
INTRODUCTION
The receptor tyrosine kinases (RTKs) Tie2 and Tie1 and their angiopoietin (Ang) endothelial growth factor ligands (in the human, Ang1–Ang4) are known to be involved in the formation of blood vessels—both developmental and pathological [1–6]. In studies to delineate the exact roles of these kinases and their ligands, the greater part of the research effort to date has been directed to Tie2 and Ang1 and Ang2 [7, 8]. It is now known that Ang1 mediates endothelial cell growth, proliferation, and neovascularization through binding and activation of Tie2 [9]. The role of Ang2 appears to be somewhat more complex: in some cases it serves as a “decoy” to prevent Ang1-Tie2 binding, whereas in others it forms a complex with Tie2 to facilitate angiogenesis [4, 7, 8].
Ang1 and Ang2 are multimerized by their coiled-coil and N-terminal domains, with their receptor binding activity residing in their carboxy-terminal fibrinogen-like domains [10–13]. Upon binding to Ang1 multimers, the Tie2 receptor oligomerizes, bringing its kinase domains into close proximity and thereby enabling the kinase domains to phosphorylate each other to promote cell proliferation, migration, and sprouting [14]. Activation of Tie2 via tyrosine phosphorylation initiates the phosphatidylinositol 3-kinase (PI3K)-Akt pathway, which leads to Akt and MAPK/ERK signaling and hence to both endothelial cell survival and migration phenotypes [15, 16].
Accumulating evidence has revealed a significant correlation between the expression of Ang1 and Ang2 in tumor invasion and metastasis in a variety of human cancers [17–19]. Similarly, Tie2 overexpression has been implicated in breast, ovarian, and liver cancers and in glioblastomas [20], with Tie2 reaching its highest levels in the peripheral neovascular endothelium of invasive tumors [21]. In light of the above, it seems likely that molecules targeting Tie2 would show promise as cancer therapeutics and diagnostics and also as tools to study Tie2 activation and inhibition during angiogenesis. Nonetheless, the work that has been done on molecules targeting the Tie2 receptor has met with very little success: there are currently no FDA-approved therapeutics that selectively target Tie2, and there are only a few Tie2-kinase domain inhibitors in pre-clinical trials, probably due to the toxicity or off-target effects of these inhibitors [22]. In a different approach, angiopoietin antagonists, including the soluble recombinant extracellular domains of the Tie2 receptor, have been investigated as potential anti-tumor therapeutic agents [23–25]. For example, blocking angiopoietin function by binding the ligand to a soluble Tie2 ectodomain (Tie2-Fc) inhibited tumorigenesis and neovascularization in postnatal rats, presumably due to depletion of the available ligand. However, given that multiple angiopoietin isoforms (i.e., Ang1–4) can bind to Tie2, it is not clear whether this effect is mediated by the inhibition of multiple isoforms or of a single isoform, and the former scenario may lead to unwanted effects. Yet another approach is based on therapeutic monoclonal antibodies and antibody fragments that target growth factors and their receptors. These strategies have indeed met with clinical success, but they are not without their limitations. To date, there is only one peptide-Fc fusion protein (peptibody) that neutralizes the interaction between Ang1 and Ang2 with Tie2; this peptibody has exhibited potency in systemic xenograft models and in a rat corneal model of angiogenesis [26, 27]. Similarly, there are only a few human monoclonal antibodies targeting the Ang2 ligand in early-stage clinical trials [28–30].
In light of the research described above, it seems that the approaches that hold the most promise for developing therapeutics are those aimed at inhibiting ligand-mediated receptor activation by receptor antagonists derived from natural ligands [31–42]. Our strategy is therefore to use natural ligands as the basis for engineering protein-protein interactions, since natural effectors will interact with ligand-binding residues on the receptors, without the limitations associated with antibodies and peptibodies, which would not necessarily bind these functionally important epitopes. An additional advantage of ligand-based antagonists is that they are inherently likely to be more specific than kinase domain small molecule inhibitors; this advantage is conferred by their large interaction surface, which involves both the highly conserved active site and different surrounding target-interacting residues. However, the less than optimal ligand binding affinity and specificity, expression yield, and stability have impeded the development of ligand-based antagonists as cancer therapeutics: to date, only a few such therapeutics have advanced to clinical trials [43, 44].
Previous work has, however, provided an indication as to how the potential of ligand-based antagonists could be realized: In particular, a rational engineering approach was applied to transform the natural multimeric Ang1 agonist into a monomeric high-affinity Tie2 antagonist [9]. The ligand multimerization was abolished by mutating two amino acids on residues critical for Ang1 multimerization in the superclustering domain (SCD) of the molecules (i.e., C41S and C54S). The Ang1C41S,C54S variant bound Tie2 well but did not multimerize properly and, consequently, could not activate Tie2 [9]. Since the diminished affinity of Ang1C41S,C54S (relative to wild-type Ang1) for Tie2 presumably resulted in a low inhibitory effect of the mutant in Ang1-mediated processes [3,45], we reasoned that the development of efficacious monomeric Ang-based antagonists would be contingent on improving their affinity for Tie2.
Taking advantage of the high sequence and structural similarities between Ang1 and the natural Tie2 antagonist, Ang2, we began by constructing a monomeric version of Ang2, namely, the Ang2-binding domain (Ang2-BD). The next step in our strategy was to engineer the natural monomeric Ang2-BD for higher affinity binding and subsequently improved antagonism to Tie2. To this end, by using yeast surface display (YSD) to randomly screen Ang2 variants [46], with increasingly stringent sorts against Tie2, we identified mutants with the highest stabilities and target affinities. The final steps were to demonstrate that these high-affinity Tie2 antagonists are potent inhibitors of Tie2 signaling in vitro and are able to inhibit angiogenesis in cell-based models.
RESULTS
Affinity maturation of Ang2-BD YSD libraries
Wild-type Ang2-BD (Ang2-BDWT) was created as the starting point for affinity maturation towards recombinant human (rh)Tie2. It was first necessary to test the compatibility of Ang2-BD with the YSD system that was to be used subsequently as a platform for the creation of the Ang2-BD library and affinity maturation towards Tie2. To this end, Ang2-BD was cloned into a YSD plasmid (pCTCON) and presented on the yeast cell surface as a fusion to agglutinin proteins. High yeast display and Tie2 binding levels were detected for Ang2-BD by staining with fluorescently labeled antibodies as compared to unstained controls. A 12-amino-acid linker (LPDKPLAFQDPS) was added between the cMyc tag and Ang2-BD to prevent steric hindrance between the two antibodies (Supplementary Figure 1). A yeast-displayed library in which random mutations were introduced to the Ang2-BD gene was generated using error-prone PCR, with 2–9 mutations per clone and a yield of approximately 6 × 106 transformants. This Ang2-BD first-generation library, enriched for expression, was subjected to four additional rounds of sorting with decreasing concentrations of Tie2 (Figure 1A–1D).The sorting gates are shown in Figure 1C for the selection of clones with high affinity relative to their expression. The expression and binding of the YSD library at the beginning and the end of the sorting process are shown in Figure 1C and 1D, respectively.
