
Overview of Dox-induced cardiotoxicity
Doxorubicin (Dox), a non-selective class I anthracycline antibiotic, is a potent chemotherapeutic agent which is used for the treatment of numerous cancers [1]. The use of Dox is limited, however, due to serious side effects; the most prominent is cardiotoxicity which can manifest acutely as well as years after treatment has been discontinued leading to left ventricular dysfunction, dilated cardiomyopathy and heart failure [2-4]. Toxic effects of Dox include cardiomyocyte damage and apoptotic and necrotic cell death [5]. The severity of heart disease is linked to accumulated Dox dosage during the course of the anticancer therapy ranging from 3-5% in patients that received a cumulative dose of 400mg/m2 to 18-48% in patients receiving 700mg/m2 (and 100% of mice receiving 71mg/ m2) [1, 6, 7].
Dox is a mitochondrial toxin, and mitochondrial damage is central to Dox-induced cardiac dysfunction and cell death [8]. Cardiomyocytes require large numbers of healthy-functioning mitochondria to ensure sufficient ATP production for contractile function and cell survival. Dox-induced over-production of reactive oxygen and nitrogen species (ROS and RNS) such as superoxide, hydrogen peroxide, hydroxyl radical and peroxynitrite has been proposed to be the main cause of Dox-induced acute cardiomyocyte toxicity and cell death by damaging various molecular constituents and organelles [9-12]. A detailed description of the pathways by which Dox induces production of ROS and RNS can be found in references [13, 14]. Nitric oxide synthase (NOS) and nicotinamide adenine dinucleotide phosphate-oxidase (NOX) enzymes play an important role in Dox-induced production of ROS [15]. Dox can be reduced to a semiquinone by NOX and/or NOS leading to the production of superoxide and hydrogen peroxide, and subsequently the highly reactive hydroxyl radical [14]. NOS enzymes catalyze the production of nitric oxide from N-arginine; superoxide can react with nitric oxide producing peroxynitrite which is highly reactive oxidizing DNA, proteins, and peroxidizing lipids. Both endothelial NOS (eNOS) and inducible NOS (iNOS) isoforms have been implicated in mediating Dox toxicity via the production of RNS [16]. Deletion or overproduction of eNOS in mice was shown, respectively, to improve or worsen cardiac outcome post-Dox compared to wild type groups [17]. There is also some evidence that iNOS, which is increased by Dox administration, contributes to the production of RNS; iNOS-knockout mice showed protection from Dox-induced toxicity [18]. Dox also interacts with iron, and Dox-iron complexes contribute to further hydroxyl radical production. Hydroxyl radical damages DNA and proteins and extensively peroxidizes lipids which contribute to major cellular damage and death [19].
Dox has a robust attraction to negatively charged membranes such as the inner mitochondrial membrane causing lipid peroxidation [10, 20]. Dox-induced peroxidation of cardiolipin uncouples respiratory chain complexes at mitochondria, resulting in reduced ATP and increased ROS production [21-24], and promoting formation of the of mitochondrial permeability transition pore, mPTP. Formation of mPTP causes cytochrome C release and induction of an intrinsic pathway of apoptosis; it is also an important feature of necrotic cell death [25]. Despite multiple studies and strong evidence from many experimental models that anti-oxidant therapies can decrease or prevent Dox-induced cardiotoxicity, results have not been as promising in the clinical setting [26-28]. Additional mechanisms of Dox-toxicity need to be considered and targeted.
Dox kills rapidly proliferating cancer cells by intercalating with their DNA and forming covalent adducts which lead to the inhibition of DNA polymerase and nucleic acid synthesis. Additionally, Dox cross-links Topoisomerase IIα (TOPIIα) to the DNA, forming a TOPIIα-Dox-DNA complex resulting in DNA breakage and cell death, a major mechanism for killing cancer cells which express high levels of TOPIIα [29-33]. Unlike tumor cells, cardiomyocytes do not express TOPIIα, rather they express topoisomerase IIβ (TOPIIβ) which can also form DNA-TOPIIβ-Dox complexes, cause double-strand breaks and cell death [34, 35]. Dox-induced DNA damage, mediated by TOPIIβ significantly alters the nuclear and mitochondrial transcriptome and markedly decreases mitochondrial biogenesis [36, 37]. There is strong evidence that TOPIIβ is a major contributor to Dox-induced cardiotoxicity; transgenic mice lacking TOPIIβ were not susceptible to Dox cardiotoxicity in an acute as well as chronic setting [37]. Interestingly, the compound dexrazoxane which is in clinical use for preventing Dox-induced cardiomyopathy due to its iron-chelating and anti-oxidant properties[38], belongs to the molecular category of TOPII poisons. It has been suggested that the TOPIIβ inhibitory activity, rather than the iron-chelating action, of dexrazoxane confers protection of cardiomyocytes from Dox [39].
Dox induced DNA damage leads to the activation of the Ataxia-Telengiectasia Mutated protein which binds to DNA break sites and upregulates and activates the tumor suppressor protein p53 [40]. Upregulation of p53 by Dox has been linked to increased ROS and double strand DNA damage, and there is considerable support to the notion that p53 protein is mediating Dox-induced apoptotic cell death of cardiomyocytes in vitro and in vivo [41-43]. Anthracycline-induced p53 up-regulation is reported to suppress transcription of GATA-4, a cardioprotective transcription factor [44]. Nevertheless, p53-independent pathways to Dox-induced deleterious effects in cardiomyocytes, such as apoptotic cell death, oxidative and nitrosative stress, and cardiac fibrosis have also been documented, in a mouse model of cardiomyocyte-specific p53 knockout [45]. In addition, using a juvenile model of Dox toxicity, Zhu and colleagues reported antithetical roles for p53 in the acute versus chronic response to Dox: the expression of dominant-interfering p53 in the heart resulted in short-term (1 week) protection against Dox, but worse outcome, compared to wild type mice at 13 weeks post -Dox [46]. Overall, it would appear that the role of p53 in mediating the multiple Dox-induced deleterious effects is context dependent, and likely depends on the model used and the timing of the assessment.
An important effector of Dox-induced cell death is Bnip3 (BH3-only protein Bcl-2-like 19 kDa-interacting protein-3), a member of the Bcl-2 family of proteins. There is evidence that Dox-induced cardiotoxicity is mediated by upregulation of Bnip3 and induction of necrosis in cardiomyocytes [47]. A similar protein, Bnip3L/NIX, an effector of apoptosis, was also found to be upregulated in the heart and myocytes by Dox, in vivo and in vitro, in response to Dox-induced downregulation of miR-30 [48]. To show the crucial role of Bnip3, transgenic mice were created expressing a mutant form of Bnip3 which is unable to insert to mitochondrial membranes; these mice were resistant to Dox induced cardiotoxicity [47]. Bnip3 causes depolarization of mitochondria by promoting mPTP formation which leads to cell death [47]. Both p53 and Bnip3 are also considered to regulate autophagy [47], as will be discussed in the following section. Dox treatment promotes premature ageing of cardiomyocytes, in vitro (long-term cultures) and in vivo [49, 50]. It is of interest that ageing is characterized by decreased potential for autophagy and mitophagy, and this in turn contributes to the pathology of ageing [51]. One might therefore surmise that Dox-induced chronic changes would, as in ageing, also include reduced ability for autophagy. This was shown to be the case by Hoshino and colleagues [52], who reported decreased autophagic elimination of damaged mitochondria (mitophagy) in models of ageing as well as during Dox-induced cardiotoxicity in vitro and in vivo. Figure 1 shows a broad overview of the mechanisms implicated in Dox-cardiotoxicity
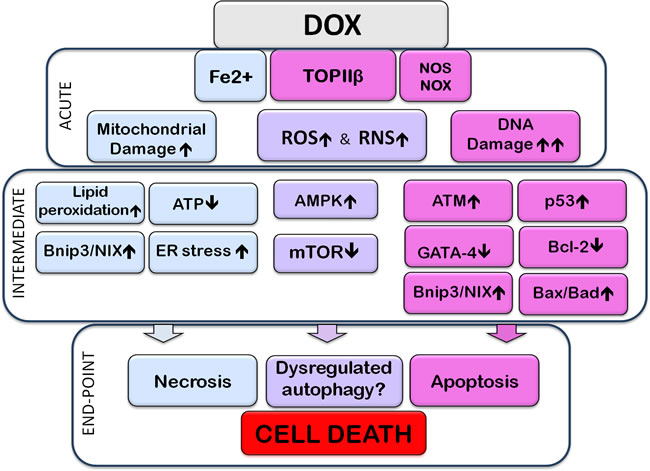
Figure 1: Subcellular events associated with Doxorubicin-induced cardiotoxicity. The image highlights acute, intermediate, and end-point (cell death) events resulting from exposure to Doxorubicin (Dox). The acute section illustrates the direct interaction of Dox with subcellular entities; the intermediate section illustrates direct consequences of these interactions. Events within the acute, or intermediate sections are likely to occur simultaneously and cross-talk with each other. The end-point section is meant to show that the preceding events lead to apoptotic, necrotic, and/or dysregulated autophagy-associated cell death. Acute Events: Dox, upon entering the cell, interacts directly with molecules and organelles: interaction with topoisomerase-IIβ (TOPIIβ) leads to DNA damage. Interaction with nitric oxide synthase (NOS), nicotinamide adenine dinucleotide phosphate-oxidase (NOX) and Fe2+, promotes reactive oxygen or nitrogen species stress (ROS or RNS, respectively), contributing to further DNA damage, oxidation and nitrosylation of proteins and peroxidation of lipids. Dox binds to mitochondrial DNA and impairs the electron transport chain resulting in production of ROS and decreased ATP. Fe2+-Dox complexes are toxic to mitochondria and the endoplasmic reticulum (ER), by causing, for example, lipid (including cardiolipin) peroxidation. The DNA damage response activates the ataxia telangiectasia mutated (ATM) protein which upregulates and activates the tumor suppressor p53. P53 upregulates expression of pro-apoptotic members of the Bcl-2 family such as Bax and Bad; it also increases expression of Bcl2/adenovirus E1B 19 kDa protein-interacting protein 3 (Bnip3), which can cause mitochondrial damage and necrotic cell death, as well as initiate mitophagy. DNA damage and increased levels of ROS lead to downregulation of the transcription factor GATA-4 which decreases expression of the anti-apoptotic, and anti-autophagy-initiation protein, Bcl-2. P53 can also inhibit the activity of mammalian target of Rapamycin (mTOR) signaling, thus dis-inhibiting autophagy initiation. Some studies have indicated that Dox can elicit AMP-activated kinase (AMPK) activation. Activation of AMPK, resulting from reduced ATP levels, can inactivate mTOR and initiate autophagy. Dox-induced effects on ROS and RNS production, mitochondrial and ER damage, DNA and gene expression, culminate in the promotion of apoptotic and necrotic cell death. Dysregulation of the autophagy/mitophagy processes are also linked to Dox-induced cell death. Signals/events associated mostly, although not exclusively, with apoptotic or necrotic cell death are included, respectively, in pink or pale-blue boxes. Pale-purple boxes contain signals/pathways associated with various types of subcellular dysfunction, including autophagic dysregulation.
