
Hepatocellular carcinoma (HCC) is the most common form of liver cancer (70%–90%). As the 2nd leading cause of global cancer mortality, HCC endangers over 780,000 new patients per year [1]. The etiological factors of HCC consist of, but not limited to, viral infections (hepatitis B and C viruses), alcohol intake, smoking, and many host factors such like cirrhosis, hemochromatosis, non-alcoholic steatohepatitis as well as diabetes [1–4]. Current options for HCC treatment include hepatectomy, transarterial chemoembolization, thermal or chemical ablation, liver transplantation, radiation and chemotherapy [4–6]. However, HCC behaves highly refractory to most anti-cancer therapies. Five year survival rate of HCC patients remains dismal [7].
HCC appears frequently as multiple nodules which are resulted from either intrahepatic metastasis or independent multicentric development [8]. Albeit both normal liver and HCC are highly vascularized, rapid growth of tumor cells within these nodules scavenges a substantial amount of oxygen, therefore often producing a hypoxic microenvironment. Indeed, HCC is one of the most hypoxic tumors with median oxygen level as low as 0.8% [9]. Inadequate intratumoral oxygen level is known to trigger a vast array of molecular and cellular responses which will influence tumor aggressiveness and therapeutic response. Hypoxia inducible factors (HIFs) are critical to sense intratumoral oxygen tension and mediate subsequently the activation of hypoxia response, thus representing as potential anti-cancer targets [10].
In this review, we are going to discuss the functional relevance of hypoxia and HIFs in HCC, and summarize recent progresses in therapeutic targeting of hypoxia pathway in this deadly disease.
Hypoxia signaling pathway
HIF system is implemented in hypoxia-responsive pathway. This system is composed of α-subunits (HIFα, including HIF1α, HIF2α/EPAS1 and HIF3α) and β-subunits (HIFβ, including HIF1β/ARNT1, ARNT2 and ARNT3). Among these proteins, the function and activity of HIF1α, HIF2α and HIF1β are relatively well-studied. Under normoxia (normal oxygen supply), HIFα is constitutively degraded and maintained at very low basal activities. Prolyl hydroxylation of HIFα (e.g. Pro 402 and 564 in human HIF1α) by prolyl hydroxylase domain-containing proteins (PHD1, PHD2 and PHD3) potentiates its subsequent recognition, ubiquitination and proteasomal degradation by an E3 ligase, von Hippel-Lindau tumor suppressor protein (pVHL). Moreover, asparaginyl hydroxylation of HIFα (e.g. Asn 803 in human HIF1α) by factor inhibiting HIF (FIH) blocks its interaction with transcriptional co-activators, CREB-binding protein (CBP) and p300 [11, 12]. Under hypoxia, hydroxylation and proteasomal degradation of HIFα are impaired due to lack of sufficient oxygen. Stabilized HIFα is then translocates into nucleus, hetero-dimerizes with HIFβ and binds core hypoxia-response element (HRE, 5′-(A/G)CGTG-3′) [13] (Figure 1). HIF1α binds preferentially to permissive chromatins where are positive for histone acetylation, H3K4me3, BRD4 and RNA-Pol2 signals [14, 15]. HIF1α utilizes CDK8-Mediator for its interaction with super-elongation-complex, thus activating the paused RNA-Pol2 and elevating the expression of HIF target genes [14]. Recently, Perez-Perri JI, et al. identified TIP60 complex as an additional co-activator to facilitate HIF1α-dependent chromatin modification and RNA-Pol2 activation [16]. Moreover, the activation of HIF1α transcriptional potency is associated with its interaction with CH1 domains of CBP and p300 [17, 18]. Loss of these CH1 domains in mouse embryonic fibroblasts showed deficiency of CBP or p300 loading on HIF targeted genes and affected the expression of 35%–50% HIF responsive genes [19]. Interestingly, while PHD proteins-dependent hydroxylation primes HIFα for pVHL-mediated destruction, HIF1α induces pVHL, PHD2, and PHD3, suggesting a feedback regulatory mechanism [20–26].
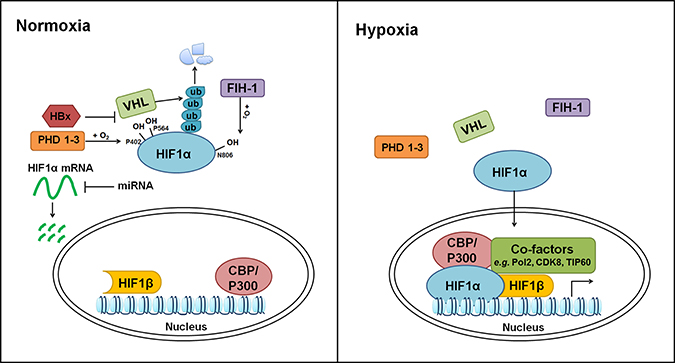
Figure 1: Regulation of hypoxia pathway. HIF1α and HIF1β are used as examples. At post-transcriptional level, HIF1α mRNA is repressed by miR-199a-5p, miR-338-3p, miR-93 and miR-122. Under normoxia, HIF1α protein is hydroxylated at P402 and P564 by PHDs and subsequently degraded by pVHL through the Ubiquitin–Proteasome Pathway. Additionally, asparaginyl hydroxylation of HIF1α by FIH-1 at N806 impairs its interaction with CBP/P300. Hypoxia blocks the hydroxylation and proteasomal degradation of HIF1α, leading to its stabilization and nuclear translocation. Within nucleus, HIF1α forms heterodimer with HIF1β, and activate the expression of hypoxia responsive genes with the help from additional transcriptional co-factors, such as CBP/P300, Pol2, CDK8, and TIP60.
HIF activation has been associated with transcriptional induction of its target genes in most studies. However, using mouse hepatoma cells, Johnson AB. et al. reported that hypoxia may induce a general transcriptional repression via altering histone modifications [27]. To date, although transcriptome analysis of hypoxia response in HCC tissues/cells is limited, accumulating evidences support that hypoxia regulates cancer epigenetics. In HepG2, a human HCC cell line, hypoxia increased overall methylation of H3K4, H3K9 and H3K27. Meanwhile, several jumonji-domain histone demethylases, such as JMJD1A, JMJD2B and JMJD2C, were identified as HIF1α targets and were highly induced under hypoxia. The upregulation of histone demethylases may serve as an adaptive strategy to compensate the hypoxic stress and maintain the methylation homeostasis in HCC cells [28]. In parallel to histone modification, hypoxia also downregulated the overall level of 5-hydroxymethylcytosine (5-hmC) in HepG2 and Hep3B cells [29]. Since the Tet methylcytosine dioxygenase (TET) proteins convert 5-methylcytosine (5-mC) to 5-hmC, it would be interesting to study the connections among hypoxia, TET proteins and 5-hmC in HCC.
Expression of HIFs in normal liver and HCC
In murine liver and hepatoma cells, Hif1a transcript containing exon 1.2 but not exon 1.1 was selectively expressed [30]. Diethyl nitrosamine (DEN) and palmitic acid elevate the transcription of HIF1α in primary hepatocytes [31]. In murine liver, nuclear HIF1α can be detected in normal hepatocytes under normoxic condition, and protein levels of HIF1α, HIF2α and HIF1β are hypoxia-inducible [32, 33]. The induction of HIF1α protein was also seen in the liver of Hepatitis B virus X (HBx) transgenic mice [34].
In human HCC samples, the protein level of HIF1α is significantly elevated and associated with worse prognosis [35–37]. Moreover, HIF1α expression in primary HCC tumors is an independent prognosis factor for overall survival of patients after receiving abdominal metastatic lymph node external beam radiotherapy [38]. However, the expression of HIF1α mRNA, HIF2α protein and HIF3α protein shows some variation [37, 39, 40] (Table 1).