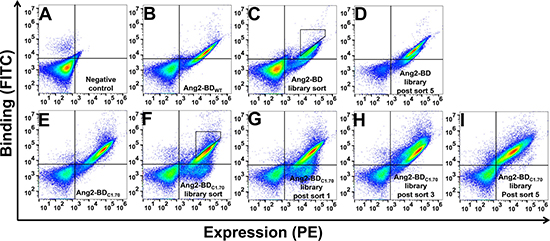
Figure 1: Screening of first- and second-generation Ang2-BD libraries against soluble Tie2. Shown is a FACS analysis of yeast expressing Ang2-BD. (A) Negative control. (B) Ang2-BDWT expression and binding of Tie2 (10 nM). (C) Ang2-BD library expression and binding of Tie2 (10 nM). (D) Ang2-BD library expression and binding of Tie2 (10 nM) after five rounds of sorting. (E) Ang2-BDC1.70 expression and binding of Tie2 (5 nM). (F) Ang2-BDC1.70 library sort expression and binding of Tie2 (5 nM). (G–I) Ang2-BDC1.70 library expression and binding of Tie2 (5 nM) after sorts 1, 3 and 5, respectively. Sorts 2–5 were conducted using gates similar to the one shown in panel F.
Isolation of clones from the first-generation library with improved binding affinity towards Tie2
To identify specific Ang2-BD variants with improved Tie2 binding affinity, 70 individual clones were isolated from the fifth sort of the affinity maturation. Most of the clones showed a 50% increase in affinity relative to Ang2-BDWT, with clone C1.70 showing the highest (2.5-fold) increase in affinity (Supplementary Figure 2). Not surprisingly, sequencing analysis of individual clones isolated from this first-generation library revealed mutations, such as K432N, I434T, N467K, F469L, N470D and Y475H, that are located within the Ang2-BD/Tie2 binding interface. In particular, in clone 70 (C1.70), which had the highest affinity towards Tie2, there were three mutations in its binding interface and one additional mutation in close proximity to the Ang2-BD-Tie2 interface (Supplementary Table 1).
Isolation from the second-generation library of clones showing further improvement in binding affinity towards Tie2
Screening a second-generation library based on the Ang2-BDC1.70 variant (Figure 1E) resulted in a diversity of approximately 8 × 106 transformants. Five rounds of sorting, starting with library expression enrichment and followed by target screening at decreasing concentrations of Tie2, were performed (Figure 1F–1I). Following the sorting, a significant shift of the library towards high affinity binding was clearly evident (Figure 1I). With the aim to isolate variants with improved binding affinities from the second-generation library, 60 individual clones were isolated, sequenced and tested for their binding affinity towards Tie2 (Supplementary Table 2). Based on the sequencing results and the increase in Tie2 binding affinity, two clones, Ang2-BDC1.70 and Ang2-BDC2.36, were chosen for their increased binding affinity (2.5- and 17-fold as compared to Ang2-BDWT, respectively) towards Tie2 (Supplementary Figure 3). Figure 2 shows the positions that were mutated in wild-type Ang2-BD to generate Ang2-BDC2.36. Ang2-BDC1.70 and Ang2-BDC2.36 were purified and used in the subsequent experiments.
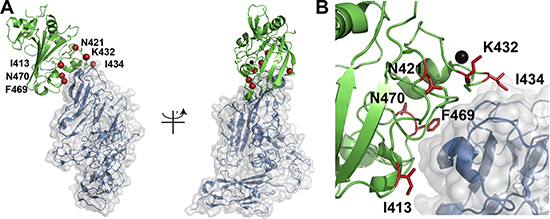
Figure 2: Localization of Ang2-BD mutations. (A) The Ang2/Tie2 complex (PDB 2GY7) is shown in cartoon format with Ang2 in green and Tie2 in blue. The location of mutations in Ang2-BDC2.36 are shown as red spheres at the Cα atom and labelled accordingly. The panel on the left is rotated 90 degrees along the y-axis relative to the image on the right. (B) A close-up view of the Ang2/Tie2 binding interface highlighting the location of the six mutations. A calcium atom is shown in space filling format while the mutations are in ball-and-stick.
Production and biochemical evaluation of soluble Ang2-BD variants
Ang2-BDWT and the mutant variants were produced recombinantly in Pichia pastoris GS115 strain and purified using affinity chromatography, followed by treatment with endoglycosidase H (Endo Hf) to remove all N-linked carbohydrates (Supplementary Figure 4) and size-exclusion chromatography (SEC) (Supplementary Figure 5). Circular dichroism (CD) spectra revealed no change in structure of Ang2-BDC1.70 and Ang2-BDC2.36 variants in both their glycosylated and non-glycosylated forms in comparison to Ang2-BDWT (Supplementary Figure 6A). Thermal denaturation of the purified proteins revealed that the melting temperature (Tm) of Ang2-BDWT and Ang2-BDC1.70 was 49°C, and that of Ang2-BDC2.36 was 47°C (Supplementary Figure 6B). The binding kinetics of Ang2-BD variants to Tie2 were determined by surface plasmon resonance (SPR) (Figure 3A). The KD values were found to be: Ang2-BDWT 434±37 nM, Ang2-BDC1.70 71.25±2.34 nM, and Ang2-BDC2.36 42.61±0.77 nM and 51.45±1.36 nM for the non-glycosylated and the glycosylated forms, respectively (Table 1). The KD values demonstrate an improvement in binding to Tie2 of 6-fold for the first-generation variant (Ang2-BDC1.70) and 10-fold for the second-generation variant (Ang2-BDC2.36). The kinetic parameters for the first- and second-generation variants are shown in Table 1. No significant difference in binding affinity was observed between the glycosylated and the non-glycosylated forms of Ang2-BDC2.36. The affinity of the purified Ang2-BD variants to Tie2 showed the same trend as that observed with the yeast displayed Ang2-BD variants, demonstrating the utility of YSD in quantitatively discriminating between clones that differ by as little as 6-fold (Ang2-BDC1.70 vs. Ang2-BDWT) in binding affinity to the desired target.
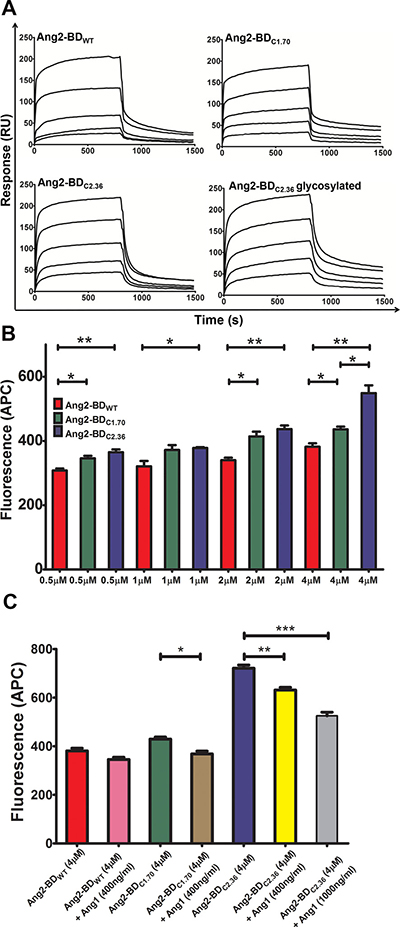
Figure 3: Binding of Ang2-BD variants to recombinant and cell-expressed human Tie2. (A) Representative SPR sensorgrams of binding of Ang2-BD variants to immobilized Tie2. The ranges of protein concentrations analyzed are indicated in parentheses: Ang2-BDWT (18.75 nM – 300 nM); Ang2BDC1.70 (6.25 nM – 100 nM); Ang2-BDC2.36 (5 nM – 80 nM); Ang2-BDC2.36 glycosylated (6.25 nM – 100 nM). (B) Binding of Ang2-BD variants to TIME cell line: 1 ×105 cells were incubated with indicated proteins (Ang2-BDWT, Ang2-BDC1.70 and Ang2-BDC2.36, red, green and blue, respectively) for 2 h at 4°C with a gentle agitation. Mean fluorescence values were determined by flow cytometry using a fluorescently labeled antibody against a FLAG epitope tag. Data shown is the average of triplicate experiments, and error bars represent standard error of the mean. *indicates P value < 0.05 for comparison of results between Ang2-BD variants at the same concentration. (C) Competitive binding assay of Ang2-BDWT, Ang2-BDC1.70 and Ang2-BDC2.36, without Ang1 (red, green and blue, respectively) and with 400 ng/ml Ang1 (pink, brown and yellow, respectively) and 1000 ng/ml Ang1 for Ang2-BDC2.36 competition (grey). *indicates P value <0.05 for comparison of results between Ang2-BD variants with and without Ang1. Data shown is the average of triplicate experiments, and error bars represent standard error of the mean.