Autophagy
Autophagy is a conserved process aimed at maintaining cell and tissue homeostasis under normal as well as stress conditions, including nutrient starvation, changes in metabolism, and energy and oxygen status. Autophagy can be subdivided to micro-autophagy, chaperone mediated autophagy, and macro-autophagy; the latter refers to the sequestration of cytosolic cargo by double-membrane bound vesicles, with subsequent fusion with lysosomes for degradation [53]. The term autophagy as used here refers to macro-autophagy. The overall Dox-induced cellular changes such as the accumulation of oxidized and damaged macromolecules and organelles as well as significantly reduced mitochondrial capacity for ATP production would be expected to trigger autophagy.
The autophagy process is composed of several steps. Initiation of autophagy describes the formation of the isolation membrane and phagophore, which then expands to engulf the cargo (protein aggregates or damaged organelles), thus forming the autophagosome. The process is completed by autophagosome clearance which occurs after fusion with lysosomes enabling degradation of cargo by lysosomal enzymes as illustrated in Figure 2. Degradation by-products, such as amino acids, can then be re-used for the building of new macromolecules or for meeting metabolic demands [54].
Several autophagy related genes (Atg) and signal transduction pathways have been implicated in autophagy initiation and progression, and have been described in some detail in recent reviews [55]. Autophagic initiation requires the phosphorylation of the protein Beclin-1 by ULK-1 (activated unc-51-like autophagy activating kinase 1), with subsequent activation of VPS34 (vesicle-mediated vacuolar protein sorting 34) complexes. VPS34 is a class II PI3 kinase, increasing phosphatidyl inositol phosphate (PI3P), involved in the recruitment of other Atg proteins, phagophore formation and elongation [56-58]. The LC3 (light chain 3 microtubule associated protein) is converted to LC-3I after proteolytic cleavage of its C-terminus; LC3-I is then lipidated, becoming LC3-II, which associates with the double membrane of the autophagosome. LC3-II acts as a ‘hook’ for proteins possessing an LC3-II-interacting domain (LIR), recruiting them to the autophagosome. One of these proteins is the scaffold protein p62/ SQSTM1, which possesses both a LIR domain and ubiquitin-binding domain [59]. p62/SQSTM1 attracts ubiquitinated aggregates to the autophagosome, and therefore facilitates their eventual degradation. Completion of the autophagy process, autophagosome clearance following fusion with the lysosome, results in the degradation of p62/ SQSTM1 by lysosomal enzymes. Increased accumulation of p62/ SQSTM1, on the other hand, is linked to the inhibition of autophagic flux/clearance [59]. Other proteins can act in a manner similar to p62/ SQSTM1. These include the “neighbor of BRCA1 gene 1” (NBR1) [60]; the TNF-receptor-associated factor 2 (TRAF2) [61], and the Smad ubiquitin regulatory factor 1, SMURF1 [62].
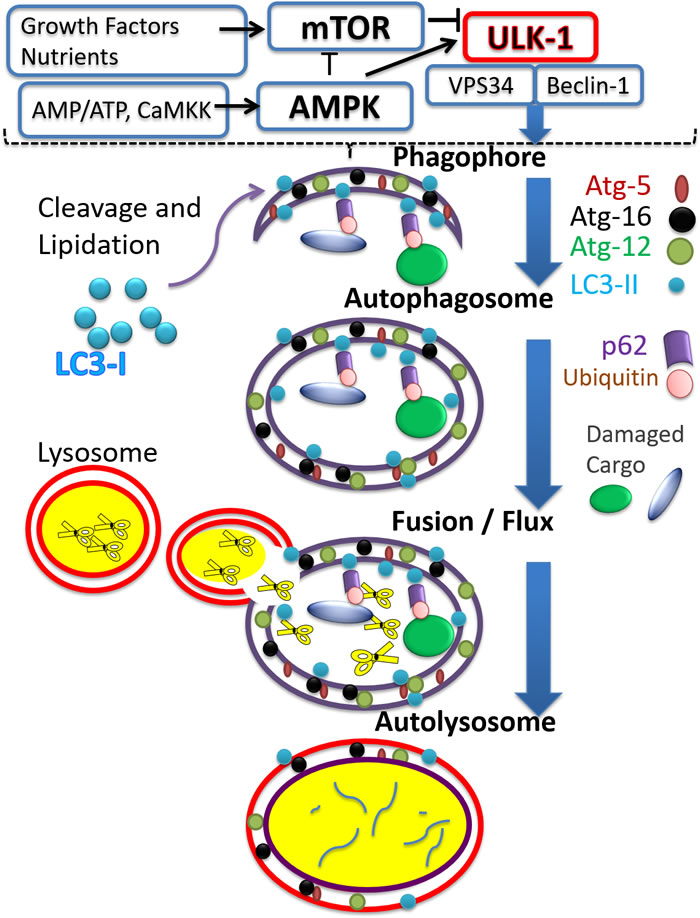
Figure 2: The autophagy process. The figure illustrates major steps constituting the process of autophagy, aiming at recycling targeted cargo. Vertical blue arrows point to a progression from autophagy-initiation via the activated unc-51-like autophagy activating kinase 1 (ULK-1) complex, to the formation/elongation of membranous entities such as the phagophore; to the formation of the autophagosome around the cargo, followed by fusion with the lysosomes and formation of autolysosomes where cargo is degraded. Major signaling pathways regulating the initiation of autophagy are included in the upper portion, separated from the rest of the figure by the broken line, and include the mammalian target of rapamycin (mTOR) and AMP activated kinase (AMPK) pathways. Activators of mTOR, include growth factors and high nutrient status, while activation of AMPK occurs when AMP/ATP ratio increases, and also upon increased calcium activating calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK). The figure shows the antithetical action of mTOR versus AMPK on autophagy initiation, as manifested by ULK-1 activation. ULK-1 mediated activation of Beclin-1 and vesicle-mediated vacuolar protein sorting 34 (VPS34) complex initiates a cascade of events leading to phagophore formation. A few representative proteins associated with the autophagy process are included for each step. Autophagy related gene (Atg) proteins 5, 12, and 16 incorporation elongates the formed phagophore. LC3-II is formed by proteolytic cleavage and lipidation of microtubule-associated proteins light chain 3B (LC3-II) which anchors it to the phagophore. Ubiquitination tags damaged cargo which can then interact with the ubiquitin binding protein, p62/SQSTM1, and, via interaction of the latter with LC3-II, cargo is engulfed within the autophagosome. For completion of the process, fusion of autophagosomes with lysosomes is required, to form autolysosomes. In the autolysosomes cargo and associated proteins are digested by the various degradative lysosomal enzymes, symbolized by scissor images.
Lysosomal associated membrane proteins (LAMPs) are essential for recognition and fusion with autophagosomes; mutations in LAMP2 (Danon’s disease) are associated with incomplete autophagy and accumulation of autophagosomes, resulting in systemic problems, including hypertrophic cardiomyopathy in patients [63].
The most upstream signaling component of the autophagy process is represented by the ULK-1 kinase complex [64]. This complex is under tight control of two major signaling pathways, mammalian target of Rapamycin (mTOR) and AMP activated protein kinase (AMPK). While mTOR inhibits, AMPK activates ULK-1 [58]. Both of these pathways are affected by Dox, although there are some conflicting data, especially regarding the role of AMPK activation.
AMPK is a central sensor of cell energetic status [65]. Low cellular energy levels, increased ROS, and elevated intracellular Ca2+ lead to the phosphorylation and activation of AMPK [66]. Additionally, some cytokines (such as adiponectin) [67, 68] and growth factors (such as vascular endothelial growth factor, VEGF) have been shown to activate AMPK, by activating an upstream phosphorylating enzyme, LKB-1 (Liver Kinase B-1) [68, 69]. Activated AMPK is capable of inducing autophagy by de-repressing mTOR-mediated autophagy inhibition [70].
The serine/threonine kinase mTOR, activated downstream of growth factors and receptor tyrosine kinase signaling, is a highly conserved kinase considered to act as a master regulator of cell growth, metabolism, and protein synthesis, and to inhibit autophagy initiation. The mTOR forms two different signaling complexes (mTORC1 and mTORC2) [71]. The mTORC1 is a point of convergence for autophagy regulatory signals: it binds to the ULK-1 complex (phosphorylating ULK-1 at serine 757) and prevents the ULK-1-mediated phosphorylation of Beclin-1. As mentioned earlier, Beclin-1 phosphorylation is required for autophagy initiation; thus mTORC1 negatively regulates autophagy initiation [57, 58]. Activated AMPK can phosphorylate ULK-1 complex at Ser317 and Ser 777. These phosphorylations are required for autophagy initiation [70]. A schematic representation of the process of autophagy is shown in Figure 2.
DOX and the autophagy process
Stimulation of autophagy prior to Dox is protective
A number of studies have indicated that boosting autophagy prior to the administration of Dox can protect from Dox toxicity. Administration of Rapamycin, an mTOR inhibitor which is used to stimulate autophagy, rescued mice from Dox-induced cardiomyopathy [72]. In aging rats, moderate caloric restriction in combination with resveratrol administration prior to Dox (6 weeks) stimulated autophagy, and protected against subsequent Dox toxicity [73]. Moderate diet restriction in rats, a process expected to stimulate autophagy, protected from acute Dox-injury [74]; in mice, starvation for 2 days upregulated autophagy and protected against Dox [75]. It is anticipated that modest activation of autophagy would eliminate dysfunctional mitochondria which are responsible for increased ROS production, remove any toxic aggregates and generally boost cellular health before Dox insult.
Dox-induced effects on the autophagy process are deleterious
While the boosting of autophagy before Dox appears to be cardioprotective, post-Dox autophagy-associated signaling likely contributes to Dox-induced toxicity and presents a complex, not fully understood picture. Nevertheless, strategies aimed at preventing post-Dox autophagy-initiation and autophagosome formation have been shown to be protective. Pharmacologic inhibition of post-Dox autophagy by 3-methyladenine (a class III PI3-kinase inhibitor) or the silencing of Beclin-1 or Atg-5 protected cardiac cells from Dox toxicity [11, 76-80].