Table 1: Expression of HIFs in HCC and their association with clinical outcomes
Gene |
Expression |
Prognostic potential |
Reference |
|---|---|---|---|
HIF1α |
Predominantly expressed in tumor tissues (72/126) while less stained in peritumoral tissues (7/126) |
Positively associated with worse disease-free survival and overall survival |
[35] |
Highly expressed at both mRNA (42/110) and protein (39/110) levels |
Positively associated with worse disease-free survival and overall survival after surgery |
[37] |
|
Positively associated with HBx protein in HCC samples |
Positively associated with worse disease-free survival and overall survival after surgery |
[41] |
|
Positive in HCC samples (32/60) |
Positively associated with shorter disease-free survival |
[42] |
|
Positive in HCC samples (212/406) |
Positively associated with higher probability of disease recurrence and worse overall survival after surgery. |
[43] |
|
Highly expressed in 30/69 HCC samples (11/30 samples show nuclear staining) |
Positively associated with the responses of abdominal metastatic lymph nodes to external beam radiotherapy, local recurrence and cancer-specific deaths |
[38] |
|
HIF2α |
Expressed in peritumoral regions (60/126) while less expressed in tumor tissues (17/126) |
No correlation |
[35] |
Positive in HCC and adjacent noncancerous tissues |
Positively associated with shorter overall survival |
[36] |
|
Lower in HCC on average |
Negatively associated with worse survival |
[44] |
|
HIF3α |
Inconsistently expressed between HCC and adjacent tissues |
No correlation |
[45] |
The expression of HIFs is governed by both transcriptional and post-transcriptional mechanisms. In HCC cells, NF-κB subunits p50 and p65, but not c-Rel, bound the HIF1α promoter and elevated HIF1α transcription [46]. In addition to well-established oxygen-sensitive regulation of HIFs, HBx stabilized HIF1α through inhibiting its binding with pVHL [47]. According to recent studies, HBx protein also bridged the interaction between HIF1α and MTA1/HDAC complex, leading to deacetylation of HIF1α. The deacetylation of HIF1α interfered with its binding to PHDs and VHL, and subsequently stabilized/activated HIF1α protein in HBV-associated HCC cells [34, 41]. Furthermore, microRNA network appears as a new regulatory layer of HIF turnover (see review [48]). For example, miR-199a-5p, miR-338-3p, miR-93 and miR-122 have been shown to repress the expression of HIF1α in HCC cells [46, 49–51] (Figure 1).
Functional relevance of HIFs in HCC
Cell line models of both human HCC and murine hepatoma origins have been applied directly to evaluate the function of HIFs in HCC. In human HepG2 and SK-Hep-1 HCC cells, HIF1α silencing strongly inhibited their anchorage independent growth, but did not affect their basal proliferation [52]. siRNA-mediated knockdown of HIF2α in HepG2 cells impaired cell cycle progression in the presence of CoCl2 and reduced cell proliferation both in vitro and in vivo [53]. In spheroid culture condition, dual-silencing of HIF1α and HIF2α diminished the growth of HepG2 cells, whereas knockdown of either HIF1α or HIF2α increased spheroid size and decreased caspase-3 activity [54]. Rat hepatoma cells with deficiency in HIF1β subunit showed much reduced tumorigenic ability in athymic mice, when compared with their wildtype counterparts [55].
Genetic data from mouse models also support the functional importance of of HIFs in HCC (Table 2). By using liver-specific transposon-based insertional mutagenesis method, Hif1a genomic region was identified with recurrent insertions in murine HCC models [56], indicating the potential involvement of aberrant HIF1α expression in HCC development. Knockout of Hif1a sensitized hepatoma cells to etoposide treatment in a transgenic murine model with hepatocyte-specific expression of SV40 large T oncogene, but did not affect the initiation and progression of murine HCCs [52]. Interestingly, liver-specific HIF1α overexpressing potentiated the development of HCC-promoting M2 macrophages [31]. Besides, myeloid HIF2α appeared to be required for liver tumor progression. Mice lacking HIF2α in myeloid cells showed decreased infiltration of tumor-associated macrophages in HCC and delayed tumor progression [57].
Table 2: Functional study of HIFs in mouse HCC models
Approach |
Genetic background |
Cells |
Results |
Reference |
|---|---|---|---|---|
Transposon-based insertional mutagenesis |
Mixed |
Hepatocyte |
Recurrent insertions in Hif1a genomic region |
[56] |
Hif1α knockout |
C57Bl/6J |
Hepatoma cells with SV40 large T antigen expression |
Sensitized the cells to etoposide treatment |
[52] |
HIF1α overexpression |
C57BL/6 |
Hepatocyte |
Increased percentage of M2 macrophages |
[31] |
HIF2α deficiency |
Mixed |
Myeloid cell |
Decreased infiltration of TAM in diethylnitrosamine (DEN) induced hepatocellular carcinoma |
[57] |
HIF1α has been shown to promote HCV replication in hepatocytes and to potentiate the migration of hepatoma cells [58]. HIF-induced VEGF expression promoted HCV entry by causing depolarization and reducing tight junction in hepatocytes [59, 60].
HIF target genes in HCC and their functional contributions
To date, albeit the direct genetic evidences supporting the involvement of HIFs await further characterization, a growing body of literatures has reported the identification and function of HIF target genes in HCC. The functional relevance of hypoxia/HIF target genes have been implicated in most cancer hallmarks (Figure 2).
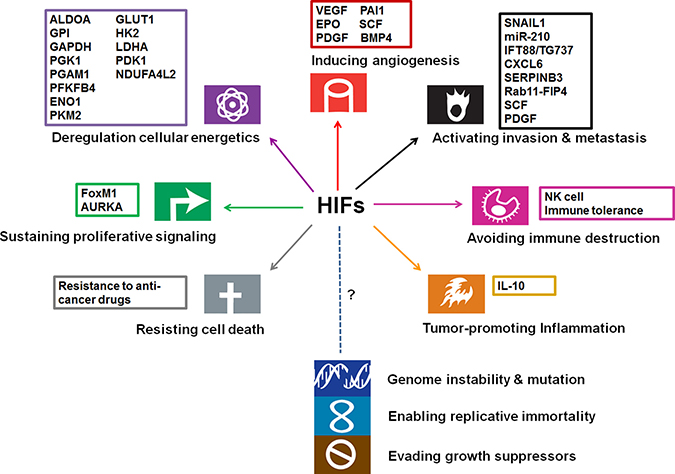
Figure 2: Involvement of HIFs and their targets in cancer hallmarks (modified from the original figure from Hanahan and Weinberg [113]). The function of HIFs has been implicated in promoting angiogenesis, invasion/metastasis, proliferation, glycolysis, therapeutic resistance, inflammation, and immune evasion.
Angiogenesis
HCC is highly vascularized as a result of up-regulation of angiogenic factors, such as VEGF, bone morphogenetic protein 4 (BMP4), plasminogen activator inhibitor-1 (PAI-1), and stem cell factor (SCF) [61–65]. As a well-characterized direct target of HIFs, VEGF stimulated the growth and migration of endothelial cells and was required for blood vessel formation [66–69]. In line with the observation that both mRNA and protein levels of VEGF were significantly induced by hypoxia in HCC cells, high VEGF expression was evident near necrotic/hypoxic regions in primary HCC samples [70]. Elevated expression of VEGF in HCC samples was correlated with early relapse and shorter survival [71, 72]. Interestingly, HIF1β only contributed partially to the hypoxic induction of VEGF in murine hepatoma cells [73], suggesting that hypoxia may employ additionally a HIF1β-independent mechanism to promote VEGF expression. Xiao H, et al. recently reported that H2AX phosphorylation (γ-H2AX) was required for EGFR- and HIF1α-mediated VEGF induction under hypoxia [74]. Combined evaluation of γ-H2AX, HIF1α and EGFR showed a powerful prognostic value for HCC after liver transplantation. In addition, hypoxia has been shown to induce HIF1α-dependent expression of BMP4 and HIF2α-dependent expression of PAI-1 and SCF to enhance HCC angiogenesis. Of note, despite lack of direct experimental evidence, hypoxia/HIFs-stimulated expression of erythropoietin (EPO) and platelet-derived growth factor (PDGF) may also promote HCC angiogenesis [75–78]. Therefore, HIF-mediated proangiogenic phenotypes greatly contribute to HCC angiogenesis.