Table 1: Equilibrium binding affinities and kinetic rate constants for Ang2-BD variants to immobilized Tie2
Variant |
SPR (immobilized Tie2) |
|||||
|---|---|---|---|---|---|---|
Steady state |
Two state binding model |
|||||
KD ± SEM, nM |
KD ± SEM, nM |
Kon1 ( M-1 s-1) × 105 |
Koff1 (s-1) × 10-2 |
Kon2 (s-1) × 10-3 |
Koff2 (s-1) × 10-4 |
|
Ang2-BDWT |
434 ± 37 |
- |
- |
- |
- |
- |
Ang2-BDC1.70 |
* |
71.25 ± 2.34 |
17.5 ± 0.01 |
12.1 ± 0.06 |
1.43 ± 0.01 |
5.62 ± 0.02 |
Ang2-BDC2.36 |
* |
42.61 ± 0.77 |
7.71 ± 0.02 |
3.16 ± 0.01 |
1.23 ± 0.01 |
15.22 ± 0.06 |
Ang2-BDC2.36glycosylated |
* |
51.45 ± 1.36 |
5.59 ± 0.01 |
2.71 ± 0.01 |
1.73 ± 0.01 |
7.39 ± 0.03 |
-Values could not be accurately determined due to inadequate fit to two state binding model.
*Values could not be accurately determined from steady state model.
Ang2-BD variants bind to cell-expressed Tie2
Binding of Ang2-BD variants to cell-expressed Tie2 was evaluated using the human telomerase-immortalized microvascular endothelium (TIME) cell line. These cells expressed Tie2 on their surface (Supplementary Figure 7), and Ang2-BD variants were found to bind to these cells in a dose-response manner (Figure 3B). Ang2-BDC2.36 exhibited the highest affinity towards TIME cells relative to the other Ang2-BD variant and to Ang2-BDWT. A competitive binding assay using Ang1 demonstrated that the Ang2-BD variants bind to Tie2 at the same epitope as does Ang1 (Figure 3C).
Ang2-BD variants inhibit Tie2 phosphorylation of TIME cells
To test the ability of the Ang2-BD variants to inhibit Tie2 phosphorylation, we used a system exploiting the endogenous expression of basal levels of Ang1, which induce phosphorylation of Tie2, in TIME cells. This phosphorylation is enhanced by the addition of soluble full-length Ang1 to the cell culture [8]. The results obtained demonstrate that the Ang2-BD variants could significantly inhibit Tie2 phosphorylation induced by both endogenous and soluble Ang1 and thus act as functional antagonists. In particular, the engineered high-affinity binder Ang2-BDC2.36 was found to be a more potent antagonist than Ang2-BDWT (Figure 4).
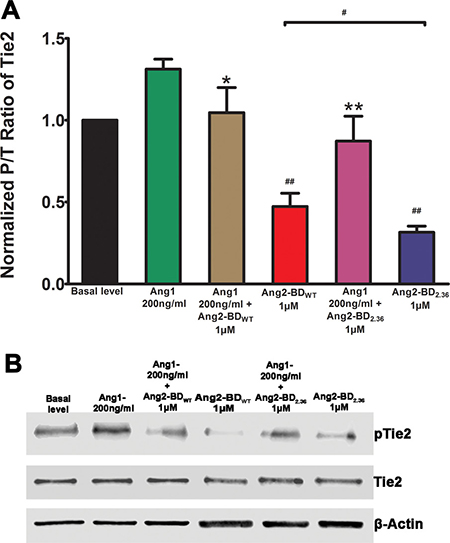
Figure 4: Inhibition of Tie2 phosphorylation by Ang2-BD variants. (A) TIME cells were treated with control buffer (basal level, black), 200 ng/ml of Ang1 (green), 200 ng/ml of Ang1 and 1 μM Ang2-BDWT (brown), 1 μM Ang2-BDWT(red), 200 ng/ml of Ang1 and 1 μM Ang2-BDc2.36 (blue) and 1 μM Ang2-BDC2.36 (purple) for 15 min for Tie2 phosphorylation. (B) Cell lysates were analyzed by western blot using antibodies against pTie2, Tie2 and β-actin. # indicates P value < 0.05 for comparison of results between Ang2-BDWT and Ang2-BDC2.36; ## indicates P value < 0.01 for comparison of results between Ang1 + Ang2-BDWT and Ang2-BDWT and between Ang1 + Ang2-BDC2.36 and Ang2-BDC2.36; *indicates P value < 0.05 for comparison of results between Ang1 and Ang1 + Ang2-BDWT; **indicates P value < 0.01 for comparison of results between Ang1 and Ang1 + Ang2-BDC2.36. Data shown is the average of triplicate experiments, and error bars represent standard error of the mean.
Ang2-BD variants inhibit tube formation and invasiveness of endothelial cells
Ang2-BD variants were tested for their ability to inhibit capillary tube formation by TIME cells grown on Matrigel, an extracellular basement membrane matrix. The Ang2-BD variants inhibited tube formation in a dose-dependent manner, and Ang2-BDC2.36 was found to be a potent inhibitor in comparison to Ang2-BDWT (Figure 5). When Ang2-BD variants were tested for their ability to inhibit endothelial cells invasiveness, it was found that Ang2-BDC2.36 was superior to Ang2-BDWT in inhibiting the invading cells (Figure 6).