There is emerging consensus that Dox induces cellular changes consistent with autophagy initiation and autophagosome formation in cardiac cells, shown by a variety of approaches. Transmission electron microscopy evaluation in H9C2 cells indicated that Dox increases prevalence of autophagic vacuoles [11]. Dox-induced accumulation of autophagic vacuoles has been shown in mouse [75] and rat hearts [81]. Increased LC3-II levels and upregulation of autophagy related genes have been reported [11, 68, 76-80, 82-85]. Immunofluorescence-based detection of ‘dots’ (autophagosomes) containing either endogenous LC3-II, or overexpressed GFP-LC3 also pointed to the upregulation of autophagosome formation or accumulation, in in vitro and in vivo studies [11, 68, 77, 79, 82, 85]. In some models, however, decreased relative levels of lipidated LC3-II in response to Dox were reported [86-88]. Dox induced up-regulation of Beclin-1 and Atg-5 has been corroborated by several studies [68, 77, 80]. Atg-5 upregulation in mouse hearts and primary cardiomyocytes furthermore is attributed to the overproduction of 4-Hydroxynonenal, 4-HNE through lipid peroxidation, [76].
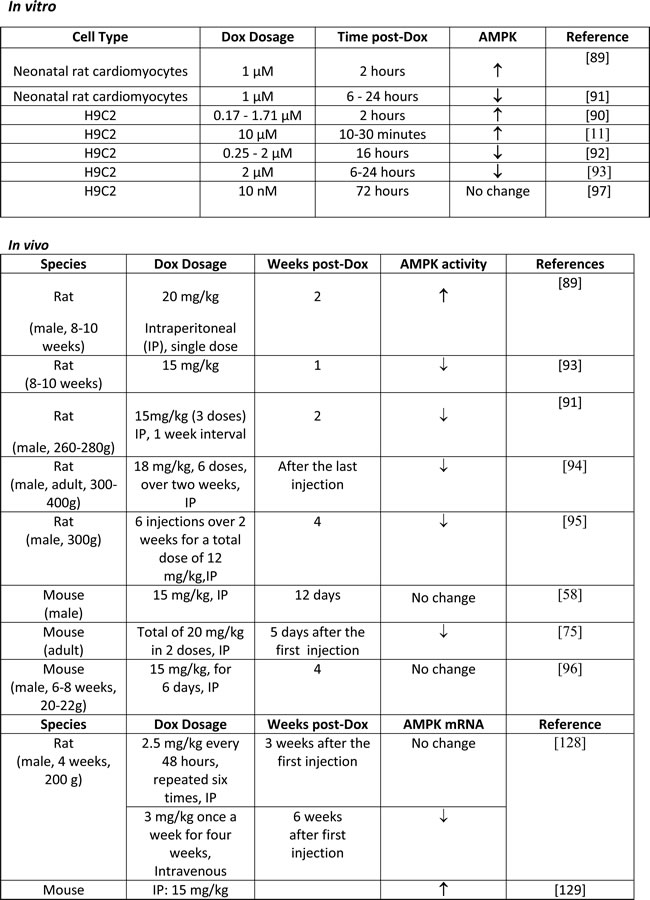
The effect of Dox on specific signals associated with various steps of the autophagy process has been addressed in several studies. AMPK and mTOR signals are considered to play antithetical roles, with AMPK triggering and mTOR inhibiting autophagy initiation [66, 71]. Conflicting reports exist on the effect of Dox on AMPK activation, summarized in Table 1. Several groups, using in vitro and in vivo systems, have reported AMPK activation post-Dox [11, 58, 89, 90]. Decreased cardiac AMPK activity post-Dox has also been described [75, 91-95], while no changes in AMPK activation post-Dox have also been reported [96, 97]. It is possible that the effect of Dox on AMPK is transient or cyclical, and dependent on a multiplicity of parameters including dosage and duration of treatment with Dox and experimental models used. The effects of age, sex and even circadian rhythm may also need to be considered [98].
Table 1: Doxorubicin induced effects on AMPK.
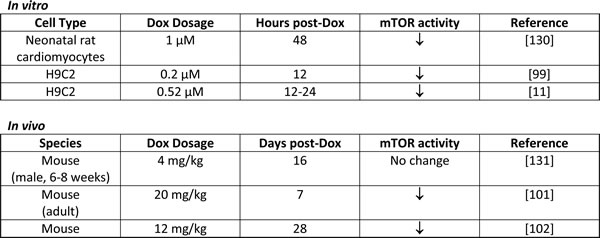
There is strong evidence based on in vitro and in vivo studies in rat, mouse and rabbit models that Dox inhibits mTOR, an inhibition that is expected to contribute to cardiomyocyte injury [11, 90, 99-102], possibly by causing an exacerbated autophagy-initiation response, summarized in Table 2. Dox-induced mTOR inhibition has been linked to p53 upregulation. Mice expressing cardiomyocyte-restricted dominant-interfering p53 are resistant to Dox induced cardiotoxicity, and retain normal levels of active mTOR, at least in the acute setting. Additionally, mice expressing cardiomyocyte-restricted constitutively active mTOR were found to be resistant to Dox insult [101]. Another group reported that p53 upregulation led to increased Bnip3 in cardiomyocytes [43]; knock-down of Bnip3 or dominant-negative inhibition of Bnip3 abrogated p53-induced autophagy.
Table 2: Doxorubicin effects on mTOR.
Another pathway by which Dox can promote autophagy initiation may be through the p53-mediated suppression of the transcription factor GATA-4 and the resulting down-regulation of the pro-survival protein Bcl-2. Bcl-2 binds to Beclin-1 and thus prevents it from interacting with VPS34, and from initiating autophagy [56, 79]. Dox can also promote Bcl-2 phosphorylation which inhibits the Bcl-2/Beclin1 interaction again facilitating autophagy initiation [42].
Overall Dox affects a number of signaling pathways converging to a robust initiation of autophagy and stimulation of autophagosome formation. For the autophagy process to elicit a protective effect, however, it is important that the process is completed through autophagosome clearance. In this context, cargo recognition/tagging (by ubiquitin) and lysosomal degradation are important steps. Thus it is important to examine how Dox affects ubiquitination, the p62/ SQSTM1protein, and the lysosomes.
It has been widely confirmed that Dox significantly increases the total levels of ubiquitinated proteins in cardiomyocytes [72, 75, 78, 82, 85, 86]. The ubiquitinated cargo can interact with p62/ SQSTM1 and facilitate lysosomal targeting. Whether Dox-induced hyper-ubiquitination is due to massive damage to intracellular proteins, dysregulation in signaling pathways of the ubiquitination process, or because of decreased proteasomal degradation is still to be elucidated [103]. It is interesting to note that Dox affects various types of ubiquitin ligases in heart cells. DOX upregulates the muscle specific ubiquitine ligase MuRF 1 (Muscle RING finger 1), and also muscle atrophy F-box (MAFbx)/atrogin-1 which contributes to its cardiotoxic properties [104], while decreasing the expression of Parkin, another E3 ubiquitin ligase which is important for mitophagy [84].
Several groups have examined the effect of Dox on the accumulation of p62/ SQSTM1, with conflicting results. Increased p62/ SQSTM1 levels in the hearts of rat and mouse models post-Dox (1-6 days) have been reported [73, 75, 105]; a decrease in p62/ SQSTM1 has been observed post-Dox [77, 84]. A biphasic response has also been described, consisting of increases in p62/ SQSTM1 at earlier time points post-Dox, followed by decreased or baseline levels at later time points [85, 106].These differences may reflect differences in Dox dosage, sex, different strains of animals, different cell types for in vitro studies, and most importantly, different treatment duration and end-points. This issue needs to be investigated further, because measuring p62/ SQSTM1 levels post-Dox is used to document effects on autophagy clearance/flux and completion of autophagy.
There is an emerging consensus, strengthened by recent studies, that Dox inhibits autophagic flux, by inhibiting lysosomal biogenesis and/or lysosomal function [72, 75, 88, 105, 106]. Bartlett and colleagues demonstrated that Dox decreases expression of TFEB (transcription factor EB) which regulates the expression of genes related to autophagy and lysosomal biogenesis [88]. Decreased TFEB expression in cardiomyocytes led to proteotoxicity and cell death; restoration of TFEB on the other hand restored flux and prevented cell death[88]. Using an experimental model of moderate Dox-induced toxicity, Li and colleagues showed that Dox-induced early upregulation of LC3-II was caused by inhibition of autophagic flux; furthermore, Dox was shown to promote autolysosome accumulation and prevention of lysosomal acidification in vivo [106]. Using tandem green fluorescence (GFP)-red fluorescence (RFP)-LC3 constructs in rat neonatal ventricular cardiomyocytes, the same groups showed that Dox inhibited autophagic flux in vitro. It is of note that while both reports implicated the lysosomes in causing inhibition of autophagic flux, one group did not see any changes in lysosome number per se [106], while the other reported decreased lysosome accumulation due to Dox [88]. This difference likely reflects methodological issues with the use of Lyso-Tracker dye which detects acidic organelles; the increase in lysosomal pH caused by Dox [106] may have led to an underestimation of lysosomes, and autolysosomes, by Lyso-Tracker Red [88, 106]. Regardless of the precise mechanism, impaired autophagic flux caused by Dox, would be expected to promote increased ROS and proteotoxicity.
By attenuating autophagy-initiation through the use of Beclin-1 haplo- insufficient mice Li and colleagues [106] were able to prevent Dox-induced toxicity, illustrating the detrimental role of autophagy initiation post-Dox. An important observation from the work by Li and colleagues was that Dox-induced inhibition of autophagic flux was observed only within a certain time period post-Dox, and not at later time points such as 4 weeks post-Dox [106]. Thus discrepancies between studies regarding the effects of Dox on autophagy may be cause by “lack of evaluation of autophagy as a process of flux”, using instead “snapshot-in time” approaches [106]. Tandem GFP-RFP-LC3 (“green” and “red”)fluorescence reporter approaches are increasingly used to monitor the movement of LC3-II from autophagosome to autolysosome, based on differences in fluorescence caused by differences in pH. The neutral environment of the autophagosome would allow both GFP and RFP fluorescence, while the acidic environment of autolysosome would only allow RFP, “red”, fluorescence. Because, however, Dox was shown to inhibit lysosomal functionality by preventing acidification, reliance on the tandem GFP-RFP-LC3 approach may lead to underestimation of lysosomal, and autolysosomal presence post-Dox [88, 106]. Thus complementary approaches are required to monitor the various steps of the autophagy process after Dox administration.