Metabolism
HIFs actively regulate a series of glycolytic genes to promote glycolysis and to favor tumor cell adaption to hypoxic stress. Many critical enzymes involved in glycolysis have been shown to be direct HIF1α targets in HCC cells, including ALDOA, GPI, GAPDH, HK2, LDHA, PGK1, PGAM1, PFKFB4, ENO1 and PKM2 [28, 79]. Moreover, glucose transporter 1 (GLUT1), responsible for cellular glucose uptake, was directly upregulated by HIF1α [80] and highly expressed in HCC cells and patient samples when compared with primary hepatocytes. High expression of GLUT1 was also associated with enhanced proliferation, poor differentiation and advanced histological stages [81]. Hexokinase 2 (HK2) and lactate dehydrogenase A (LDHA), both of which enhance glycolytic switch from glucose to pyruvate, are also direct targets of HIFs [82–84]. Moreover, HIF1α was required for the expression of pyruvate dehydrogenase kinase 1 (PDK1) which suppresses the tricarboxylic acid cycle. Ectopic PDK1 expression not only rescued the hypoxia-induced cell apoptosis, but also reduced hypoxic ROS production and restored the ATP production in HIF1α-deficient cells [85]. NDUFA4L2 was also induced by hypoxia and HIF1α. Overexpression of NDUFA4L2 was strongly associated with tumor microsatellite formation, absence of tumor encapsulation, and poor overall survival in HCC patients. Inhibition of HIF1α/NDUFA4L2 enhanced oxygen consumption and mitochondrial activity, resulting in ROS accumulation and apoptotic cell death. Depletion of NDUFA4L2 suppressed HCC xenograft growth and metastasis [86]. Interestingly, in addition to oxygen, 2-oxoglutarate, Fe (II) and ascorbate are required for proper hydroxylation of HIFα by hydroxylase, linking HIF activation to metabolic stress responses (see review [87]).
Migration, invasion and metastasis
In HCC cells, epithelial mesenchymal transition (EMT) can be induced under hypoxia condition through the activation of Wnt/β-catenin pathway or PI3K/AKT pathway [88–90]. SNAIL1, which harbors two HREs in its promoter, can be greatly induced by HIF1α under hypoxia [91]. Hypoxia-induced miR-210 was associated with HCC cell invasion and migration through downregulating vacuole membrane protein 1 (VMP1) [92]. Hypoxia also led to the downregulation of IFT88/TG737 and promoted cell migration and invasion, partially through IFT88-mediated effects on the expression of polycystin-1, IL-8, and TGF-β1 [93]. Additional factors such as CXCL6 (HIF1α target) and SERPINB3 (HIF2α target) can promote the migratory and metastatic potentials of HCC cells [40, 94]. Moreover, Rab11-FIP4 (HIF1α target) and SCF(HIF2α target), both of which were positively associated with worse survival of HCC patients, can promote HCC cell migration and invasion in vitro and metastasis in vivo [63, 95, 96].
Tumor microenvironment and tumor stromal cells
Hypoxic microenvironment remodels the tumor-stromal interactions in HCC. Monocytes/macrophages were recruited to the hypoxic regions of tumor tissues and subsequently up-regulated TIE2 expression [97]. These TIE2-positive monocytes/macrophages (CD14+CD16+) were proangiogenic, and their frequency in either blood or tumors correlated significantly with microvessel density in HCC [98, 99]. Moreover, hypoxia has been shown to accelerate murine HCC development by HIF1α-induced expression of IL-10 which favored the intratumoral macrophage converting from M1 to M2 type [31]. Cross-talk between hepatocyte-hepatic satellite cells also generated a proangiogenic and proinflammatory microenvironment [100]. Hypoxia-induced up-regulation of PDGF-BB in hepatic satellite cells activated the PI3K/Akt pathway in HCC cells and enhanced cell proliferation, migration, and resistance to bile acid-induced apoptosis [101].
Cell proliferation, survival and drug/therapy response
HIF1α can directly enhance the expression of crucial oncogenes involved in cell replication in HCC, such as FoxM1 and AURKA [43, 102]. Moreover, hypoxia confers resistance to various anticancer drugs in HCC cells, including etoposide, sorafenib, SN38, cisplatin and doxorubicin [52, 78, 103–107]. Sorafenib resistant HCC patients showed higher intratumoral hypoxia [108]. Hypoxia-activated YAP and TGF-α/EGFR pathways blunted the response of HCC cells to sorafenib. Sorafenib inhibited HIF1α synthesis whereas up-regulated the expression of HIF2α, shifting hypoxic responses from HIF1α- to HIF2α-dependent pathways. Silencing of HIF2α synergized with the sorafenib to block the proliferation of HCC cells under hypoxia and the growth of xenograft tumors [109]. In addition, hypoxia-induced NDRG-1 and CBR1 can render HCC cells resistance to doxorubicin [106, 107]. Silencing of either HIF1α or HIF2α has been shown to improve the efficacy of doxorubicin in HCC models by inhibiting cell proliferation, tumor angiogenesis and enhancing cell apoptosis [53, 110]. Besides, HIF1β-deficient murine hepatoma cells were more sensitive to radiotherapy [111], suggesting that hypoxia activation may protect tumor cells from radiation. Furthermore, intratumoral gene transfer of antisense HIF1α triggered a NK cell-dependent rejection of small (0.1 cm in diameter) EL-4 tumors in mice. Combination of antisense HIF1α and B7-1-mediated immunotherapy led to a strong synergistic effect in inducing NK cell- and CD8 T cell-dependent rejection of larger EL-4 tumors (0.4 cm in diameter) [112], highlighting the promise of targeting hypoxia pathway together with cancer immunotherapy in HCC treatment.
Therapeutic targeting of hypoxia pathway in HCC
Tremendous efforts have been made to develop effective inhibitors for HCC treatment. However, to date, sorafenib is the only FDA-approved first-line drug for advanced HCC treatment [114]. Targeted therapy for HCC is still very limited.
Given the functional importance of hypoxia pathway in HCC, targeting hypoxia related molecules may be beneficial (Table 3). Recently, several inhibitors targeting hypoxia downstream signaling (e.g. VEGF-VEGFR system) have entered phase 3 clinical trials for HCC treatment. Alternatively, the strategy of targeting HIF expression or activation is actively tested in either preclinical studies or in trials. RO7070179 and EZN-2968 are oligonucleotide inhibitors which inhibit the synthesis of HIF1α [115]. Since HIF1α translation is dependent on PI3K-AKT-mTOR pathway [116], targeted inhibition of PI3K-AKT-mTOR activity (e.g. Bufalin) effectively suppressed HIF1α expression in HCC cells [117]. ENMD-1198 (a microtubule destabilizing agent) and Metformin (an established antidiabetic drug) have also been shown to downregulate the expression of HIF1α in HCC models [118, 119]. Besides, Acriflavine which inhibits HIF1 dimerization showed anti-tumor activity in HCC models [120, 121]. Two novel small-molecule inhibitors targeting HIF2α (PT2385 and PT2977) have been evaluated in phase 1 trials in advanced clear cell renal cell carcinoma and other solid tumors [122, 123]; however, their activities against HCC cells need to be examined.
Table 3: Clinical trials related to targeting hypoxia pathway in HCC
Name |
Mechanism of action |
Disease |
Clinical Stage |
ClinicalTrials.gov |
|---|---|---|---|---|
Apatinib |
VEGFR-2 inhibitor |
Hepatocellular Carcinoma |
Phase 3 |
NCT03046979 |
Regorafenib |
Inhibitor of multiple kinases, including VEGFR, PDGFR and FGFR |
Hepatocellular Carcinoma |
Phase 3 |
NCT01774344 |
Lenvatinib |
Inhibitor of multiple kinases, including VEGFR |
Hepatocellular Carcinoma |
Phase 3 |
NCT01761266 |
Cabozantinib |
VEGFR2 and MET |
Hepatocellular Carcinoma |
Phase 3 |
NCT01908426 |
Ramucirumab |
VEGFR2 |
Hepatocellular Carcinoma |
Phase 3 |
NCT02435433 |
RO7070179 |
HIF1α mRNA Antagonist |
Hepatocellular Carcinoma |
Phase 1 |
NCT02564614 |
EZN-2968 |
HIF1α antisense oligonucleotide inhibitor |
Advanced Solid Tumors/ Lymphoma/Advanced Solid Tumors With Liver Metastases |
Phase 1 completed |
NCT02564614 |
OXY111A |
Anti-hypoxic molecule |
Hepato-Pancreato-Biliary Neoplasm |
Phase 1 and 2 |
NCT02528526 |
TH-302 |
Hypoxia-Activated Prodrug |
Advanced Kidney Cancer or Liver Cancer |
Phase 1 and 2 suspend |
NCT01497444 |
Hepatocellular Carcinoma |
Phase 1 |
NCT01721941 |
||
Tirapazamine |
Hypoxia-Activated Prodrug |
Hepatocellular Carcinoma Combined with Transarterial embolization |
Phase 1 |
NCT02174549 |
Another strategy is to target hypoxia itself. OXY111A, a synthetic allosteric effector of hemoglobin to promote normoxia in hypoxic tumors, has been shown to prevent HIF1α stabilization as well as VEGF production in tumor masses [124]. OXY111A is currently under phase 1 and 2 clinical trials in patients with malignancies of the liver, pancreas and biliary tract. Moreover, hypoxia can be harnessed to selectively activate cytotoxic pro-drugs such as tirapazamine (TPZ) and TH-302 [125, 126]. By using a HBx-transgenic murine model, TPZ co-operated with arterial embolization to induce tumor necrosis without affecting normal liver cells [125]. Of note, Q6, a novel pro-drug activated under hypoxia condition, showed a more potent anti-proliferative effect than TPZ, and induced apoptosis of HCC cells. Interestingly, Q6 can also promote HIF1α degradation through autophagy pathway [127].