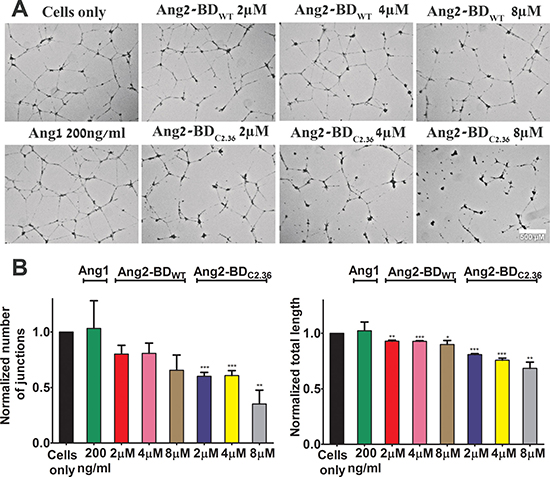
Figure 5: Inhibition of tube formation in endothelial cells by Ang2-BD variants. (A) TIME cells were treated with the indicated proteins. (B) Control buffer (cells only, black), 200 ng/ml of Ang1 (green), 2 μM, 4 μM and 8 μM Ang2-BDWT (red, pink and brown respectively), 2 μM, 4 μM and 8 μM Ang2-BDC2.36 (blue, yellow and grey respectively). Tube structures were analyzed for the number of generated junctions and the total tube length. *indicates P value < 0.05 for comparison of results between cells alone and tested proteins. Data shown is the average of triplicate experiments, and error bars represent standard error of the mean. Scale bar, 500 μm.
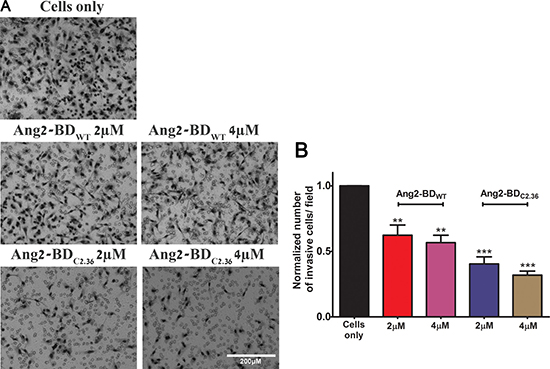
Figure 6: Inhibition of endothelial cells invasiveness by Ang2-BD variants. (A) TIME cells were treated with indicated proteins in Boyden chambers. (B) Control buffer (cells only, black), 2 μM and 4 μM Ang2-BDWT (red and pink, respectively), 2 μM and 4 μM Ang2-BDC2.36 (blue and brown, respectively). The invading cells accumulated on the bottom of membrane were counted in 16 frames for each membrane and analyzed for the number of cells. *indicates P value < 0.05 for comparison of results between cells only and cells + tested proteins. Data shown is the average of triplicate experiments, and error bars represent standard error of the mean. Scale bar, 200 μm.
DISCUSSION
The success story of protein-based therapeutics centers on monoclonal antibodies [47, 48] that exert their activity either through immune-related effector functions or by inhibiting dysregulated ligand-receptor interactions. The above notwithstanding, the development of monoclonal antibodies that target Ang/Tie2 interactions has met with little success, and only a few human monoclonal antibodies that target the angiopoietin ligand (3.19.3 and MEDI3617) have entered early-stage clinical trials [28, 29]. The current study opens the way to a more promising approach, namely, a combinatorial methodology for engineering natural ligands to function as alternatives to antibodies. We have shown that this method is indeed an effective strategy for creating ligand-based RTK receptor inhibitors and molecular tools to study the effects of growth factor/RTK recognition on receptor activation and cell function.
Some success has indeed been achieved by applying rational methods relying on natural ligand-receptor interactions to the engineering of growth factor-based RTK antagonists [38, 44]. Nonetheless, despite the relative simplicity of these methods, the development of first-generation agents as therapeutics has met with several difficulties in terms of improving binding affinity, stability and expression. For example, a study designed to engineer vascular endothelial growth factor-A (VEGFA) from its natural dimeric form into a monomer showed that the binding affinity of the monomer for the ligand’s VEGFR2 receptor was markedly less (by three orders of magnitude) than that of natural dimer [49]. Similarly, a number of studies have shown significantly decreased stability of single domain ligands engineered in isolation from their stabilizing domains [50, 51]. Finally, low levels of recombinant expression have been reported for multidomain mammalian proteins, such as growth factor ligands and RTKs with clinical potential [52, 53].
In a similar vein, a previous approach for transforming a multimeric Ang1 agonist into an antagonist, having two amino acid mutations corresponding to residues critical for Ang1 multimerization at the supercluster domain (SCD) of the molecules (i.e., C41S and C54S), produced an Ang1 variant that could not multimerize and was unable to activate Tie2, although it did bind well to Tie2 [3, 9, 45]. In addition, this recombinant monomeric Ang1 variant exhibited low expression yields and low stability [3, 45]. To overcome these limitations, particularly to generate new Ang-based variants with improved expression, stability and affinity to Tie2, we utilized combinatorial Ang2-BD libraries for generating protein diversity and YSD as a quantitative selection platform for enhancing expression, affinity and stability.
Sequencing of 70 and 60 clones selected from the final sorting rounds of the first and second generation Ang2-BD libraries, yielded 5 and 14 unique sequences for each library, respectively. None of the first generation Ang2-BDC1.70 mutations either reverted to wild-type residues or was replaced with other mutations during the second-generation library screens, demonstrating the strength and effectiveness of the YSD system in affinity maturation.
We found that two mutations, namely N467K and F469L, in the affinity-matured clones were common to the clones selected from the two independent libraries. This result supports the idea that these mutations, which are located in the Ang2-BD/Tie2 binding interface, are indeed involved in the improved affinity between Ang2-BD and Tie2, through direct binding interactions. The fact that three mutations were observed for position 467 (N467Y, N467H and N467K) further suggests that this position stabilizes the Ang2-BD/Tie2 complex. Another common mutation (already identified from the first-generation screens), namely, I413T, was located very close to the Ang2-BD/Tie2 binding interface [11] and could therefore also contribute to the conformational stability of the binding site (Figure 2).
The most commonly observed mutations fall into two classes: those located within the Ang2-BD/Tie2 binding interface (residues 421, 432, 434, 436, 467, 469, 470 and 475), and those located along the Ang2-BD backbone (residues 302, 304, 324, 330, 343, 353, 359, 386, 389, 393, 402, 407, 413 and 415). The most marked changes in amino acid properties were evident in mutations K432N, N467K, N467H, N470D, Y475H, and smaller changes, in N421Y, I434T and N467Y. The above mutations manifested as electrostatic changes (K432N, N467K, N467H, N470D and Y475H), as changes in the hydrophobicity (N421Y and N467Y), or as a combination of altered size and altered hydrophobicity (I434T).
Mutations K432N and I434T were clustered together in the first-generation clone Ang2-BDC1.70. It is possible that these mutations together contribute to binding. In the second-generation sorts, K436R, which lies in close proximity to K432N and I434T, was added to this cluster in clone Ang2-BDC2.19. Importantly, a comparison of the second-generation sequences allowed us to identify unique mutations that independently enhance the binding of Ang2-BD to Tie2. These include N421Y, N467K, N467Y and F469L; all had an individual effect on binding affinity.
Steady state and two-state binding models were used to obtain the best fit for the experimental SPR data for the respective Ang2-BDWT and the engineered high affinity variants. The differences between the observed binding mechanisms may result from mutations within the calcium-binding loop (Figure 2), which may affect calcium binding and, as a result, the mode of interaction of Ang2-BD/Tie2 [11]. The binding of the engineered second-generation Ang2-BD mutant Ang2-BDC2.36 to Tie2 was approximately twice as strong as that of the first generation mutant Ang2-BDC1.70 and 10-fold stronger than that of the parental Ang2-BDWT. Glycosylation of Ang2-BDC2.36 did not have any influence on its affinity for Tie2, suggesting that the interaction between this variant and Tie2 was mediated solely by the amino acid residues of Ang2-BDC2.36. This finding is also in agreement with previous reported data for Ang2-BDWT protein [11].