The use of inhibitors such as Bafilomycin A1 (prevents fusion of autophagosome to the lysosome) and Chloroquine (inhibits lysosomal activity), introduces additional interpretation issues. By blocking lysosomal degradation of cargo these reagents can indicate whether changes in the levels of ‘classic’ autophagy markers (LC3-II and p62/ SQSTM1 for example) are due to an effect on de novo autophagosome formation or on autophagosome cargo degradation [59]. However, the timing of administration, as well as dosage used (sufficient for inhibition but not toxic), need to be standardized, and off-target effects need to be taken into account for meaningful comparison between studies. A detailed description of problems associated the use of lysosomal inhibitors can be found in [104].
Liposomal formulations of Dox have been used to reduce Dox accumulation and toxicity on healthy tissues, and there is evidence from clinical applications that this approach may reduce heart injury [107, 108]. To our knowledge, there are no reports as to how liposomal Dox would affect autophagy in the heart. One may speculate that since heart muscle- associated Dox concentration is reduced when Dox is administered in liposomes [109], Dox-induced deleterious effects, including dysregulated autophagy, would be reduced.
Mitophagy and Dox
Myocardial contractile function and cell survival depend on a high content of healthy mitochondria to ensure sufficient ATP production through oxidative phosphorylation. Defective mitochondria are targeted for autophagic elimination (mitophagy), a process which contributes to the mitochondrial quality control system. Mitophagy plays a fundamental role in cardiomyocyte survival under various cellular stresses. Additional and possibly overlapping processes involved in mitochondrial quality control include the ubiquitin/proteasome-mediated elimination of mitochondrial proteins; and the elimination of toxic mitochondrial contents via formation of mitochondrial-derived vesicles (MDV) [110]. The latter is reported to occur more frequently than mitophagy under baseline conditions, and to represent a fast response to Dox-induced oxidative stress, preceding mitophagy [86].
Various mechanisms mediating the mitophagy process have been described, and reviewed recently in references [111-114]. Specific proteins at the mitochondrial outer membrane can act as selective mitophagy ‘receptors’, by possessing the LIR motif which binds LC3-II and enables mitochondrial engulfment by the autophagosome [115]. Two major pathways to mitophagy have been described for cardiomyocytes, the PINK1/Parkin pathway, and a Parkin-independent pathway, the Bnip3/Nix pathway. Several Parkin-independet mitophagy pathways have been described for various cell types but have not been identified in the cardiac system.
PINK1/Parkin
Parkin-mediated mitophagy occurs in damaged/depolarized mitochondria: depolarization of the outer mitochondrial membrane promotes the stabilization of PINK1, a mitochondrial serine/threonine kinase, at the mitochondrial outer membrane. PINK1 binds to Parkin, facilitating its translocation to mitochondria. Moreover, PINK1 phosphorylates ubiquitin, which is required for the ubiquitin ligase activity of Parkin [113]. Once recruited to mitochondria, Parkin ubiquitinates mitochondrial surface proteins such as Mitofusin 1, 2 (Mfn1, 2), voltage dependent anion channel-1 (VDAC1), and MIRO, a GTPase enzyme facilitating mitochondrial transport [116]. Parkin-dependent ubiquitination of Mfn and MIRO leads to proteasome degradation of these proteins and prevents mitochondrial re-fusion, keeping mitochondria in a fragmented state, stopping mitochondrial movement and promoting mitochondrial autophagic removal [113, 114, 117].
Expression levels of Parkin/PINK1, taken as indicators of mitophagy potential, were reported to show a biphasic response post-Dox in mouse hearts [118]: decreased Parkin/PINK1, at 5 days post Dox suggested decreased Parkin-mediated mitophagy at this time point. At 2 weeks post-Dox, however, expression of these proteins bounced back and reached levels even higher than those of untreated controls. Increased expression of a protein, Fis1, associated with mitochondrial fission and increased mitophagy, was also observed [118]. There is some evidence that the mitochondrial fission and mitophagy inhibitor peptide mdivi-1 prevented the deleterious effects of Dox[119], which would suggest that excessive mitophagy contributes to Dox-toxicity.
Another group has reported that the Dox-induced increase in p53, which can interact with and sequester Parkin to the cytosol, resulted in decreased Parkin translocation to mitochondria and decreased mitophagic potential, which was proposed to contribute to Dox-toxicity by allowing the accumulation of damaged mitochondria. This effect of Dox was not observed in p53-defficient mice, and could be counter-acted by over-expression of Parkin in neonatal rat cardiomyocytes [52]. It should be noted that Hoshino and colleagues [52] measured the mitophagy response after stimulation with carbonyl cyanide m-chlorophenylhydrazone (CCCP), which represent potential for, rather than endogenous, mitophagy.
Bnip3 and Bnip3L/NIX
Two members of BH3-only family proteins, Bnip3 and Bnip3L/NIX which have been associated with apoptotic and necrotic cell death, and the induction of permeability transition, can also act as mitophagy receptors. These proteins contain an LIR motif which enables them to interact with LC3-II on the surface of the autophagosomes. Unlike Parkin, the translocation of Bnip3 to mitochondria does not require loss of membrane potential. The ability of Bnip3 to induce permeability transition pore opening and cell death can be distinct from its ability to promote autophagy; these outcomes are likely modulated by distinct phosphorylation states. The phosphorylation of serines 23 and 17, adjacent to the LIR domain, was reported to promote mitophagy [120]; on the other hand, phosphorylation of C-terminal Bnip3 residues can block cell death, without affecting autophagic elimination [120]. Currently, it is not known how Dox affects Bnip3 or Bip3L/NIX- dependent mitophagy in cardiomyocytes. Following Dox, Bnip3 is reported to translocate to mitochondria and promote permeability transition and depolarization [47]. Depolarization would be expected to recruit Parkin to mitochondria. In fact, the Bnip3-mediated mitophagy is reported to depend on Parkin recruitment in cardiomyocytes [121].
FUNDC1, an outer mitochondrial membrane protein with an LIR motif enabling it to interact with LC3-II has also been linked to mitophagy, although only hypoxia-induced mitophagy has been studied; the role of Dox on FUNDC1 mediated mitophagy remains to be determined [113, 114]. Cardiolipin, which is induced by Dox to translocate to the outer mitochondrial membrane, could also serve as another mitophagy ‘receptor’. An illustration of the various mitophagy receptors is shown in Figure 3. A member of the Bcl-2 family of proteins, Bcl-2 like-13 , possessing a LIR motif, was also identified in mammalian cells as a mitophagy receptor [122].
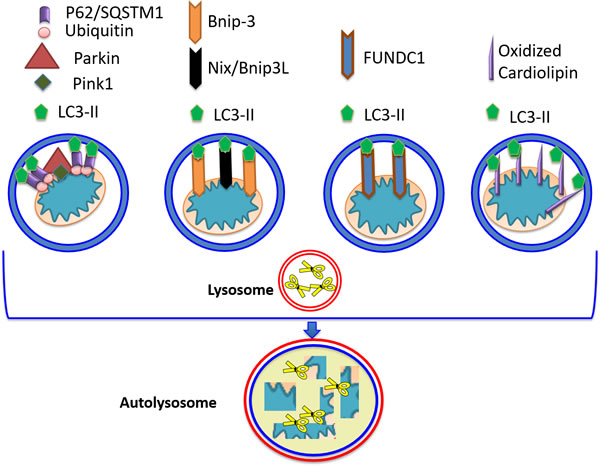
Figure 3: Mitophagy receptors. Four pathways leading to targeting/ recognition of mitochondria for autophagic elimination have been described. The Parkin/PINK1 pathway, operating on depolarized mitochondria, consists of PINK1 stabilization, enabling interaction with Parkin and its translocation to the outer mitochondrial membrane, OMM. Parkin can ubiquitinate various OMM proteins enabling recognition and interaction with p62/SQSTM1. LC3-II can interact with p62/SQSTM1 allowing autophagosomal engulfment of mitochondria, and subsequent degradation via fusion with the lysosome. In the second pathway, Bcl2/adenovirus E1B 19 kDa protein-interacting protein 3 (Bnip3) can also act as mitophagy receptor, as it possesses the LC3-II recognition motif, as well as a transmembrane domain which anchors it to mitochondria. Nix/Bnip3L (the other member of Bnip3 family) acts in a similar fashion. Translocation of Bnip3 to mitochondria does not require loss of membrane potential. Additional molecules possessing LC3-II-interacting ability include FUN14 Domain Containing 1 (FUNDC1), and peroxidized cardiolipin.
There is currently some controversy as to the relevance of the PINK1/Parkin pathway in cardiac baseline or stress-induced mitophagy [123, 124]. A notion that may explain differences between studies is the possibility that different mitophagy receptors could target different mitochondrial populations within the myocytes. Adult cardiomyocytes possess subsarcolemmal (SSM) and interfibrillar mitochondria (IFM), with distinct morphological and functional properties as well as distinct sensitivities to calcium overload [125]. There is currently limited information regarding how these different mitochondrial populations may be targeted for mitophagy, before or after Dox. Recently, a group studied the effects of Dox on SSM and IFM in adult female rats [126]. It was reported that SSM accumulated more Dox than IFM; furthermore, Dox resulted in an increase in PINK1 and a decrease in p62/SQSTM1 in the IFM population only, while Parkin content was not changed in either SSM or IFM. The authors interpreted the results as Dox-induced upregulation of Parkin mediated mitophagy in IFM [126]. However, one can speculate that Dox mediates inhibition of Parkin translocation to the IFM with impaired subsequent p62/SQSTM1 translocation as has also been described in [52].
The relative contribution of the various mitophagy receptors to baseline and Dox-associated mitophagy remains to be determined. One promising approach would be to use the Keima-mouse, expressing a mitochondria-targeted form of the fluorescent protein Keima [127], to examine how mitophagy is affected post-Dox. Fluorescence of the Keima protein is pH sensitive and changes color in the acidic environment of the lysosomes. This mouse model has already demonstrated that baseline levels of cardiac mitophagy are high relative to other organs [127], and could provide valuable information about Dox-associated mitophagy over time, within myocytes and in the various regions of the heart. The mt-Keima mouse could also address the effect of various pre-conditioning, cardioprotective manipulation on the mitophagy process before and after Dox-administration.