Conclusions and future perspectives
As summarized above, hypoxia pathway and HIFs are involved extensively in HCC development. Although many aspects await further exploration, hypoxia pathway appears to be functionally relevant and therapeutically targetable in HCC. Further efforts can be made to characterize the mechanism of HIFs activation and putative roles of HIFs in HCC as outlined below.
As transcription factors, transactivation of HIFs in response to oxygen tension modulates a vast array of hypoxia responsive genes. However, to date, hypoxia-responsive transcriptome and the contribution of individual HIF to hypoxia response remain largely unknown in HCC. Moreover, genome-wide comparative study of various HIFs, the connections between HIFs binding and gene expression, and the contribution of epigenetic alternations in hypoxia response in HCC need to be addressed further.
Additional functional studies, including genetic models, are essential to dissect further the roles of HIFs in HCC development. Upon HIF1α inactivation, alternative pathways such as HIF2α may compensate for the HIF1α loss. HIF2α-dependent network seems to be associated more with therapy-resistance and tumor aggressiveness [108]. When compared to HIF1α, HIF2α and HIF3α remain less-well characterized in HCC. Whether HCC cells activate preferentially certain HIFs during progression is not clear, thus a more thorough investigation of the unique roles of each HIF in HCC is warranted. For example, liver-specific inactivation of HIFs using transgenic animal models with various oncogenic backgrounds may provide more insights into the function of HIFs and their interplays with different oncogenic pathways. In addition, as exemplified from HBx studies, potential impacts of hepatitis virus infection and other carcinogens on HIFs may be a fertile ground of study.
Targeting hypoxia holds a promise for HCC treatment. However, more inhibitors of HIFs and/or their co-factors need to be developed. The potential combination of hypoxia/HIF inhibitors and immunotherapy will be an exciting and active area of investigation. Further, identification of biomarkers associated with hypoxia-targeted therapy will be very valuable and helpful.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by the Scientific Research Project from Department of Education of Zhejiang Province (Y201636292).
CONFLICTS OF INTEREST
None.
REFERENCES
1. American Cancer Society. Global Cancer Facts & Figures 3rd Edition. 2015.
2. Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology. 2004; 127:S35–50.
3. Hassan MM, Hwang LY, Hatten CJ, Swaim M, Li D, Abbruzzese JL, Beasley P, Patt YZ. Risk factors for hepatocellular carcinoma: synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology. 2002; 36:1206–13. doi: 10.1053/jhep.2002.36780.
4. Waller LP, Deshpande V, Pyrsopoulos N. Hepatocellular carcinoma: A comprehensive review. World J Hepatol. 2015; 7:2648–63. doi: 10.4254/wjh.v7.i26.2648.
5. Bruix J, Llovet JM. Prognostic prediction and treatment strategy in hepatocellular carcinoma. Hepatology. 2002; 35:519–24. doi: 10.1053/jhep.2002.32089.
6. El-Serag HB, Marrero JA, Rudolph L, Reddy KR. Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology. 2008; 134:1752–63. doi: 10.1053/j.gastro.2008.02.090.
7. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017; 67:7–30. doi: 10.3322/caac.21332.
8. Schlageter M, Terracciano LM, D’Angelo S, Sorrentino P. Histopathology of hepatocellular carcinoma. World J Gastroenterol. 2014; 20:15955–64. doi: 10.3748/wjg.v20.i43.15955.
9. McKeown SR. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br J Radiol. 2014; 87:20130676. doi: 10.1259/bjr.20130676.
10. Blagosklonny MV. Hypoxia-inducible factor: Achilles’ heel of antiangiogenic cancer therapy (review). International Journal of Oncology. 2001; 19:257–62.
11. Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001; 15:2675–86. doi: 10.1101/gad.924501.
12. McNeill LA, Hewitson KS, Claridge TD, Seibel JF, Horsfall LE, Schofield CJ. Hypoxia-inducible factor asparaginyl hydroxylase (FIH-1) catalyses hydroxylation at the beta-carbon of asparagine-803. Biochem J. 2002; 367:571–5. doi: 10.1042/BJ20021162.
13. Semenza GL. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology (Bethesda). 2004; 19:176–82. doi: 10.1152/physiol.00001.2004.
14. Galbraith MD, Allen MA, Bensard CL, Wang X, Schwinn MK, Qin B, Long HW, Daniels DL, Hahn WC, Dowell RD, Espinosa JM. HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell. 2013; 153:1327–39. doi: 10.1016/j.cell.2013.04.048.
15. Xia X, Kung AL. Preferential binding of HIF-1 to transcriptionally active loci determines cell-type specific response to hypoxia. Genome Biol. 2009; 10:R113. doi: 10.1186/gb-2009-10-10-r113.
16. Perez-Perri JI, Dengler VL, Audetat KA, Pandey A, Bonner EA, Urh M, Mendez J, Daniels DL, Wappner P, Galbraith MD, Espinosa JM. The TIP60 Complex Is a Conserved Coactivator of HIF1A. Cell Rep. 2016; 16:37–47. doi: 10.1016/j.celrep.2016.05.082.
17. Freedman SJ, Sun ZY, Poy F, Kung AL, Livingston DM, Wagner G, Eck MJ. Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1 alpha. Proc Natl Acad Sci USA. 2002; 99:5367–72. doi: 10.1073/pnas.082117899.
18. Dames SA, Martinez-Yamout M, De Guzman RN, Dyson HJ, Wright PE. Structural basis for Hif-1 alpha /CBP recognition in the cellular hypoxic response. Proc Natl Acad Sci USA. 2002; 99:5271–6. doi: 10.1073/pnas.082121399.
19. Kasper LH, Boussouar F, Boyd K, Xu W, Biesen M, Rehg J, Baudino TA, Cleveland JL, Brindle PK. Two transactivation mechanisms cooperate for the bulk of HIF-1-responsive gene expression. EMBO J. 2005; 24:3846–58. doi: 10.1038/sj.emboj.7600846.
20. Demidenko ZN, Blagosklonny MV. The purpose of the HIF-1/PHD feedback loop: to limit mTOR-induced HIF-1alpha. Cell cycle. 2011; 10:1557–62. doi: 10.4161/cc.10.10.15789.
21. Karhausen J, Kong T, Narravula S, Colgan SP. Induction of the von Hippel-Lindau tumor suppressor gene by late hypoxia limits HIF-1 expression. Journal of cellular biochemistry. 2005; 95:1264–75. doi: 10.1002/jcb.20489.
22. del Peso L, Castellanos MC, Temes E, Martin-Puig S, Cuevas Y, Olmos G, Landazuri MO. The von Hippel Lindau/hypoxia-inducible factor (HIF) pathway regulates the transcription of the HIF-proline hydroxylase genes in response to low oxygen. The Journal of biological chemistry. 2003; 278:48690–5. doi: 10.1074/jbc.M308862200.
23. Marxsen JH, Stengel P, Doege K, Heikkinen P, Jokilehto T, Wagner T, Jelkmann W, Jaakkola P, Metzen E. Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by induction of HIF-alpha-prolyl-4-hydroxylases. The Biochemical journal. 2004; 381:761–7. doi: 10.1042/BJ20040620.