Ang2-BDC2.36 bound more strongly than Ang2-BDWT to endothelial cells, with the binding being dose dependent. This variant was able to compete with full-length Ang1 for binding to endothelial cells, demonstrating direct interaction with cellular Tie2. Not surprisingly, the ability of Ang2-BDWT and its Ang2-BDC2.36 variant to inhibit Tie2 phosphorylation in TIME cells was highly correlated with their affinity for recombinant soluble Tie2 and for cell-surface-expressed Tie2. As expected, both Ang2-BDWT and its Ang2-BDC2.36 variant inhibited Tie2 phosphorylation, with effect of the variant being stronger. A similar inhibitory trend (but a weaker effect) was observed in the presence of exogenous agonistic Ang1, which served as a Tie2 inducer.
Having established that the affinity-matured Ang2-BD variants do indeed antagonize Tie2 in endothelial cells, we examined the effect of the Ang2-BD variants on the angiogenesis process in these cells. An in vitro endothelial tube formation assay of endothelial cells incubated with Ang2-BD variants on an extracellular basement membrane matrix showed Ang2-BDC2.36 to be a potent inhibitor of the formation of capillary-like structures. In a different assay, the Ang2-BD variants were tested for their ability to inhibit the invasiveness of endothelial cells. Here again, the ability of Ang2-BDWT and the affinity-matured variant Ang2-BDC2.36 to inhibit the formation of capillary-like structures by endothelial cells and to inhibit endothelial cell invasiveness was correlated with the affinity of the variants to Tie2. The results of the phosphorylation and tube formation assays thus further define the roles of Ang1 and Ang2-BD as Tie2 agonists and antagonists, respectively.
In conclusion, our combinatorial engineering strategy has provided both new tools for studying the molecular mechanisms that mediate Ang- and Tie2-dependent angiogenesis and further insight into the sequence-structure-function triad of the antagonistic Ang mutants. Perhaps even more importantly, the promising findings support the use of our strategy as a template for engineering high-affinity agents from natural protein ligands to create functional protein antagonists against biomedical targets.
MATERIALS AND METHODS
Preparation of YSD Ang2-BD constructs and libraries
The construct for Ang2-BDWT (amino acids 281 to 496) was obtained by custom gene synthesis (Integrated DNA Technologies). Amplification of the gene was performed using primers containing NheI and BamHI restriction sites and a 12-amino-acid linker (LPDKPLAFQDPS) to connect the C-terminus of the Ang2-BDWT protein with a c-Myc epitope. The amplified gene was then introduced into the pCTCON yeast display vector (a generous gift from the laboratory of Dane Wittrup, MIT). The first-generation library was prepared using the Ang2-BDWT construct as the template, and the library was generated by error-prone PCR and homologous recombination into Saccharomyces cerevisiae EBY100 cells, as previously described [54]. The library size was 6 × 106 transformants, as estimated by dilution plating on selective SDCAA medium (2% dextrose, 1.47% sodium citrate, 0.429% citric acid monohydrate, 0.67% yeast nitrogen base and 0.5% casamino acids, pH 4.5). The second-generation library was prepared as described above using the Ang2-BDC1.70 clone, isolated from first-generation library sort 5, as the template. The library size was about 8 × 106 transformants, as estimated by dilution plating on selective SDCAA medium.
Screening of YSD Ang2-BD libraries
The yeast-displayed Ang2-BD libraries growing in selective SDCAA medium were induced for expression with 2% w/v galactose at 20°C overnight until an OD of 3.0 was reached, according to established protocols [54]. The first-generation library underwent five rounds of screening using high-throughput flow cytometric sorting to isolate clones with high affinity for recombinant human Tie2 (Ala23-Lys745). Initial and final sorts were performed using 100 nM and 5 nM Tie2-Fc, respectively. A diagonal sorting gate including 1% of the entire yeast pull was used to select Ang2-BD mutants that bind strongly to Tie2, relative their expression. For each round of sorting, yeast cells of approximately 10 times the library size were labeled as described below to facilitate fluorescent detection by flow cytometry. Binding of the yeast-displayed Ang-BD library to Tie2 was detected using soluble Tie2-Fc (R&D Systems) in Tie2 binding buffer [20 mM Hepes, pH 7.0, 150 mM NaCl, and 1% bovine serum albumin (BSA)], and Ang-BD expression levels were detected using 1:50 dilution of mouse anti c-Myc 9E10 antibody (Abcam) in a 1-h reaction at room temperature. Cells were washed and resuspended in ice-cold PBSA (phosphate buffered saline + 1% BSA) containing a 1:50 dilution of anti-human Fc fluorescein isothiocyanate (FITC) conjugated antibody (Sigma) and a 1:50 dilution of phycoerythrin (PE) conjugated anti-mouse IgG (Sigma). After 25 min on ice, yeast cells were washed in PBSA and sorted using iCyt Synergy FACS (fluorescence-activated cell sorting ) [Proteomics Unit, National Institute for Biotechnology in the Negev (NIBN), Ben-Gurion University of the Negev (BGU)]. Plasmid DNA was extracted from the yeast clones using a Zymoprep kit (Zymo Research) and transformed into electrocompetent Escherichia coli cells for plasmid miniprep (RBC Bioscience Corp, Taiwan) and DNA sequencing (DNA Microarray and Sequencing Unit, NIBN, BGU). The second-generation library was subjected to five rounds of sorting using the method described above, where the initial and final sorts were performed with 20 nM and 500 pM Tie2, respectively. Sixty clones from the two final sorts were sequenced (DNA Microarray and Sequencing Unit, NIBN, BGU) and evaluated for their binding affinity towards Tie2-Fc by dividing the mean fluorescence intensity (MFI) of the Tie2 binding signal by the MFI of expression levels. The values obtained were normalized to Ang2-BDWT.
Purification of Ang2-BD variants
The Multi-Copy Pichia Expression Kit (Invitrogen K1750–01) was used to produce the soluble proteins, as previously described [55]. The Ang2-BD variants were cloned into the pPIC9K vector for expression in P. pastoris yeast strain GS115 using EcoRI and AvrII restriction sites. Plasmid DNA (approximately 20 μg) was linearized by digestion with SacI (New England Biolabs) and electroporated into P. pastoris. Proteins were prepared with an N-terminal FLAG epitope tag and a C-terminal hexahistidine tag as handles for cell binding studies and protein purification, respectively. Transformed yeast cells were allowed to recover on RDB plates (18.6% sorbitol, 2% agar, 2% dextrose, 1.34% yeast nitrogen base, 0.001% biotin and 0.005%
Far-UV circular dichroism spectroscopy
CD spectra were recorded on a Jasco J-715 spectropolarimeter over a range of 185–260 nm in 20 mM Hepes, pH 7.0, 150 mM NaCl buffer using a quartz cuvette with a path length of 1 mm. Protein spectra were collected at a scanning speed of 50 nm/min and a data interval of 1 nm. Four scans of 30 μM protein solutions were averaged to obtain smooth data. All spectra were background corrected with respect to 20 mM Hepes, pH 7.0, 150 mM NaCl buffer and converted to units of mean residue ellipticity. For thermal denaturation studies, ellipticity was monitored at 230 nm using a 1°C∕min scan rate.