Concluding remarks
There is no doubt that a systematic approach will be required to elucidate the role of autophagy and mitophagy in Dox-induced pathology, taking into account potential sex and ageing- related effects. Our overall conclusions based on available evidence are summarized in Figure 4. One can surmise that Dox leads to an over-activation of autophagy initiation while at the same time preventing autophagy completion due to deleterious effects on lysosomes. The resulting accumulation of un-degraded cargo which promotes ROS overproduction would be expected to be toxic to the cells, contributing to cell death. It would seem that Dox dysregulates endogenous mitophagy, and MDV production, in a manner that contributes to Dox toxicity. Since Dox promotes Parkin depletion in a manner similar to the ageing process one can conclude that Dox may selectively diminish Parkin/PINK1-mediated mitophagy. At the same time, Dox-induced Bnip3, and Bnip3L/NIX upregulation would suggest that Bnip3/NIX receptor mediated mitophagy takes over, contributing to Dox-cardiotoxicity.
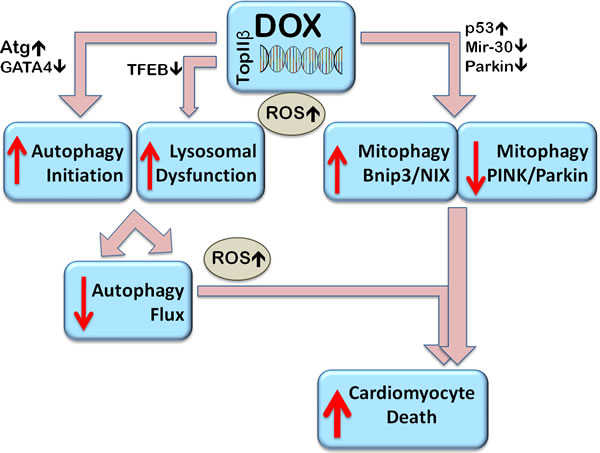
Figure 4: Proposed mechanisms of Dox-induced dysregulation of autophagy and mitophagy resulting in cell death. Dox stimulates autophagy initiation (by upregulating Atg genes, for example) and at the same time, by decreasing expression of the master transcription factor EB (TFEB), impairs function of available lysosomes and prevents their biogenesis resulting in autophagosome accumulation and inhibition of flux. Autophagosome accumulation contributes to the accumulation of reactive oxygen species (ROS). Dox induced changes in gene expression include the downregulation of Parkin and via p53, in impairment of its translocation to mitochondria, thus inhibiting Parkin/PINK1 mediated mitophagy. Parkin-independent mitophagy, such as Bcl-2-like 19 KDa-interacting protein-3 (Bnip3) - mediated mitophagy can be upregulated. Overall, the dysregulated autophagy and impaired mitophagy can induce cardiomyocyte death.
Abbreviations
Dox: Doxorubicin; ROS: reactive oxygen species ; RNS: reactive nitrogen species ; NOS: nitric oxide synthase; iNOS: inducible nitric oxide synthase; eNOS: endothelial nitric oxide synthase; NOX: nicotinamide adenine dinucleotide phosphate-oxidase; mPTP: mitochondrial permeability transition pore; TOPIIα: Topoisomerase-II α; TOPIIβ: Topoisomerase-II β; ATM: ataxia telangiectasia mutated; Bnip3: Bcl2/adenovirus E1B 19 kDa protein-interacting protein 3; Atg: autophagy related genes; ULK-1: unc-51-like autophagy activating kinase 1; VPS34: vesicle-mediated vacuolar protein sorting 34; PI3P: phosphatidyl inositol phosphate; LC3: light chain 3 microtubule associated protein; p62/SQSTM1: p62/Sequestosome-1; NBR1: neighbor of BRCA1 gene 1; TRAF2: TNF-receptor-associated factor 2; SMURF1: Smad ubiquitin regulatory factor 1; LAMP: Lysosomal associated membrane protein; mTOR: mammalian target of Rapamycin; AMPK: AMP activated protein kinase; VEGF: vascular endothelial growth factor; MuRF: Muscle RING finger ; MAFbx: muscle atrophy F-box; TFEB: transcription factor EB; MDV: mitochondrial-derived vesicles; Mfn: Mitofusin; VDAC1: voltage dependent anion channel-1; MIRO: Mitochondrial Rho GTPase; CCCP: carbonyl cyanide m-chlorophenylhydrazone; SSM: subsarcolemmal mitochondria; IFM: interfibrillar mitochondria; mt-Keima: mitochondria keima.
Acknowledgments
We are thankful to Dr. Barbara E. Nickel for editorial help.
Conflicts of interest
The authors declare no conflicts of interest.
Funding
This work was funded through an operating research grant granted by the Canadian Institutes for Health Research (FRN-74733). NK is the recipient of the Alfred E. Deacon doctoral scholarship, Institute of Cardiovascular Sciences, St. Boniface Hospital Albrechtsen Research Centre.
References
1. Lipshultz SE, Miller TL, Lipsitz SR, Neuberg DS, Dahlberg SE, Colan SD, Silverman LB, Henkel JM, Franco VI, Cushman LL, Asselin BL, Clavell LA, Athale U, et al. Continuous Versus Bolus Infusion of Doxorubicin in Children With ALL: Long-term Cardiac Outcomes. Pediatrics. 2012; 130: 1003-11. doi: 10.1542/peds.2012-0727.
2. Lipshultz SE, Scully RE, Lipsitz SR, Sallan SE, Silverman LB, Miller TL, Barry EV, Asselin BL, Athale U, Clavell LA, Larsen E, Moghrabi A, Samson Y, et al. Assessment of dexrazoxane as a cardioprotectant in doxorubicin-treated children with high-risk acute lymphoblastic leukaemia: long-term follow-up of a prospective, randomised, multicentre trial. Lancet Oncol. 2010; 11: 950-61. doi: 10.1016/s1470-2045(10)70204-7.
3. Lipshultz SE, Miller TL, Scully RE, Lipsitz SR, Rifai N, Silverman LB, Colan SD, Neuberg DS, Dahlberg SE, Henkel JM, Asselin BL, Athale UH, Clavell LA, et al. Changes in cardiac biomarkers during doxorubicin treatment of pediatric patients with high-risk acute lymphoblastic leukemia: associations with long-term echocardiographic outcomes. J Clin Oncol. 2012; 30: 1042-9. doi: 10.1200/jco.2010.30.3404.
4. Colombo A, Cipolla C, Beggiato M, Cardinale D. Cardiac toxicity of anticancer agents. Curr Cardiol Rep. 2013; 15: 362. doi: 10.1007/s11886-013-0362-6.
5. Wang EY, Biala AK, Gordon JW, Kirshenbaum LA. Autophagy in the heart: too much of a good thing? J Cardiovasc Pharmacol. 2012; 60: 110-7. doi: 10.1097/FJC.0b013e31824cc427.
6. Zamorano JL, Lancellotti P, Rodriguez Munoz D, Aboyans V, Asteggiano R, Galderisi M, Habib G, Lenihan DJ, Lip GY, Lyon AR, Lopez Fernandez T, Mohty D, Piepoli MF, et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur J Heart Fail. 2017; 19: 9-42. doi: 10.1002/ejhf.654.
7. Desai VG, Herman EH, Moland CL, Branham WS, Lewis SM, Davis KJ, George NI, Lee T, Kerr S, Fuscoe JC. Development of doxorubicin-induced chronic cardiotoxicity in the B6C3F1 mouse model. Toxicol Appl Pharmacol. 2013; 266: 109-21. doi: 10.1016/j.taap.2012.10.025.
8. Zhang YW, Shi J, Li YJ, Wei L. Cardiomyocyte death in doxorubicin-induced cardiotoxicity. Arch Immunol Ther Exp (Warsz). 2009; 57: 435-45. doi: 10.1007/s00005-009-0051-8.
9. Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Naga Prasad SV, Mutharasan RK, Naik TJ, Ardehali H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest. 2014; 124: 617-30. doi: 10.1172/jci72931.
10. Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013; 65: 157-70. doi: 10.1111/j.2042-7158.2012.01567.x.
11. Wang X, Wang XL, Chen HL, Wu D, Chen JX, Wang XX, Li RL, He JH, Mo L, Cen X, Wei YQ, Jiang W. Ghrelin inhibits doxorubicin cardiotoxicity by inhibiting excessive autophagy through AMPK and p38-MAPK. Biochem Pharmacol. 2014; 88: 334-50. doi: 10.1016/j.bcp.2014.01.040.
12. Guo R, Lin J, Xu W, Shen N, Mo L, Zhang C, Feng J. Hydrogen sulfide attenuates doxorubicin-induced cardiotoxicity by inhibition of the p38 MAPK pathway in H9c2 cells. Int J Mol Med. 2013; 31: 644-50. doi: 10.3892/ijmm.2013.1246.
13. Simunek T, Sterba M, Popelova O, Adamcova M, Hrdina R, Gersl V. Anthracycline-induced cardiotoxicity: overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol Rep. 2009; 61: 154-71.
14. Angsutararux P, Luanpitpong S, Issaragrisil S. Chemotherapy-Induced Cardiotoxicity: Overview of the Roles of Oxidative Stress. Oxid Med Cell Longev. 2015; 2015: 795602. doi: 10.1155/2015/795602.
15. Damiani RM, Moura DJ, Viau CM, Caceres RA, Henriques JA, Saffi J. Pathways of cardiac toxicity: comparison between chemotherapeutic drugs doxorubicin and mitoxantrone. Arch Toxicol. 2016; 90: 2063-76. doi: 10.1007/s00204-016-1759-y.
16. Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol. 2012; 52: 1213-25.
17. Neilan TG, Blake SL, Ichinose F, Raher MJ, Buys ES, Jassal DS, Furutani E, Perez-Sanz TM, Graveline A, Janssens SP, Picard MH, Scherrer-Crosbie M, Bloch KD. Disruption of nitric oxide synthase 3 protects against the cardiac injury, dysfunction, and mortality induced by doxorubicin. Circulation. 2007; 116: 506-14.
18. Mukhopadhyay P, Rajesh M, Batkai S, Kashiwaya Y, Hasko G, Liaudet L, Szabo C, Pacher P. Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am J Physiol Heart Circ Physiol. 2009; 296: H1466-83.
19. Miura T, Muraoka S, Ogiso T. Adriamycin-Fe3+-induced mitochondrial protein damage with lipid peroxidation. Biol Pharm Bull. 1995; 18: 514-7.
20. Nohl H. Identification of the site of adriamycin-activation in the heart cell. Biochem Pharmacol. 1988; 37: 2633-7.
21. de Wolf FA. Binding of doxorubicin to cardiolipin as compared to other anionic phospholipids--an evaluation of electrostatic effects. Biosci Rep. 1991; 11: 275-84.