24. Aprelikova O, Chandramouli GV, Wood M, Vasselli JR, Riss J, Maranchie JK, Linehan WM, Barrett JC. Regulation of HIF prolyl hydroxylases by hypoxia-inducible factors. Journal of cellular biochemistry. 2004; 92:491–501. doi: 10.1002/jcb.20067.
25. Minamishima YA, Moslehi J, Padera RF, Bronson RT, Liao R, Kaelin WG Jr. A feedback loop involving the Phd3 prolyl hydroxylase tunes the mammalian hypoxic response in vivo. Molecular and cellular biology. 2009; 29:5729–41. doi: 10.1128/MCB.00331-09.
26. Henze AT, Riedel J, Diem T, Wenner J, Flamme I, Pouyseggur J, Plate KH, Acker T. Prolyl hydroxylases 2 and 3 act in gliomas as protective negative feedback regulators of hypoxia-inducible factors. Cancer research. 2010; 70:357–66. doi: 10.1158/0008-5472.CAN-09-1876.
27. Johnson AB, Denko N, Barton MC. Hypoxia induces a novel signature of chromatin modifications and global repression of transcription. Mutat Res. 2008; 640:174–9. doi: 10.1016/j.mrfmmm.2008.01.001.
28. Xia X, Lemieux ME, Li W, Carroll JS, Brown M, Liu XS, Kung AL. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc Natl Acad Sci USA. 2009; 106:4260–5. doi: 10.1073/pnas.0810067106.
29. Thienpont B, Steinbacher J, Zhao H, D’Anna F, Kuchnio A, Ploumakis A, Ghesquiere B, Van Dyck L, Boeckx B, Schoonjans L, Hermans E, Amant F, Kristensen VN, et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature. 2016; 537:63–8. doi: 10.1038/nature19081.
30. Wenger RH, Rolfs A, Spielmann P, Zimmermann DR, Gassmann M. Mouse hypoxia-inducible factor-1alpha is encoded by two different mRNA isoforms: expression from a tissue-specific and a housekeeping-type promoter. Blood. 1998; 91:3471–80.
31. Ambade A, Satishchandran A, Saha B, Gyongyosi B, Lowe P, Kodys K, Catalano D, Szabo G. Hepatocellular carcinoma is accelerated by NASH involving M2 macrophage polarization mediated by hif-1alphainduced IL-10. Oncoimmunology. 2016; 5:e1221557. doi: 10.1080/2162402X.2016.1221557.
32. Stroka DM, Burkhardt T, Desbaillets I, Wenger RH, Neil DA, Bauer C, Gassmann M, Candinas D. HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. FASEB J. 2001; 15:2445–53. doi: 10.1096/fj.01-0125com.
33. Wiesener MS, Jurgensen JS, Rosenberger C, Scholze CK, Horstrup JH, Warnecke C, Mandriota S, Bechmann I, Frei UA, Pugh CW, Ratcliffe PJ, Bachmann S, Maxwell PH, et al. Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. 2003; 17:271–3. doi: 10.1096/fj.02-0445fje.
34. Yoo YG, Na TY, Seo HW, Seong JK, Park CK, Shin YK, Lee MO. Hepatitis B virus X protein induces the expression of MTA1 and HDAC1, which enhances hypoxia signaling in hepatocellular carcinoma cells. Oncogene. 2008; 27:3405–13. doi: 10.1038/sj.onc.1211000.
35. Yang SL, Liu LP, Jiang JX, Xiong ZF, He QJ, Wu C. The correlation of expression levels of HIF-1alpha and HIF-2alpha in hepatocellular carcinoma with capsular invasion, portal vein tumor thrombi and patients’ clinical outcome. Jpn J Clin Oncol. 2014; 44:159–67. doi: 10.1093/jjco/hyt194.
36. Bangoura G, Liu ZS, Qian Q, Jiang CQ, Yang GF, Jing S. Prognostic significance of HIF-2alpha/EPAS1 expression in hepatocellular carcinoma. World J Gastroenterol. 2007; 13:3176–82.
37. Dai CX, Gao Q, Qiu SJ, Ju MJ, Cai MY, Xu YF, Zhou J, Zhang BH, Fan J. Hypoxia-inducible factor-1 alpha, in association with inflammation, angiogenesis and MYC, is a critical prognostic factor in patients with HCC after surgery. BMC Cancer. 2009; 9: 418. doi: 10.1186/1471-2407-9-418.
38. Xiang ZL, Zeng ZC, Fan J, Tang ZY, He J, Zeng HY, Chang JY. The expression of HIF-1alpha in primary hepatocellular carcinoma and its correlation with radiotherapy response and clinical outcome. Mol Biol Rep. 2012; 39:2021–9. doi: 10.1007/s11033-011-0949-1.
39. Bjornson E, Mukhopadhyay B, Asplund A, Pristovsek N, Cinar R, Romeo S, Uhlen M, Kunos G, Nielsen J, Mardinoglu A. Stratification of Hepatocellular Carcinoma Patients Based on Acetate Utilization. Cell Rep. 2015; 13:2014–26. doi: 10.1016/j.celrep.2015.10.045.
40. Cannito S, Turato C, Paternostro C, Biasiolo A, Colombatto S, Cambieri I, Quarta S, Novo E, Morello E, Villano G, Fasolato S, Musso T, David E, et al. Hypoxia up-regulates SERPINB3 through HIF-2alpha in human liver cancer cells. Oncotarget. 2015; 6:2206–21. doi: 10.18632/oncotarget.2943.
41. Liu LP, Hu BG, Ye C, Ho RL, Chen GG, Lai PB. HBx mutants differentially affect the activation of hypoxia-inducible factor-1alpha in hepatocellular carcinoma. Br J Cancer. 2014; 110:1066–73. doi: 10.1038/bjc.2013.787.
42. Wada H, Nagano H, Yamamoto H, Yang Y, Kondo M, Ota H, Nakamura M, Yoshioka S, Kato H, Damdinsuren B, Tang D, Marubashi S, Miyamoto A, et al. Expression pattern of angiogenic factors and prognosis after hepatic resection in hepatocellular carcinoma: importance of angiopoietin-2 and hypoxia-induced factor-1 alpha. Liver Int. 2006; 26:414–23. doi: 10.1111/j.1478-3231.2006.01243.x.
43. Xia L, Mo P, Huang W, Zhang L, Wang Y, Zhu H, Tian D, Liu J, Chen Z, Zhang Y, Hu H, Fan D, Nie Y, et al. The TNF-alpha/ROS/HIF-1-induced upregulation of FoxMI expression promotes HCC proliferation and resistance to apoptosis. Carcinogenesis. 2012; 33:2250–9. doi: 10.1093/carcin/bgs249.
44. Sun HX, Xu Y, Yang XR, Wang WM, Bai H, Shi RY, Nayar SK, Devbhandari RP, He YZ, Zhu QF, Sun YF, Hu B, Khan M, et al. Hypoxia inducible factor 2 alpha inhibits hepatocellular carcinoma growth through the transcription factor dimerization partner 3/ E2F transcription factor 1-dependent apoptotic pathway. Hepatology. 2013; 57:1088–97. doi: 10.1002/hep.26188.
45. Liu P, Fang X, Song Y, Jiang JX, He QJ, Liu XJ. Expression of hypoxia-inducible factor 3alpha in hepatocellular carcinoma and its association with other hypoxia-inducible factors. Exp Ther Med. 2016; 11:2470–6. doi: 10.3892/etm.2016.3193.
46. Jiang Y, Zhu Y, Wang X, Gong J, Hu C, Guo B, Zhu B, Li Y. Temporal regulation of HIF-1 and NF-kappaB in hypoxic hepatocarcinoma cells. Oncotarget. 2015; 6:9409–19. doi: 10.18632/oncotarget.3352.
47. Moon EJ, Jeong CH, Jeong JW, Kim KR, Yu DY, Murakami S, Kim CW, Kim KW. Hepatitis B virus X protein induces angiogenesis by stabilizing hypoxia-inducible factor-1alpha. FASEB J. 2004; 18:382–4. doi: 10.1096/fj.03-0153fje.
48. Li Y, Wei Y, Guo J, Cheng Y, He W. Interactional role of microRNAs and bHLH-PAS proteins in cancer (Review). Int J Oncol. 2015; 47:25–34. doi: 10.3892/ijo.2015.3007.