Surface plasmon resonance experiments
The binding interactions of Tie2 to Ang2-BDWT, Ang2-BDC1.70, Ang2-BDC2.36 (glycosylated and non-glycosylated) were analyzed (Proteomics Unit, NIBN, BGU) in real-time by SPR using a ProteOn XPR36 instrument (Bio-Rad). A ProteOn GLC sensor chip (Bio-Rad) was air initialized, and PBST (PBS×1, 0.005% Tween) buffer was flushed through the instrument prior to binding measurements. The rhTie2 extracellular domain (Sino Biological Inc.) was immobilized on the surface of a GLC sensor chip by using the amine coupling reagents N-hydroxysuccinimide (0.1 M; sulfo-NHS) and 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (0.4 M; EDC; Bio-Rad). rhTie2 (0.8 μg) in 10 mM sodium acetate, pH 5.0, was allowed to flow over an activated GLC sensor chip channel surfaces, respectively, at a flow rate of 30 μL/min until the target immobilization level (2100 RU) was reached. BSA (3 μg) in 10 mM sodium acetate, pH 4.5, was then allowed to flow over the activated surfaces of a control GLC sensor chip channel at a flow rate of 30 μl/min until the target immobilization level (3000 RU) was reached. After protein immobilization, the chip surface was treated with 1 M ethanolamine HCl at pH 8.5 to deactivate excess reactive esters. All binding experiments were performed at 25°C in degassed Tie2 binding buffer (20 mM Hepes, pH 7.0, 150 mM NaCl, 1 mM CaCl2). A range of concentrations (18.7 nM to 300 nM for Ang2-BDWT, 6.25 nM to 100 nM for Ang2-BDC1.70, 5 nM to 80 nM for non-glycosylated Ang2-BDC2.36 and 6.25 nM to 100 nM for glycosylated Ang2-BDC2.36) of the protein analytes were allowed to flow over the surface-immobilized rhTie2 at a flow rate of 30 μl/min for 13.3 min, and the binding interactions were monitored. Following association, the dissociation of the various ligand-receptor complexes was monitored for 5 min. After the dissociation of each analyte, a regeneration step with 50 mM NaOH at a flow rate of 100 μl/min was performed. Each analyte sensorgram run was normalized by subtracting the BSA-immobilized channel and the zero analyte concentration runs. The binding constant (KD) was determined from the sensorgram of the equilibrium binding phase for Ang2-BDWT. Binding kinetics of Ang2-BDC1.70, glycosylated Ang2-BDC2.36 and non-glycosylated Ang2-BDC2.36 were analyzed by fitting to a two-state model.
Cell binding assays
TIME cells (ATCC) were cultured in growth-factor-depleted Vascular Cell Basal Medium (ATCC) supplemented with 2% FBS and growth factor supplements (ATCC). For binding assays, 105 cells were suspended in different concentrations of Ang2-BD variants in a total volume of 200 μl of PBSA (PBS and 0.1% BSA), followed by incubation at 4°C for 2 h with gentle agitation. Cell suspensions were centrifuged at 150 g at 4°C for 5 min and washed in 100 μl of PBSA followed by centrifugation at 150 g at 4°C for 5 min for two additional times. Cells were then resuspended in 100 μl of PBSA containing a 1:200 dilution of allophycocyanin (APC)-conjugated anti-FLAG antibody (Biolegend). After 20 min on ice, cells were washed twice in PBSA and analyzed by flow cytometry with a BD Accuri C6 flow cytometer (BD Biosciences). Mean fluorescence values were generated using FlowJo software (Treestar). For receptor level detection, 105 cells were harvested, resuspended in 100 μl of PBSA with 1:100 APC-labeled anti-human Tie2 antibody (Biolegend), incubated at 4°C for 30 min, and then analyzed by flow cytometry. The data for the cellular assays was analyzed for column statistics with GraphPad Prism version 5.00 for Windows (La Jolla, CA, USA). Data shown is the average of triplicate experiments, and error bars represent standard error of the mean. Statistical significance was determined by column statistics and t-test analysis. P value < 0.05 was considered statistically significant.
Tie2 phosphorylation assays
Confluent TIME cells were cultured in growth-factor-depleted Vascular Cell Basal Medium supplemented with 0.5% fetal bovine serum for 12 h at 37°C/5% CO2 prior to experimentation. Cells were then washed with PBS, and the medium was exchanged to fresh Vascular Cell Basal Medium depleted of growth factors and serum. After pretreatment with 1 mM sodium orthovanadate (Na3VO4, Sigma) for 15 min, cells were co-incubated for 15 min at 37°C with either commercial full-length rhAng1 as a positive control (R&D Systems) or a combination of full-length rhAng1 and the Ang2-BD variants. Since rhAng1 exists in different oligomeric states, the rhAng1 concentration was reported in mass concentration units instead of molar concentration units. Unstimulated cells were used as the negative control. Cells were then washed twice with PBS plus 1 mM Na3VO4 and lysed in ice-cold lysis buffer [20 mM HEPES, pH 7.4, 150 mM NaCl, 1% TritonX-100, 1 mM Na3VO4, and 1× complete protease inhibitor cocktail tablet (Roche)]. Cells were scraped from the culture plate wells, and the lysates were clarified by centrifugation (13,000 rpm for 30 min at 4°C). Protein concentration was measured by the BCA assay (Thermo Fisher Scientific), and equivalent amounts of each lysate sample were analyzed by duplicate 10% SDS-PAGE and transferred to duplicate PVDF membranes (Biorad). Blots were blocked (5% BSA, 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Tween 20) for 1 h at room temperature and probed with a phospho-Tie2 specific rabbit polyclonal antibody (1:500 dilution; Y992–Tie2, R&D Systems) and a specific Tie2 rabbit monoclonal antibody (1:1000 dilution; Tie2 (D9D10) rabbit mAb, Cell Signaling Technology) overnight at 4°C. Membranes were washed three times with TBST (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Tween 20) and probed with anti-rabbit, HRP-linked antibody (1:1000 dilution, Cell Signaling Technology) for 1 h at room temperature. Membranes were washed three times with TBST and then visualized and quantified using chemiluminescence (ECL, Biological Industries) and ImageJ software, respectively. The intensities of the phospho-Tie2 bands were adjusted for the expression of total Tie2 for each experiment. Blots were stripped and re-probed with anti-actin antibody for further normalization. Tie2 phosphorylation assay data was analyzed with GraphPad Prism version 5.00 for Windows (La Jolla, CA, USA). Data shown is the average of triplicate experiments, and error bars represent standard error of the mean. Statistical significance was determined by column statistics and t test analysis. P value < 0.05 was considered statistically significant.
Endothelial cell tube formation assay
Serum-reduced Matrigel (10 mg/ml; BD Biosciences) was thawed overnight at 4°C, and 150 μl were added to each well of a 48-well microtiter plate and allowed to solidify for 1 h at 37°C. Wells were incubated with 3.25 × 104 TIME cells with 2 μM, 4 μM and 8 μM of Ang2-BDWT and Ang2-BDC2.36; rhAng1, 200 ng/ml, was added as the positive control. Cells were incubated for 16–18 h at 37°C/5% CO2. Cells were then washed twice in HBSS (Hanks’ balanced salt solution, Sigma), and capillary tube formation was observed using EVOS Cell Imaging Systems microscope (Thermo Fisher Scientific). Images were taken with EVOS 2× Objective, phase-contrast. Total length and number of junctions of the tubes were quantified by analysis of digitized images using ImageJ software and the Angiogenesis Analyzer plugin of the capillary-like structures. Tube formation assay data was analyzed with GraphPad Prism version 5.00 for Windows (La Jolla, CA, USA). Data shown is the average of triplicate experiments, and error bars represent standard error of the mean. Statistical significance was determined by column statistics and t test analysis. P value < 0.05 was considered statistically significant.