22. Robinson NC. Functional binding of cardiolipin to cytochrome c oxidase. J Bioenerg Biomembr. 1993; 25: 153-63.
23. Parker MA, King V, Howard KP. Nuclear magnetic resonance study of doxorubicin binding to cardiolipin containing magnetically oriented phospholipid bilayers. Biochim Biophys Acta. 2001; 1514: 206-16.
24. Cheung KG, Cole LK, Xiang B, Chen K, Ma X, Myal Y, Hatch GM, Tong Q, Dolinsky VW. Sirtuin-3 (SIRT3) Protein Attenuates Doxorubicin-induced Oxidative Stress and Improves Mitochondrial Respiration in H9c2 Cardiomyocytes. J Biol Chem. 2015; 290: 10981-93. doi: 10.1074/jbc.M114.607960.
25. Galluzzi L, Kepp O, Trojel-Hansen C, Kroemer G. Mitochondrial control of cellular life, stress, and death. Circ Res. 2012; 111: 1198-207. doi: 10.1161/circresaha.112.268946.
26. Unverferth DV, Jagadeesh JM, Unverferth BJ, Magorien RD, Leier CV, Balcerzak SP. Attempt to prevent doxorubicin-induced acute human myocardial morphologic damage with acetylcysteine. J Natl Cancer Inst. 1983; 71: 917-20.
27. Broeyer FJ, Osanto S, Suzuki J, de Jongh F, van Slooten H, Tanis BC, Bruning T, Bax JJ, Ritsema van Eck HJ, de Kam ML, Cohen AF, Mituzhima Y, Burggraaf J. Evaluation of lecithinized human recombinant super oxide dismutase as cardioprotectant in anthracycline-treated breast cancer patients. Br J Clin Pharmacol. 2014; 78: 950-60. doi: 10.1111/bcp.12429.
28. Vincent DT, Ibrahim YF, Espey MG, Suzuki YJ. The role of antioxidants in the era of cardiooncology. Cancer Chemother Pharmacol. 2013; 72: 1157-68. doi: 10.1007/s00280-013-2260-4.
29. Green H, Stal O, Bachmeier K, Backlund LM, Carlsson L, Hansen J, Lagerlund M, Norberg B, Franzen A, Aleskog A, Malmstrom A. Pegylated liposomal doxorubicin as first-line monotherapy in elderly women with locally advanced or metastatic breast cancer: novel treatment predictive factors identified. Cancer Lett. 2011; 313: 145-53. doi: 10.1016/j.canlet.2011.07.017.
30. Jones SE, Collea R, Paul D, Sedlacek S, Favret AM, Gore I Jr, Lindquist DL, Holmes FA, Allison MA, Brooks BD, Portillo RM, Vukelja SJ, Steinberg MS, et al. Adjuvant docetaxel and cyclophosphamide plus trastuzumab in patients with HER2-amplified early stage breast cancer: a single-group, open-label, phase 2 study. Lancet Oncol. 2013; 14: 1121-8. doi: 10.1016/s1470-2045(13)70384-x.
31. Press MF, Sauter G, Buyse M, Bernstein L, Guzman R, Santiago A, Villalobos IE, Eiermann W, Pienkowski T, Martin M, Robert N, Crown J, Bee V, et al. Alteration of topoisomerase II-alpha gene in human breast cancer: association with responsiveness to anthracycline-based chemotherapy. J Clin Oncol. 2011; 29: 859-67. doi: 10.1200/jco.2009.27.5644.
32. O’Malley FP, Chia S, Tu D, Shepherd LE, Levine MN, Huntsman D, Bramwell VH, Andrulis IL, Pritchard KI. Topoisomerase II alpha protein and responsiveness of breast cancer to adjuvant chemotherapy with CEF compared to CMF in the NCIC CTG randomized MA.5 adjuvant trial. Breast Cancer Res Treat. 2011; 128: 401-9. doi: 10.1007/s10549-011-1511-5.
33. Burgess DJ, Doles J, Zender L, Xue W, Ma B, McCombie WR, Hannon GJ, Lowe SW, Hemann MT. Topoisomerase levels determine chemotherapy response in vitro and in vivo. Proc Natl Acad Sci U S A. 2008; 105: 9053-8. doi: 10.1073/pnas.0803513105.
34. Lyu YL, Kerrigan JE, Lin CP, Azarova AM, Tsai YC, Ban Y, Liu LF. Topoisomerase IIbeta mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007; 67: 8839-46. doi: 10.1158/0008-5472.can-07-1649.
35. Deng S, Yan T, Jendrny C, Nemecek A, Vincetic M, Godtel-Armbrust U, Wojnowski L. Dexrazoxane may prevent doxorubicin-induced DNA damage via depleting both topoisomerase II isoforms. BMC Cancer. 2014; 14: 842. doi: 10.1186/1471-2407-14-842.
36. Tokarska-Schlattner M, Lucchinetti E, Zaugg M, Kay L, Gratia S, Guzun R, Saks V, Schlattner U. Early effects of doxorubicin in perfused heart: transcriptional profiling reveals inhibition of cellular stress response genes. Am J Physiol Regul Integr Comp Physiol. 2010; 298: R1075-88. doi: 10.1152/ajpregu.00360.2009.
37. Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, Yeh ET. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012; 18: 1639-42. doi: 10.1038/nm.2919.
38. Hasinoff BB, Hellmann K, Herman EH, Ferrans VJ. Chemical, biological and clinical aspects of dexrazoxane and other bisdioxopiperazines. Curr Med Chem. 1998; 5: 1-28.
39. Vavrova A, Jansova H, Mackova E, Machacek M, Haskova P, Tichotova L, Sterba M, Simunek T. Catalytic inhibitors of topoisomerase II differently modulate the toxicity of anthracyclines in cardiac and cancer cells. PLoS One. 2013; 8: e76676.
40. Yoshida M, Shiojima I, Ikeda H, Komuro I. Chronic doxorubicin cardiotoxicity is mediated by oxidative DNA damage-ATM-p53-apoptosis pathway and attenuated by pitavastatin through the inhibition of Rac1 activity. J Mol Cell Cardiol. 2009; 47: 698-705. doi: 10.1016/j.yjmcc.2009.07.024.
41. Chang YL, Lee HJ, Liu ST, Lin YS, Chen TC, Hsieh TY, Huang HS, Huang SM. Different roles of p53 in the regulation of DNA damage caused by 1,2-heteroannelated anthraquinones and doxorubicin. Int J Biochem Cell Biol. 2011; 43: 1720-8. doi: 10.1016/j.biocel.2011.08.006.
42. Velez JM, Miriyala S, Nithipongvanitch R, Noel T, Plabplueng CD, Oberley T, Jungsuwadee P, Van Remmen H, Vore M, St Clair DK. p53 Regulates oxidative stress-mediated retrograde signaling: a novel mechanism for chemotherapy-induced cardiac injury. PLoS One. 2011; 6: e18005. doi: 10.1371/journal.pone.0018005.
43. Wang EY, Gang H, Aviv Y, Dhingra R, Margulets V, Kirshenbaum LA. p53 mediates autophagy and cell death by a mechanism contingent on Bnip3. Hypertension. 2013; 62: 70-7. doi: 10.1161/hypertensionaha.113.01028.
44. Park AM, Nagase H, Liu L, Vinod Kumar S, Szwergold N, Wong CM, Suzuki YJ. Mechanism of anthracycline-mediated down-regulation of GATA4 in the heart. Cardiovasc Res. 2011; 90: 97-104.
45. Feridooni T, Hotchkiss A, Remley-Carr S, Saga Y, Pasumarthi KB. Cardiomyocyte specific ablation of p53 is not sufficient to block doxorubicin induced cardiac fibrosis and associated cytoskeletal changes. PLoS One. 2011; 6: e22801. doi: 10.1371/journal.pone.0022801.
46. Zhu W, Zhang W, Shou W, Field LJ. P53 inhibition exacerbates late-stage anthracycline cardiotoxicity. Cardiovasc Res. 2014; 103: 81-9. doi: 10.1093/cvr/cvu118.
47. Dhingra R, Margulets V, Chowdhury SR, Thliveris J, Jassal D, Fernyhough P, Dorn GW 2nd, Kirshenbaum LA. Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling. Proc Natl Acad Sci U S A. 2014; 111: E5537-44. doi: 1414665111 [pii] 10.1073/pnas.1414665111.
48. Roca-Alonso L, Castellano L, Mills A, Dabrowska AF, Sikkel MB, Pellegrino L, Jacob J, Frampton AE, Krell J, Coombes RC, Harding SE, Lyon AR, Stebbing J. Myocardial MiR-30 downregulation triggered by doxorubicin drives alterations in beta-adrenergic signaling and enhances apoptosis. Cell Death Dis. 2015; 6: e1754. doi: 10.1038/cddis.2015.89.
49. Maejima Y, Adachi S, Ito H, Hirao K, Isobe M. Induction of premature senescence in cardiomyocytes by doxorubicin as a novel mechanism of myocardial damage. Aging Cell. 2008; 7: 125-36.
50. Xu X, Pang J, Chen Y, Bucala R, Zhang Y, Ren J. Macrophage Migration Inhibitory Factor (MIF) Deficiency Exacerbates Aging-Induced Cardiac Remodeling and Dysfunction Despite Improved Inflammation: Role of Autophagy Regulation. Sci Rep. 2016; 6: 22488.
51. Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK, Sadoshima J. Aging and Autophagy in the Heart. Circ Res. 2016; 118: 1563-76.
52. Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, Ikeda K, Ogata T, Matoba S. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. 2013; 10:322-37.
53. Terman A, Brunk UT. Autophagy in cardiac myocyte homeostasis, aging, and pathology. Cardiovasc Res. 2005; 68: 355-65. doi: 10.1016/j.cardiores.2005.08.014.
54. Kim KH, Lee MS. Autophagy-a key player in cellular and body metabolism. Nat Rev Endocrinol. 2014. doi: 10.1038/nrendo.2014.35.
55. Wesselborg S, Stork B. Autophagy signal transduction by ATG proteins: from hierarchies to networks. Cell Mol Life Sci. 2015; 72: 4721-57. doi: 10.1007/s00018-015-2034-8.
56. Fu LL, Cheng Y, Liu B. Beclin-1: autophagic regulator and therapeutic target in cancer. Int J Biochem Cell Biol. 2013; 45: 921-4. doi: 10.1016/j.biocel.2013.02.007.