49. Zhang LF, Lou JT, Lu MH, Gao C, Zhao S, Li B, Liang S, Li Y, Li D, Liu MF. Suppression of miR-199a maturation by HuR is crucial for hypoxia-induced glycolytic switch in hepatocellular carcinoma. EMBO J. 2015; 34:2671–85. doi: 10.15252/embj.201591803.
50. Xu H, Zhao L, Fang Q, Sun J, Zhang S, Zhan C, Liu S, Zhang Y. MiR-338-3p inhibits hepatocarcinoma cells and sensitizes these cells to sorafenib by targeting hypoxia-induced factor 1alpha. PLoS One. 2014; 9:e115565. doi: 10.1371/journal.pone.0115565.
51. Csak T, Bala S, Lippai D, Satishchandran A, Catalano D, Kodys K, Szabo G. microRNA-122 regulates hypoxia-inducible factor-1 and vimentin in hepatocytes and correlates with fibrosis in diet-induced steatohepatitis. Liver Int. 2015; 35:532–41. doi: 10.1111/liv.12633.
52. Daskalow K, Rohwer N, Raskopf E, Dupuy E, Kuhl A, Loddenkemper C, Wiedenmann B, Schmitz V, Cramer T. Role of hypoxia-inducible transcription factor 1alpha for progression and chemosensitivity of murine hepatocellular carcinoma. J Mol Med (Berl). 2010; 88:817–27. doi: 10.1007/s00109-010-0623-4.
53. He C, Sun XP, Qiao H, Jiang X, Wang D, Jin X, Dong X, Wang J, Jiang H, Sun X. Downregulating hypoxia-inducible factor-2alpha improves the efficacy of doxorubicin in the treatment of hepatocellular carcinoma. Cancer Sci. 2012; 103:528–34. doi: 10.1111/j.1349-7006.2011.02177.x.
54. Menrad H, Werno C, Schmid T, Copanaki E, Deller T, Dehne N, Brune B. Roles of hypoxia-inducible factor-1alpha (HIF-1alpha) versus HIF-2alpha in the survival of hepatocellular tumor spheroids. Hepatology. 2010; 51:2183–92. doi: 10.1002/hep.23597.
55. Jiang BH, Agani F, Passaniti A, Semenza GL. V-SRC induces expression of hypoxia-inducible factor 1 (HIF-1) and transcription of genes encoding vascular endothelial growth factor and enolase 1: involvement of HIF-1 in tumor progression. Cancer Res. 1997; 57:5328–35.
56. Keng VW, Villanueva A, Chiang DY, Dupuy AJ, Ryan BJ, Matise I, Silverstein KA, Sarver A, Starr TK, Akagi K, Tessarollo L, Collier LS, Powers S, et al. A conditional transposon-based insertional mutagenesis screen for genes associated with mouse hepatocellular carcinoma. Nat Biotechnol. 2009; 27:264–74. doi: 10.1038/nbt.1526.
57. Imtiyaz HZ, Williams EP, Hickey MM, Patel SA, Durham AC, Yuan LJ, Hammond R, Gimotty PA, Keith B, Simon MC. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest. 2010; 120:2699–714. doi: 10.1172/JCI39506.
58. Wilson GK, Brimacombe CL, Rowe IA, Reynolds GM, Fletcher NF, Stamataki Z, Bhogal RH, Simoes ML, Ashcroft M, Afford SC, Mitry RR, Dhawan A, Mee CJ, et al. A dual role for hypoxia inducible factor-1alpha in the hepatitis C virus lifecycle and hepatoma migration. J Hepatol. 2012; 56:803–9. doi: 10.1016/j.jhep.2011.11.018.
59. Mee CJ, Farquhar MJ, Harris HJ, Hu K, Ramma W, Ahmed A, Maurel P, Bicknell R, Balfe P, McKeating JA. Hepatitis C virus infection reduces hepatocellular polarity in a vascular endothelial growth factor-dependent manner. Gastroenterology. 2010; 138:1134–42. doi: 10.1053/j.gastro.2009.11.047.
60. Mee CJ, Harris HJ, Farquhar MJ, Wilson G, Reynolds G, Davis C, van ISC, Balfe P, McKeating JA. Polarization restricts hepatitis C virus entry into HepG2 hepatoma cells. J Virol. 2009; 83:6211–21. doi: 10.1128/JVI.00246-09.
61. Zhu AX, Duda DG, Sahani DV, Jain RK. HCC and angiogenesis: possible targets and future directions. Nat Rev Clin Oncol. 2011; 8:292–301. doi: 10.1038/nrclinonc.2011.30.
62. Mise M, Arii S, Higashituji H, Furutani M, Niwano M, Harada T, Ishigami S, Toda Y, Nakayama H, Fukumoto M, Fujita J, Imamura M. Clinical significance of vascular endothelial growth factor and basic fibroblast growth factor gene expression in liver tumor. Hepatology. 1996; 23:455–64. doi: 10.1053/jhep.1996.v23.pm0008617424.
63. Wang X, Dong J, Jia L, Zhao T, Lang M, Li Z, Lan C, Li X, Hao J, Wang H, Qin T, Huang C, Yang S, et al. HIF-2-dependent expression of stem cell factor promotes metastasis in hepatocellular carcinoma. Cancer letters. 2017; 393:113–24. doi: 10.1016/j.canlet.2017.01.032.
64. Geis T, Doring C, Popp R, Grossmann N, Fleming I, Hansmann ML, Dehne N, Brune B. HIF-2alpha-dependent PAI-1 induction contributes to angiogenesis in hepatocellular carcinoma. Experimental cell research. 2015; 331:46–57. doi: 10.1016/j.yexcr.2014.11.018.
65. Maegdefrau U, Amann T, Winklmeier A, Braig S, Schubert T, Weiss TS, Schardt K, Warnecke C, Hellerbrand C, Bosserhoff AK. Bone morphogenetic protein 4 is induced in hepatocellular carcinoma by hypoxia and promotes tumour progression. J Pathol. 2009; 218:520–9. doi: 10.1002/path.2563.
66. Ema M, Taya S, Yokotani N, Sogawa K, Matsuda Y, Fujii-Kuriyama Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci U S A. 1997; 94:4273–8.
67. Levy AP, Levy NS, Wegner S, Goldberg MA. Transcriptional regulation of the rat vascular endothelial growth factor gene by hypoxia. J Biol Chem. 1995; 270:13333–40.
68. Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996; 16:4604–13.
69. Liu Y, Cox SR, Morita T, Kourembanas S. Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells. Identification of a 5’ enhancer. Circ Res. 1995; 77:638–43.
70. von Marschall Z, Cramer T, Hocker M, Finkenzeller G, Wiedenmann B, Rosewicz S. Dual mechanism of vascular endothelial growth factor upregulation by hypoxia in human hepatocellular carcinoma. Gut. 2001; 48:87–96.
71. Schoenleber SJ, Kurtz DM, Talwalkar JA, Roberts LR, Gores GJ. Prognostic role of vascular endothelial growth factor in hepatocellular carcinoma: systematic review and meta-analysis. Br J Cancer. 2009; 100:1385–92. doi: 10.1038/sj.bjc.6605017.
72. Chao Y, Li CP, Chau GY, Chen CP, King KL, Lui WY, Yen SH, Chang FY, Chan WK, Lee SD. Prognostic significance of vascular endothelial growth factor, basic fibroblast growth factor, and angiogenin in patients with resectable hepatocellular carcinoma after surgery. Ann Surg Oncol. 2003; 10:355–62.
73. Gassmann M, Kvietikova I, Rolfs A, Wenger RH. Oxygen- and dioxin-regulated gene expression in mouse hepatoma cells. Kidney Int. 1997; 51:567–74.
74. Xiao H, Tong R, Ding C, Lv Z, Du C, Peng C, Cheng S, Xie H, Zhou L, Wu J, Zheng S. gamma-H2AX promotes hepatocellular carcinoma angiogenesis via EGFR/HIF-1alpha/VEGF pathways under hypoxic condition. Oncotarget. 2015; 6:2180–92. doi: 10.18632/oncotarget.2942.
75. Rankin EB, Biju MP, Liu Q, Unger TL, Rha J, Johnson RS, Simon MC, Keith B, Haase VH. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J Clin Invest. 2007; 117:1068–77. doi: 10.1172/JCI30117.
76. Rey S, Semenza GL. Hypoxia-inducible factor-1-dependent mechanisms of vascularization and vascular remodelling. Cardiovasc Res. 2010; 86:236–42. doi: 10.1093/cvr/cvq045.