Invasion assay
An in-vitro Boyden chamber assay was performed using ThinCert™ 24 well inserts (Greiner Bio-One). ThinCert cell culture insert membranes were coated with Matrigel (Corning) diluted in Vascular Cell Basal Medium (ATCC) to a 1:30 ratio. The lower compartment was filled with 600 μl of Vascular Cell Basal Medium supplemented with 2% FBS. TIME cells, 2 × 104, with or without Ang2-BD variants, were incubated in 200 μl of supplement-free Vascular Cell Basal Medium, added to the pre-coated ThinCert cell culture inserts, and incubated for 20 h at 37°C with 5% CO2. Invasive cells were stained with DippKwik stain kit (American MasterTech Scientific) and were detected by EVOS FL Cell Imaging System at ×20 magnification. Quantification was accomplished by counting 16 fields for each membrane. Analysis of digitized images was performed using ImageJ software and Cell Colony Edge Analyser. Data was analyzed with GraphPad Prism version 5.00 for Windows (La Jolla, CA, USA). Data shown is the average of triplicate experiments, and error bars represent standard error of the mean. Statistical significance was determined by column statistics and t test analysis. P value < 0.05 was considered statistically significant.
Abbreviations
PPI, protein–protein interaction; RTK, receptor tyrosine kinases; Ang, angiopoietin; Ang2-BD, angiopoetin 2 binding domain; YSD, yeast surface display; Endo Hf, endoglycosidase H; size-exclusion chromatography (SEC); CD, circular dichroism; SPR, surface plasmon resonance; TIME, telomerase-immortalized human microvascular endothelium; BSA, bovine serum albumin; FITC, fluorescein isothiocyanate; PE, phycoerythrin; FACS, fluorescence-activated cell sorting; NIBN, National Institute for Biotechnology in the Negev; BGU, Ben-Gurion University of the Negev; MFI, mean fluorescence intensity; APC, allophycocyanin; RU, response unit.
Authors’ contributions
T.S. and N.P. designed research; T.S. and L.A. performed research; T.S., L.A. and N.P. analyzed data; W.A.B generated Figure 2; T.S. and N.P. wrote the paper; All authors edited the manuscript and approved the final version.
ACKNOWLEDGMENTS
The authors thank Dr. Alon Zilka for his technical assistance and Mr. Michael Heyne for structural analysis. FACS and Proteon experiments were performed at the NIBN proteomics unit.
CONFLICTS OF INTEREST
The authors declare that they have no conflict of interest with respect to publication of this paper.
FUNDING
This work was supported by the European Research Council “Ideas program” ERC-2013-StG (contract grant number: 336041) to Niv Papo.
REFERENCES
1. Strauss LG, Koczan D, Klippel S, Pan L, Cheng C, Willis S, Haberkorn U, Dimitrakopoulou-Strauss A. Impact of angiogenesis-related gene expression on the tracer kinetics of 18F-FDG in colorectal tumors. J Nucl Med. 2008; 49:1238–44.
2. Hashizume H, Falcon BL, Kuroda T, Baluk P, Coxon A, Yu D, Bready J V, Oliner JD, McDonald DM. Complementary actions of inhibitors of angiopoietin-2 and VEGF on tumor angiogenesis and growth. Cancer Res. 2010; 70:2213–23.
3. Davis S, Papadopoulos N, Aldrich TH, Maisonpierre PC, Huang T, Kovac L, Xu A, Leidich R, Radziejewska E, Rafique A, Goldberg J, Jain V, Bailey K, et al. Angiopoietins have distinct modular domains essential for receptor binding, dimerization and superclustering. Nat Struct Biol. 2003; 10:38–44.
4. Fiedler U, Scharpfenecker M, Koidl S, Hegen A, Grunow V, Schmidt JM, Kriz W, Thurston G, Augustin HG. The Tie-2 ligand angiopoietin-2 is stored in and rapidly released upon stimulation from endothelial cell Weibel-Palade bodies. Blood. 2004; 103:4150–6.
5. Sato TN, Qin Y, Kozak CA, Audus KL. Tie-1 and tie-2 define another class of putative receptor tyrosine kinase genes expressed in early embryonic vascular system. Proc Natl Acad Sci U S A. 1993; 90:9355–8.
6. Sato TN, Tozawa Y, Deutsch U, Wolburg-Buchholz K, Fujiwara Y, Gendron-Maguire M, Gridley T, Wolburg H, Risau W, Qin Y. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature. 1995; 376:70–4.
7. Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997; 277:55–60.
8. Yuan HT, Khankin E V, Karumanchi SA, Parikh SM. Angiopoietin 2 is a partial agonist/antagonist of Tie2 signaling in the endothelium. Mol Cell Biol. 2009; 29:2011–22.
9. Kim KT, Choi HH, Steinmetz MO, Maco B, Kammerer RA, Ahn SY, Kim HZ, Lee GM, Koh GY. Oligomerization and multimerization are critical for angiopoietin-1 to bind and phosphorylate Tie2. J Biol Chem. 2005; 280:20126–31.
10. Barton WA, Tzvetkova D, Nikolov DB. Structure of the angiopoietin-2 receptor binding domain and identification of surfaces involved in Tie2 recognition. Structure. 2005; 13:825–32.
11. Barton WA, Tzvetkova-Robev D, Miranda EP, Kolev M V, Rajashankar KR, Himanen JP, Nikolov DB. Crystal structures of the Tie2 receptor ectodomain and the angiopoietin-2–Tie2 complex. Nat Struct Mol Biol. 2006; 13:524–32.
12. Yu X, Seegar TCM, Dalton AC, Tzvetkova-Robev D, Goldgur Y, Rajashankar KR, Nikolov DB, Barton WA. Structural basis for angiopoietin-1-mediated signaling initiation. Proc Natl Acad Sci U S A. 2013; 110:7205–10.
13. Macdonald PR, Progias P, Ciani B, Patel S, Mayer U, Steinmetz MO, Kammerer RA. Structure of the extracellular domain of tie receptor tyrosine kinases and localization of the angiopoietin-binding epitope. J Biol Chem. 2006; 281:28408–14.
14. Fukuhara S, Sako K, Minami T, Noda K, Kim HZ, Kodama T, Shibuya M, Takakura N, Koh GY, Mochizuki N. Differential function of Tie2 at cell-cell contacts and cell-substratum contacts regulated by angiopoietin-1. Nat Cell Biol. 2008; 10:513–26.
15. DeBusk LM, Hallahan DE, Lin PC. Akt is a major angiogenic mediator downstream of the Ang1/Tie2 signaling pathway. Exp Cell Res. 2004; 298:167–77.
16. Kim I, Kim HG, So J, Kim JH, Kwak HJ, Koh GY. Angiopoietin-1 Regulates Endothelial Cell Survival Through the Phosphatidylinositol 3-Kinase/Akt Signal Transduction Pathway. Circ Res. 2000; 1:24–9.