57. Nazarko VY, Zhong Q. ULK1 targets Beclin-1 in autophagy. Nat Cell Biol. 2013; 15: 727-8. doi: 10.1038/ncb2797.
58. Hong EG, Kim BW, Jung DY, Kim JH, Yu T, Seixas Da Silva W, Friedline RH, Bianco SD, Seslar SP, Wakimoto H, Berul CI, Russell KS, Lee KW, et al. Cardiac expression of human type 2 iodothyronine deiodinase increases glucose metabolism and protects against doxorubicin-induced cardiac dysfunction in male mice. Endocrinology. 2013; 154: 3937-46. doi: 10.1210/en.2012-2261.
59. Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012; 8: 445-544.
60. Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol. 2010; 12: 836-41. doi: 10.1038/ncb0910-836.
61. Yang KC, Ma X, Liu H, Murphy J, Barger PM, Mann DL, Diwan A. Tumor necrosis factor receptor-associated factor 2 mediates mitochondrial autophagy. Circ Heart Fail. 2015; 8: 175-87. doi: 10.1161/circheartfailure.114.001635.
62. Orvedahl A, Sumpter R Jr, Xiao G, Ng A, Zou Z, Tang Y, Narimatsu M, Gilpin C, Sun Q, Roth M, Forst CV, Wrana JL, Zhang YE, et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011; 480: 113-7. doi: 10.1038/nature10546.
63. Ruivo R, Anne C, Sagne C, Gasnier B. Molecular and cellular basis of lysosomal transmembrane protein dysfunction. Biochim Biophys Acta. 2009; 1793: 636-49. doi: 10.1016/j.bbamcr.2008.12.008.
64. Egan D, Kim J, Shaw RJ, Guan KL. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. 2011; 7: 643-4.
65. Russell RC, Yuan HX, Guan KL. Autophagy regulation by nutrient signaling. Cell Res. 2014; 24: 42-57.
66. Qi D, Young LH. AMPK: energy sensor and survival mechanism in the ischemic heart. Trends Endocrinol Metab. 2015; 26: 422-9. doi: 10.1016/j.tem.2015.05.010.
67. Konishi M, Haraguchi G, Ohigashi H, Ishihara T, Saito K, Nakano Y, Isobe M. Adiponectin protects against doxorubicin-induced cardiomyopathy by anti-apoptotic effects through AMPK up-regulation. Cardiovasc Res. 2011; 89: 309-19. doi: 10.1093/cvr/cvq335.
68. Zhang C, Liao Y, Li Q, Chen M, Zhao Q, Deng R, Wu C, Yang A, Guo Z, Wang D, He X. Recombinant adiponectin ameliorates liver ischemia reperfusion injury via activating the AMPK/eNOS pathway. PLoS One. 2013; 8: e66382. doi: 10.1371/journal.pone.0066382.
69. Zhang Y, Zhao J, Li R, Lau WB, Yuan YX, Liang B, Li R, Gao EH, Koch WJ, Ma XL, Wang YJ. AdipoRon, the first orally active adiponectin receptor activator, attenuates postischemic myocardial apoptosis through both AMPK-mediated and AMPK-independent signalings. Am J Physiol Endocrinol Metab. 2015; 309: E275-82. doi: 10.1152/ajpendo.00577.2014.
70. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011; 13: 132-41. doi: 10.1038/ncb2152.
71. Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010; 584: 1287-95. doi: 10.1016/j.febslet.2010.01.017.
72. Xu X, Bucala R, Ren J. Macrophage migration inhibitory factor deficiency augments doxorubicin-induced cardiomyopathy. J Am Heart Assoc. 2013; 2: e000439. doi: 10.1161/jaha.113.000439.
73. Dutta D, Xu J, Dirain ML, Leeuwenburgh C. Calorie restriction combined with resveratrol induces autophagy and protects 26-month-old rat hearts from doxorubicin-induced toxicity. Free Radic Biol Med. 2014; 74: 252-62.
74. Mitra MS, Donthamsetty S, White B, Latendresse JR, Mehendale HM. Mechanism of protection of moderately diet restricted rats against doxorubicin-induced acute cardiotoxicity. Toxicol Appl Pharmacol. 2007; 225: 90-101.
75. Kawaguchi T, Takemura G, Kanamori H, Takeyama T, Watanabe T, Morishita K, Ogino A, Tsujimoto A, Goto K, Maruyama R, Kawasaki M, Mikami A, Fujiwara T, et al. Prior starvation mitigates acute doxorubicin cardiotoxicity through restoration of autophagy in affected cardiomyocytes. Cardiovasc Res. 2012; 96: 456-65.
76. Sun A, Cheng Y, Zhang Y, Zhang Q, Wang S, Tian S, Zou Y, Hu K, Ren J, Ge J. Aldehyde dehydrogenase 2 ameliorates doxorubicin-induced myocardial dysfunction through detoxification of 4-HNE and suppression of autophagy. J Mol Cell Cardiol. 2014; 71:92-104.
77. Xu X, Chen K, Kobayashi S, Timm D, Liang Q. Resveratrol attenuates doxorubicin-induced cardiomyocyte death via inhibition of p70 S6 kinase 1-mediated autophagy. J Pharmacol Exp Ther. 2012; 341: 183-95. doi: 10.1124/jpet.111.189589.
78. Chen K, Xu X, Kobayashi S, Timm D, Jepperson T, Liang Q. Caloric restriction mimetic 2-deoxyglucose antagonizes doxorubicin-induced cardiomyocyte death by multiple mechanisms. J Biol Chem. 2011; 286: 21993-2006.
79. Kobayashi S, Volden P, Timm D, Mao K, Xu X, Liang Q. Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death. J Biol Chem. 2010; 285: 793-804.
80. Lu L, Wu W, Yan J, Li X, Yu H, Yu X. Adriamycin-induced autophagic cardiomyocyte death plays a pathogenic role in a rat model of heart failure. Int J Cardiol. 2009; 134: 82-90.
81. Wu S, Zhu L, Yang J, Fan Z, Dong Y, Luan R, Cai J, Fu L. Hydrogen-containing saline attenuates doxorubicin-induced heart failure in rats. Pharmazie. 2014; 69: 633-6.
82. Dimitrakis P, Romay-Ogando MI, Timolati F, Suter TM, Zuppinger C. Effects of doxorubicin cancer therapy on autophagy and the ubiquitin-proteasome system in long-term cultured adult rat cardiomyocytes. Cell Tissue Res. 2012; 350: 361-72. doi: 10.1007/s00441-012-1475-8.
83. Nordgren KK, Wallace KB. Keap1 redox-dependent regulation of doxorubicin-induced oxidative stress response in cardiac myoblasts. Toxicol Appl Pharmacol. 2014; 274: 107-16. doi: 10.1016/j.taap.2013.10.023.
84. Sishi BJ, Bester DJ, Wergeland A, Loos B, Jonassen AK, van Rooyen J, Engelbrecht AM. Daunorubicin therapy is associated with upregulation of E3 ubiquitin ligases in the heart. Exp Biol Med (Maywood). 2012; 237: 219-26. doi: 10.1258/ebm.2011.011106.
85. Zhang YY, Meng C, Zhang XM, Yuan CH, Wen MD, Chen Z, Dong DC, Gao YH, Liu C, Zhang Z. Ophiopogonin D attenuates doxorubicin-induced autophagic cell death by relieving mitochondrial damage in vitro and in vivo. J Pharmacol Exp Ther. 2015; 352: 166-74. doi: 10.1124/jpet.114.219261.
86. Sishi BJ, Loos B, van Rooyen J, Engelbrecht AM. Autophagy upregulation promotes survival and attenuates doxorubicin-induced cardiotoxicity. Biochem Pharmacol. 2013; 85: 124-34. doi: 10.1016/j.bcp.2012.10.005.
87. Dickey JS, Gonzalez Y, Aryal B, Mog S, Nakamura AJ, Redon CE, Baxa U, Rosen E, Cheng G, Zielonka J, Parekh P, Mason KP, Joseph J, et al. Mito-tempol and dexrazoxane exhibit cardioprotective and chemotherapeutic effects through specific protein oxidation and autophagy in a syngeneic breast tumor preclinical model. PLoS One. 2013; 8: e70575. doi: 10.1371/journal.pone.0070575.
88. Bartlett JJ, Trivedi PC, Yeung P, Kienesberger PC, Pulinilkunnil T. Doxorubicin Impairs Cardiomyocyte Viability by Suppressing Transcription Factor EB Expression and Disrupting Autophagy. Biochem J. 2016; 473:3769-3789.
89. Lv X, Yu X, Wang Y, Wang F, Li H, Wang Y, Lu D, Qi R, Wang H. Berberine inhibits doxorubicin-triggered cardiomyocyte apoptosis via attenuating mitochondrial dysfunction and increasing Bcl-2 expression. PLoS One. 2012; 7: e47351. doi: 10.1371/journal.pone.0047351.
90. Chen MB, Wu XY, Gu JH, Guo QT, Shen WX, Lu PH. Activation of AMP-activated protein kinase contributes to doxorubicin-induced cell death and apoptosis in cultured myocardial H9c2 cells. Cell Biochem Biophys. 2011; 60: 311-22. doi: 10.1007/s12013-011-9153-0.
91. Gao S, Li H, Feng XJ, Li M, Liu ZP, Cai Y, Lu J, Huang XY, Wang JJ, Li Q, Chen SR, Ye JT, Liu PQ. alpha-Enolase plays a catalytically independent role in doxorubicin-induced cardiomyocyte apoptosis and mitochondrial dysfunction. J Mol Cell Cardiol. 2015; 79: 92-103. doi: 10.1016/j.yjmcc.2014.11.007.
92. Wang S, Song P, Zou MH. Inhibition of AMP-activated protein kinase alpha (AMPKalpha) by doxorubicin accentuates genotoxic stress and cell death in mouse embryonic fibroblasts and cardiomyocytes: role of p53 and SIRT1. J Biol Chem. 2012; 287: 8001-12. doi: 10.1074/jbc.M111.315812.
93. Gu J, Hu W, Song ZP, Chen YG, Zhang DD, Wang CQ. Resveratrol-induced autophagy promotes survival and attenuates doxorubicin-induced cardiotoxicity. Int Immunopharmacol. 2016; 32: 1-7. doi: 10.1016/j.intimp.2016.01.002.
94. Andreadou I, Mikros E, Ioannidis K, Sigala F, Naka K, Kostidis S, Farmakis D, Tenta R, Kavantzas N, Bibli SI, Gikas E, Skaltsounis L, Kremastinos DT, et al. Oleuropein prevents doxorubicin-induced cardiomyopathy interfering with signaling molecules and cardiomyocyte metabolism. J Mol Cell Cardiol. 2014; 69: 4-16. doi: 10.1016/j.yjmcc.2014.01.007.