77. Rosmorduc O, Housset C. Hypoxia: a link between fibrogenesis, angiogenesis, and carcinogenesis in liver disease. Semin Liver Dis. 2010; 30:258–70. doi: 10.1055/s-0030-1255355.
78. Lau CK, Yang ZF, Ho DW, Ng MN, Yeoh GC, Poon RT, Fan ST. An Akt/hypoxia-inducible factor-1alpha/platelet-derived growth factor-BB autocrine loop mediates hypoxia-induced chemoresistance in liver cancer cells and tumorigenic hepatic progenitor cells. Clin Cancer Res. 2009; 15:3462–71. doi: 10.1158/1078-0432.CCR-08-2127.
79. Hamaguchi T, Iizuka N, Tsunedomi R, Hamamoto Y, Miyamoto T, Iida M, Tokuhisa Y, Sakamoto K, Takashima M, Tamesa T, Oka M. Glycolysis module activated by hypoxia-inducible factor 1alpha is related to the aggressive phenotype of hepatocellular carcinoma. Int J Oncol. 2008; 33:725–31.
80. Chen C, Pore N, Behrooz A, Ismail-Beigi F, Maity A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J Biol Chem. 2001; 276:9519–25. doi: 10.1074/jbc.M010144200.
81. Amann T, Maegdefrau U, Hartmann A, Agaimy A, Marienhagen J, Weiss TS, Stoeltzing O, Warnecke C, Scholmerich J, Oefner PJ, Kreutz M, Bosserhoff AK, Hellerbrand C. GLUT1 expression is increased in hepatocellular carcinoma and promotes tumorigenesis. Am J Pathol. 2009; 174:1544–52. doi: 10.2353/ajpath.2009.080596.
82. Gwak GY, Yoon JH, Kim KM, Lee HS, Chung JW, Gores GJ. Hypoxia stimulates proliferation of human hepatoma cells through the induction of hexokinase II expression. J Hepatol. 2005; 42:358–64. doi: 10.1016/j.jhep.2004.11.020.
83. Mathupala SP, Rempel A, Pedersen PL. Glucose catabolism in cancer cells: identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J Biol Chem. 2001; 276:43407–12. doi: 10.1074/jbc.M108181200.
84. Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996; 271:32529–37.
85. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006; 3:177–85. doi: 10.1016/j.cmet.2006.02.002.
86. Lai RK, Xu IM, Chiu DK, Tse AP, Wei LL, Law CT, Lee D, Wong CM, Wong MP, Ng IO, Wong CC. NDUFA4L2 Fine-tunes Oxidative Stress in Hepatocellular Carcinoma. Clin Cancer Res. 2016; 22:3105–17. doi: 10.1158/1078-0432.CCR-15-1987.
87. Masson N, Ratcliffe PJ. Hypoxia signaling pathways in cancer metabolism: the importance of co-selecting interconnected physiological pathways. Cancer Metab. 2014; 2: 3. doi: 10.1186/2049-3002-2-3.
88. Zhang Q, Bai X, Chen W, Ma T, Hu Q, Liang C, Xie S, Chen C, Hu L, Xu S, Liang T. Wnt/beta-catenin signaling enhances hypoxia-induced epithelial-mesenchymal transition in hepatocellular carcinoma via crosstalk with hif-1alpha signaling. Carcinogenesis. 2013; 34:962–73. doi: 10.1093/carcin/bgt027.
89. Liu L, Zhu XD, Wang WQ, Shen Y, Qin Y, Ren ZG, Sun HC, Tang ZY. Activation of beta-catenin by hypoxia in hepatocellular carcinoma contributes to enhanced metastatic potential and poor prognosis. Clin Cancer Res. 2010; 16:2740–50. doi: 10.1158/1078-0432.CCR-09-2610.
90. Yan W, Fu Y, Tian D, Liao J, Liu M, Wang B, Xia L, Zhu Q, Luo M. PI3 kinase/Akt signaling mediates epithelial-mesenchymal transition in hypoxic hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2009; 382:631–6. doi: 10.1016/j.bbrc.2009.03.088.
91. Zhang L, Huang G, Li X, Zhang Y, Jiang Y, Shen J, Liu J, Wang Q, Zhu J, Feng X, Dong J, Qian C. Hypoxia induces epithelial-mesenchymal transition via activation of SNAI1 by hypoxia-inducible factor -1alpha in hepatocellular carcinoma. BMC Cancer. 2013; 13: 108. doi: 10.1186/1471-2407-13-108.
92. Ying Q, Liang L, Guo W, Zha R, Tian Q, Huang S, Yao J, Ding J, Bao M, Ge C, Yao M, Li J, He X. Hypoxia-inducible microRNA-210 augments the metastatic potential of tumor cells by targeting vacuole membrane protein 1 in hepatocellular carcinoma. Hepatology. 2011; 54:2064–75. doi: 10.1002/hep.24614.
93. You N, Liu W, Tang L, Zhong X, Ji R, Zhang N, Wang D, He Y, Dou K, Tao K. Tg737 signaling is required for hypoxia-enhanced invasion and migration of hepatoma cells. J Exp Clin Cancer Res. 2012; 31:75. doi: 10.1186/1756-9966-31-75.
94. Tian H, Huang P, Zhao Z, Tang W, Xia J. HIF-1alpha plays a role in the chemotactic migration of hepatocarcinoma cells through the modulation of CXCL6 expression. Cell Physiol Biochem. 2014; 34:1536–46. doi: 10.1159/000366357.
95. Hu F, Deng X, Yang X, Jin H, Gu D, Lv X, Wang C, Zhang Y, Huo X, Shen Q, Luo Q, Zhao F, Ge T, et al. Hypoxia upregulates Rab11-family interacting protein 4 through HIF-1alpha to promote the metastasis of hepatocellular carcinoma. Oncogene. 2015; 34:6007–17. doi: 10.1038/onc.2015.49.
96. Wang X, Ren H, Zhao T, Chen J, Sun W, Sun Y, Ma W, Wang J, Gao C, Gao S, Lang M, Jia L, Hao J. Stem cell factor is a novel independent prognostic biomarker for hepatocellular carcinoma after curative resection. Carcinogenesis. 2014; 35:2283–90. doi: 10.1093/carcin/bgu162.
97. Murdoch C, Tazzyman S, Webster S, Lewis CE. Expression of Tie-2 by human monocytes and their responses to angiopoietin-2. Journal of immunology. 2007; 178:7405–11.
98. Matsubara T, Kanto T, Kuroda S, Yoshio S, Higashitani K, Kakita N, Miyazaki M, Sakakibara M, Hiramatsu N, Kasahara A, Tomimaru Y, Tomokuni A, Nagano H, et al. TIE2-expressing monocytes as a diagnostic marker for hepatocellular carcinoma correlates with angiogenesis. Hepatology. 2013; 57:1416–25. doi: 10.1002/hep.25965.
99. He YF, Wang CQ, Yu Y, Qian J, Song K, Sun QM, Zhou J. Tie2-Expressing Monocytes Are Associated with Identification and Prognoses of Hepatitis B Virus Related Hepatocellular Carcinoma after Resection. PloS one. 2015; 10: e0143657. doi: 10.1371/journal.pone.0143657.
100. Coulouarn C, Corlu A, Glaise D, Guenon I, Thorgeirsson SS, Clement B. Hepatocyte-stellate cell cross-talk in the liver engenders a permissive inflammatory microenvironment that drives progression in hepatocellular carcinoma. Cancer Res. 2012; 72:2533–42. doi: 10.1158/0008-5472.CAN-11-3317.
101. Cho Y, Cho EJ, Lee JH, Yu SJ, Kim YJ, Kim CY, Yoon JH. Hypoxia Enhances Tumor-Stroma Crosstalk that Drives the Progression of Hepatocellular Carcinoma. Dig Dis Sci. 2016; 61:2568–77. doi: 10.1007/s10620-016-4158-6.
102. Klein A, Flugel D, Kietzmann T. Transcriptional regulation of serine/threonine kinase-15 (STK15) expression by hypoxia and HIF-1. Mol Biol Cell. 2008; 19:3667–75. doi: 10.1091/mbc.E08-01-0042.