95. Gratia S, Kay L, Potenza L, Seffouh A, Novel-Chate V, Schnebelen C, Sestili P, Schlattner U, Tokarska-Schlattner M. Inhibition of AMPK signalling by doxorubicin: at the crossroads of the cardiac responses to energetic, oxidative, and genotoxic stress. Cardiovasc Res. 2012; 95: 290-9. doi: 10.1093/cvr/cvs134.
96. Sun A, Cheng Y, Zhang Y, Zhang Q, Wang S, Tian S, Zou Y, Hu K, Ren J, Ge J. Aldehyde dehydrogenase 2 ameliorates doxorubicin-induced myocardial dysfunction through detoxification of 4-HNE and suppression of autophagy. J Mol Cell Cardiol. 2014; 71: 92-104. doi: 10.1016/j.yjmcc.2014.01.002.
97. Kobashigawa LC, Xu YC, Padbury JF, Tseng YT, Yano N. Metformin protects cardiomyocyte from doxorubicin induced cytotoxicity through an AMP-activated protein kinase dependent signaling pathway: an in vitro study. PLoS One. 2014; 9: e104888. doi: 10.1371/journal.pone.0104888.
98. Asher G, Schibler U. Crosstalk between components of circadian and metabolic cycles in mammals. Cell Metab. 2011; 13: 125-37. doi: 10.1016/j.cmet.2011.01.006.
99. Lee BS, Oh J, Kang SK, Park S, Lee SH, Choi D, Chung JH, Chung YW, Kang SM. Insulin Protects Cardiac Myocytes from Doxorubicin Toxicity by Sp1-Mediated Transactivation of Survivin. PLoS One. 2015; 10: e0135438. doi: 10.1371/journal.pone.0135438.
100. Chahine N, Makhlouf H, Duca L, Martiny L, Chahine R. Cardioprotective effect of saffron extracts against acute doxorubicin toxicity in isolated rabbit hearts submitted to ischemia-reperfusion injury. Z Naturforsch C. 2014; 69: 459-70. doi: 10.5560/znc.2014-0124.
101. Zhu W, Soonpaa MH, Chen H, Shen W, Payne RM, Liechty EA, Caldwell RL, Shou W, Field LJ. Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation. 2009; 119: 99-106. doi: 10.1161/circulationaha.108.799700.
102. Singla DK. Akt-mTOR Pathway Inhibits Apoptosis and Fibrosis in Doxorubicin-Induced Cardiotoxicity Following Embryonic Stem Cell Transplantation. Cell Transplant. 2015; 24: 1031-42. doi: 10.3727/096368914x679200.
103. Sishi BJ, Loos B, van Rooyen J, Engelbrecht AM. Doxorubicin induces protein ubiquitination and inhibits proteasome activity during cardiotoxicity. Toxicology. 2013; 309: 23-9. doi: 10.1016/j.tox.2013.04.016.
104. da Silva MG, Mattos E, Camacho-Pereira J, Domitrovic T, Galina A, Costa MW, Kurtenbach E. Cardiac systolic dysfunction in doxorubicin-challenged rats is associated with upregulation of MuRF2 and MuRF3 E3 ligases. Exp Clin Cardiol. 2012; 17: 101-9.
105. Li S, Wang W, Niu T, Wang H, Li B, Shao L, Lai Y, Li H, Janicki JS, Wang XL, Tang D, Cui T. Nrf2 deficiency exaggerates doxorubicin-induced cardiotoxicity and cardiac dysfunction. Oxid Med Cell Longev. 2014; 2014: 748524. doi: 10.1155/2014/748524.
106. Li DL, Wang ZV, Ding G, Tan W, Luo X, Criollo A, Xie M, Jiang N, May H, Kyrychenko V, Schneider JW, Gillette TG, Hill JA. Doxorubicin Blocks Cardiomyocyte Autophagic Flux by Inhibiting Lysosome Acidification. Circulation. 2016; 133: 1668-87. doi: 10.1161/circulationaha.115.017443.
107. Rafiyath SM, Rasul M, Lee B, Wei G, Lamba G, Liu D. Comparison of safety and toxicity of liposomal doxorubicin vs. conventional anthracyclines: a meta-analysis. Exp Hematol Oncol. 2012; 1: 10.
108. Xing M, Yan F, Yu S, Shen P. Efficacy and Cardiotoxicity of Liposomal Doxorubicin-Based Chemotherapy in Advanced Breast Cancer: A Meta-Analysis of Ten Randomized Controlled Trials. PLoS One. 2015; 10: e0133569.
109. Luo R, Li Y, He M, Zhang H, Yuan H, Johnson M, Palmisano M, Zhou S, Sun D. Distinct biodistribution of doxorubicin and the altered dispositions mediated by different liposomal formulations. Int J Pharm. 2017; 519: 1-10.
110. Cadete VJ, Deschenes S, Cuillerier A, Brisebois F, Sugiura A, Vincent A, Turnbull D, Picard M, McBride HM, Burelle Y. Formation of mitochondrial-derived vesicles is an active and physiologically relevant mitochondrial quality control process in the cardiac system. J Physiol. 2016; 594: 5343-62. doi: 10.1113/jp272703.
111. Tong M, Sadoshima J. Mitochondrial autophagy in cardiomyopathy. Curr Opin Genet Dev. 2016; 38: 8-15. doi: 10.1016/j.gde.2016.02.006.
112. Hamacher-Brady A, Brady NR. Mitophagy programs: mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell Mol Life Sci. 2016; 73: 775-95.
113. Moyzis AG, Sadoshima J, Gustafsson AB. Mending a broken heart: the role of mitophagy in cardioprotection. Am J Physiol Heart Circ Physiol. 2015; 308: H183-92. doi: 10.1152/ajpheart.00708.2014.
114. Saito T, Sadoshima J. Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ Res. 2015; 116: 1477-90. doi: 10.1161/circresaha.116.303790.
115. Yamaguchi O, Murakawa T, Nishida K, Otsu K. Receptor-mediated mitophagy. J Mol Cell Cardiol. 2016; 95: 50-6. doi: 10.1016/j.yjmcc.2016.03.010.
116. Saxton WM, Hollenbeck PJ. The axonal transport of mitochondria. J Cell Sci. 2012; 125: 2095-104. doi: 10.1242/jcs.053850.
117. Wrighton KH. Organelle dynamics. Stopping mitochondria in their tracks. Nat Rev Mol Cell Biol. 2012; 13: 4-5. doi: 10.1038/nrm3251.
118. Hull TD, Boddu R, Guo L, Tisher CC, Traylor AM, Patel B, Joseph R, Prabhu SD, Suliman HB, Piantadosi CA, Agarwal A, George JF. Heme oxygenase-1 regulates mitochondrial quality control in the heart. JCI Insight. 2016; 1: e85817. doi: 10.1172/jci.insight.85817.
119. Gharanei M, Hussain A, Janneh O, Maddock H. Attenuation of doxorubicin-induced cardiotoxicity by mdivi-1: a mitochondrial division/mitophagy inhibitor. PLoS One. 2013; 8: e77713. doi: 10.1371/journal.pone.0077713.
120. Zhu Y, Massen S, Terenzio M, Lang V, Chen-Lindner S, Eils R, Novak I, Dikic I, Hamacher-Brady A, Brady NR. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem. 2013; 288: 1099-113. doi: 10.1074/jbc.M112.399345.
121. Lee Y, Lee HY, Hanna RA, Gustafsson AB. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2011; 301: H1924-31. doi: 10.1152/ajpheart.00368.2011.
122. Otsu K, Murakawa T, Yamaguchi O. BCL2L13 is a mammalian homolog of the yeast mitophagy receptor Atg32. Autophagy. 2015; 11: 1932-3. doi: 10.1080/15548627.2015.1084459.
123. Dorn GW 2nd. Parkin-dependent mitophagy in the heart. J Mol Cell Cardiol. 2016; 95: 42-9.
124. Song M, Gong G, Burelle Y, Gustafsson AB, Kitsis RN, Matkovich SJ, Dorn GW 2nd. Interdependence of Parkin-Mediated Mitophagy and Mitochondrial Fission in Adult Mouse Hearts. Circ Res. 2015; 117: 346-51.
125. Srisakuldee W, Makazan Z, Nickel BE, Zhang F, Thliveris JA, Pasumarthi KB, Kardami E. The FGF-2-triggered protection of cardiac subsarcolemmal mitochondria from calcium overload is mitochondrial connexin 43-dependent. Cardiovasc Res. 2014; 103: 72-80. doi: 10.1093/cvr/cvu066.
126. Kavazis AN, Morton AB, Hall SE, Smuder AJ. Effects of doxorubicin on cardiac muscle subsarcolemmal and intermyofibrillar mitochondria. Mitochondrion. 2016; 34:9-19.
127. Sun N, Yun J, Liu J, Malide D, Liu C, Rovira, II, Holmstrom KM, Fergusson MM, Yoo YH, Combs CA, Finkel T. Measuring In Vivo Mitophagy. Mol Cell. 2015; 60: 685-96.
128. Wu R, Wang HL, Yu HL, Cui XH, Xu MT, Xu X, Gao JP. Doxorubicin toxicity changes myocardial energy metabolism in rats. Chem Biol Interact. 2016; 244: 149-58. doi: 10.1016/j.cbi.2015.12.010.
129. Pointon AV, Walker TM, Phillips KM, Luo J, Riley J, Zhang SD, Parry JD, Lyon JJ, Marczylo EL, Gant TW. Doxorubicin in vivo rapidly alters expression and translation of myocardial electron transport chain genes, leads to ATP loss and caspase 3 activation. PLoS One. 2010; 5: e12733. doi: 10.1371/journal.pone.0012733.
130. Bian Y, Sun M, Silver M, Ho KK, Marchionni MA, Caggiano AO, Stone JR, Amende I, Hampton TG, Morgan JP, Yan X. Neuregulin-1 attenuated doxorubicin-induced decrease in cardiac troponins. Am J Physiol Heart Circ Physiol. 2009; 297: H1974-83. doi: 10.1152/ajpheart.01010.2008.
131. Sturgeon K, Schadler K, Muthukumaran G, Ding D, Bajulaiye A, Thomas NJ, Ferrari V, Ryeom S, Libonati JR. Concomitant low-dose doxorubicin treatment and exercise. Am J Physiol Regul Integr Comp Physiol. 2014; 307: R685-92. doi: 10.1152/ajpregu.00082.2014.