103. Zhou TY, Zhuang LH, Hu Y, Zhou YL, Lin WK, Wang DD, Wan ZQ, Chang LL, Chen Y, Ying MD, Chen ZB, Ye S, Lou JS, et al. Inactivation of hypoxia-induced YAP by statins overcomes hypoxic resistance tosorafenib in hepatocellular carcinoma cells. Sci Rep. 2016; 6: 30483. doi: 10.1038/srep30483.
104. Dai XY, Zhuang LH, Wang DD, Zhou TY, Chang LL, Gai RH, Zhu DF, Yang B, Zhu H, He QJ. Nuclear translocation and activation of YAP by hypoxia contributes to the chemoresistance of SN38 in hepatocellular carcinoma cells. Oncotarget. 2016; 7:6933–47. doi: 10.18632/oncotarget.6903.
105. Zhao D, Zhai B, He C, Tan G, Jiang X, Pan S, Dong X, Wei Z, Ma L, Qiao H, Jiang H, Sun X. Upregulation of HIF-2alpha induced by sorafenib contributes to the resistance by activating the TGF-alpha/EGFR pathway in hepatocellular carcinoma cells. Cell Signal. 2014; 26:1030–9. doi: 10.1016/j.cellsig.2014.01.026.
106. Jung EU, Yoon JH, Lee YJ, Lee JH, Kim BH, Yu SJ, Myung SJ, Kim YJ, Lee HS. Hypoxia and retinoic acid-inducible NDRG1 expression is responsible for doxorubicin and retinoic acid resistance in hepatocellular carcinoma cells. Cancer Lett. 2010; 298:9–15. doi: 10.1016/j.canlet.2010.05.020.
107. Tak E, Lee S, Lee J, Rashid MA, Kim YW, Park JH, Park WS, Shokat KM, Ha J, Kim SS. Human carbonyl reductase 1 upregulated by hypoxia renders resistance to apoptosis in hepatocellular carcinoma cells. J Hepatol. 2011; 54:328–39. doi: 10.1016/j.jhep.2010.06.045.
108. Liang Y, Zheng T, Song R, Wang J, Yin D, Wang L, Liu H, Tian L, Fang X, Meng X, Jiang H, Liu J, Liu L. Hypoxia-mediated sorafenib resistance can be overcome by EF24 through Von Hippel-Lindau tumor suppressor-dependent HIF-1alpha inhibition in hepatocellular carcinoma. Hepatology. 2013; 57:1847–57. doi: 10.1002/hep.26224.
109. Liu F, Dong X, Lv H, Xiu P, Li T, Wang F, Xu Z, Li J. Targeting hypoxia-inducible factor-2alpha enhances sorafenib antitumor activity via beta-catenin/C-Myc-dependent pathways in hepatocellular carcinoma. Oncol Lett. 2015; 10:778–84. doi: 10.3892/ol.2015.3315.
110. Liu F, Wang P, Jiang X, Tan G, Qiao H, Jiang H, Krissansen GW, Sun X. Antisense hypoxia-inducible factor 1alpha gene therapy enhances the therapeutic efficacy of doxorubicin to combat hepatocellular carcinoma. Cancer Sci. 2008; 99:2055–61. doi: 10.1111/j.1349-7006.2008.00905.x.
111. Williams KJ, Telfer BA, Xenaki D, Sheridan MR, Desbaillets I, Peters HJ, Honess D, Harris AL, Dachs GU, van der Kogel A, Stratford IJ. Enhanced response to radiotherapy in tumours deficient in the function of hypoxia-inducible factor-1. Radiother Oncol. 2005; 75:89–98. doi: 10.1016/j.radonc.2005.01.009.
112. Sun X, Kanwar JR, Leung E, Lehnert K, Wang D, Krissansen GW. Gene transfer of antisense hypoxia inducible factor-1 alpha enhances the therapeutic efficacy of cancer immunotherapy. Gene Ther. 2001; 8:638–45. doi: 10.1038/sj.gt.3301388.
113. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–74. doi: 10.1016/j.cell.2011.02.013.
114. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008; 359:378–90. doi: 10.1056/NEJMoa0708857.
115. Ju C, Colgan SP, Eltzschig HK. Hypoxia-inducible factors as molecular targets for liver diseases. J Mol Med (Berl). 2016; 94:613–27. doi: 10.1007/s00109-016-1408-1.
116. Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, Simons JW, Semenza GL. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer research. 2000; 60:1541–5.
117. Wang H, Zhang C, Xu L, Zang K, Ning Z, Jiang F, Chi H, Zhu X, Meng Z. Bufalin suppresses hepatocellular carcinoma invasion and metastasis by targeting HIF-1alpha via the PI3K/AKT/mTOR pathway. Oncotarget. 2016; 7:20193–208. doi: 10.18632/oncotarget.7935.
118. Moser C, Lang SA, Mori A, Hellerbrand C, Schlitt HJ, Geissler EK, Fogler WE, Stoeltzing O. ENMD-1198, a novel tubulin-binding agent reduces HIF-1alpha and STAT3 activity in human hepatocellular carcinoma(HCC) cells, and inhibits growth and vascularization in vivo. BMC Cancer. 2008; 8: 206. doi: 10.1186/1471-2407-8-206.
119. Zhou X, Chen J, Yi G, Deng M, Liu H, Liang M, Shi B, Fu X, Chen Y, Chen L, He Z, Wang J, Liu J. Metformin suppresses hypoxia-induced stabilization of HIF-1alpha through reprogramming of oxygen metabolism in hepatocellular carcinoma. Oncotarget. 2016; 7:873–84. doi: 10.18632/oncotarget.6418.
120. Lee K, Zhang H, Qian DZ, Rey S, Liu JO, Semenza GL. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc Natl Acad Sci U S A. 2009; 106:17910–5. doi: 10.1073/pnas.0909353106.
121. Lee CJ, Yue CH, Lin YY, Wu JC, Liu JY. Antitumor activity of acriflavine in human hepatocellular carcinoma cells. Anticancer Res. 2014; 34:3549–56.
122. Wallace EM, Rizzi JP, Han G, Wehn PM, Cao Z, Du X, Cheng T, Czerwinski RM, Dixon DD, Goggin BS, Grina JA, Halfmann MM, Maddie MA, et al. A Small-Molecule Antagonist of HIF2alpha Is Efficacious in Preclinical Models of Renal Cell Carcinoma. Cancer Res. 2016; 76:5491–500. doi: 10.1158/0008-5472.CAN-16-0473.
123. Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, Homayoun F, Ma Y, Patel N, Yell P, Hao G, Yousuf Q, Joyce A, et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature. 2016; 539:112–7. doi: 10.1038/nature19796.
124. Limani P, Linecker M, Kron P, Samaras P, Pestalozzi B, Stupp R, Jetter A, Dutkowski P, Mullhaupt B, Schlegel A, Nicolau C, Lehn JM, Petrowsky H, et al. Development of OXY111A, a novel hypoxia-modifier as a potential antitumor agent in patients with hepato-pancreato-biliary neoplasms - Protocol of a first Ib/IIa clinical trial. BMC Cancer. 2016; 16:812. doi: 10.1186/s12885-016-2855-3.
125. Lin WH, Yeh SH, Yeh KH, Chen KW, Cheng YW, Su TH, Jao P, Ni LC, Chen PJ, Chen DS. Hypoxia-activated cytotoxic agent tirapazamine enhances hepatic artery ligation-induced killing of liver tumor in HBx transgenic mice. Proc Natl Acad Sci USA. 2016; 113:11937–42. doi: 10.1073/pnas.1613466113.
126. Weiss GJ, Infante JR, Chiorean EG, Borad MJ, Bendell JC, Molina JR, Tibes R, Ramanathan RK, Lewandowski K, Jones SF, Lacouture ME, Langmuir VK, Lee H, et al. Phase 1 study of the safety, tolerability, and pharmacokinetics of TH-302, a hypoxia-activated prodrug, in patients with advanced solid malignancies. Clin Cancer Res. 2011; 17:2997–3004. doi: 10.1158/1078-0432.CCR-10-3425.
127. Liu XW, Cai TY, Zhu H, Cao J, Su Y, Hu YZ, He QJ, Yang B. Q6, a novel hypoxia-targeted drug, regulates hypoxia-inducible factor signaling via an autophagy-dependent mechanism in hepatocellular carcinoma. Autophagy. 2014; 10:111–22. doi: 10.4161/auto.26838.