17. Hu B, Jarzynka MJ, Guo P, Imanishi Y, Schlaepfer DD, Cheng SY. Angiopoietin 2 induces glioma cell invasion by stimulating matrix metalloprotease 2 expression through the alphavbeta1 integrin and focal adhesion kinase signaling pathway. Cancer Res. 2006; 66:775–83.
18. Ahmad SA, Liu W, Jung YD, Fan F, Wilson M, Reinmuth N, Shaheen RM, Bucana CD, Ellis LM. The Effects of Angiopoietin-1 and -2 on Tumor Growth and Angiogenesis in Human Colon Cancer. Cancer Res. 2001; 61: 1255 LP-1259.
19. Sfiligoi C, De Luca A, Cascone I, Sorbello V, Fuso L, Ponzone R, Biglia N, Audero E, Arisio R, Bussolino F, Sismonde P, De Bortoli M. Angiopoietin-2 expression in breast cancer correlates with lymph node invasion and short survival. Int J Cancer. 2003; 103:466–74.
20. Tanaka S, Sugimachi K, Yamashita Yi Y, Ohga T, Shirabe K, Shimada M, Wands JR. Tie2 vascular endothelial receptor expression and function in hepatocellular carcinoma. Hepatology. 2002; 35:861–7.
21. Peters KG, Coogan A, Berry D, Marks J, Iglehart JD, Kontos CD, Rao P, Sankar S, Trogan E. Expression of Tie2/Tek in breast tumour vasculature provides a new marker for evaluation of tumour angiogenesis. Br J Cancer. 1998; 77:51–6.
22. Luke RW, Ballard P, Buttar D, Campbell L, Curwen J, Emery SC, Griffen AM, Hassall L, Hayter BR, Jones CD, McCoull W, Mellor M, Swain ML, et al. Novel thienopyrimidine and thiazolopyrimidine kinase inhibitors with activity against Tie-2 in vitro and in vivo. Bioorg Med Chem Lett. 2009; 19:6670–4.
23. Hangai M, Moon YS, Kitaya N, Chan CK, Wu DY, Peters KG, Ryan SJ, Hinton DR. Systemically expressed soluble Tie2 inhibits intraocular neovascularization. Hum Gene Ther. 2001; 12:1311–21.
24. Lin P, Polverini P, Dewhirst M, Shan S, Rao PS, Peters K. Inhibition of tumor angiogenesis using a soluble receptor establishes a role for Tie2 in pathologic vascular growth. J Clin Invest. 1997; 100:2072–8.
25. Lin P, Buxton JA, Acheson A, Radziejewski C, Maisonpierre PC, Yancopoulos GD, Channon KM, Hale LP, Dewhirst MW, George SE, Peters KG. Antiangiogenic gene therapy targeting the endothelium-specific receptor tyrosine kinase Tie2. Proc Natl Acad Sci U S A. 1998; 95:8829–34.
26. Oliner J, Min H, Leal J, Yu D, Rao S, You E, Tang X, Kim H, Meyer S, Han SJ, Hawkins N, Rosenfeld R, Davy E, et al. Suppression of angiogenesis and tumor growth by selective inhibition of angiopoietin-2. Cancer Cell. 2004; 6:507–16.
27. Herbst RS, Hong D, Chap L, Kurzrock R, Jackson E, Silverman JM, Rasmussen E, Sun YN, Zhong D, Hwang YC, Evelhoch JL, Oliner JD, Le N, et al. Safety, pharmacokinetics, and antitumor activity of AMG 386, a selective angiopoietin inhibitor, in adult patients with advanced solid tumors. J Clin Oncol. 2009; 27:3557–65.
28. Brown JL, Cao ZA, Pinzon-Ortiz M, Kendrew J, Reimer C, Wen S, Zhou JQ, Tabrizi M, Emery S, McDermott B, Pablo L, McCoon P, Bedian V, et al. A human monoclonal anti-ANG2 antibody leads to broad antitumor activity in combination with VEGF inhibitors and chemotherapy agents in preclinical models. Mol Cancer Ther. 2010; 9:145–56.
29. Leow CC, Coffman K, Inigo I, Breen S, Czapiga M, Soukharev S, Gingles N, Peterson N, Fazenbaker C, Woods R, Jallal B, Ricketts S-A, Lavallee T, et al. MEDI3617, a human anti-angiopoietin 2 monoclonal antibody, inhibits angiogenesis and tumor growth in human tumor xenograft models. Int J Oncol. 2012; 40:1321–30.
30. Thomas M, Kienast Y, Scheuer W, Bähner M, Kaluza K, Gassner C, Herting F, Brinkmann U, Seeber S, Kavlie A, Welschof M, Ries S, Weidner KM, et al. A Novel Angiopoietin-2 Selective Fully Human Antibody with Potent Anti-Tumoral and Anti-Angiogenic Efficacy and Superior Side Effect Profile Compared to Pan-Angiopoietin-1/-2 Inhibitors. PLoS One. 2013; 8.
31. Jones DJ, Silverman AP, Cochran JR. Developing therapeutic proteins by engineering ligand-receptor interactions. Trends Biotechnol. 2008; 26:498–505.
32. Nanda S, Bathon JM. Etanercept: a clinical review of current and emerging indications. Expert Opin Pharmacother. 2004; 5:1175–86.
33. Dupont J, Rothenberg ML, Spriggs DR, Cedarbaum JM, Furfine ES, Cohen DP, Dancy I, Lee HS, Cooper W, Lockhart AC. Safety and pharmacokinetics of intravenous VEGF Trap in a phase I clinical trial of patients with advanced solid tumors. J Clin Oncol. 2005; 23:3029.
34. Cretney E, Shanker A, Yagita H, Smyth MJ, Sayers TJ. TNF-related apoptosis-inducing ligand as a therapeutic agent in autoimmunity and cancer. Immunol Cell Biol. 2006; 84:87–98.
35. Jones 2nd DS, Tsai PC, Cochran JR. Engineering hepatocyte growth factor fragments with high stability and activity as Met receptor agonists and antagonists. Proc Natl Acad Sci U S A. 2011; 108:13035–40.
36. Cochran JR, Kim YS, Lippow SM, Rao B, Wittrup KD. Improved mutants from directed evolution are biased to orthologous substitutions. Protein Eng Des Sel. 2006; 19:245–53.
37. Sarkar CA, Lowenhaupt K, Horan T, Boone TC, Tidor B, Lauffenburger DA. Rational cytokine design for increased lifetime and enhanced potency using pH-activated “histidine switching.” Nat Biotechnol. 2002; 20:908–13.
38. Fuh G, Cunningham BC, Fukunaga R, Nagata S, Goeddel D V., Wells J a. Rational Design of Potent Antagonists to the Human Growth Hormone Receptor. Science. 1992; 256:1677–80.
39. Papo N, Silverman AP, Lahti JL, Cochran JR. Antagonistic VEGF variants engineered to simultaneously bind to and inhibit VEGFR2 and {alpha}v{beta}3 integrin. Proc Natl Acad Sci U S A. 2011; 108:14067–72.
40. Kariolis MS, Miao YR, Jones II DS, Kapur S, Mathews II, Giaccia AJ, Cochran JR. An engineered Axl “decoy receptor” effectively silences the Gas6-Axl signaling axis. Nat Chem Biol. 2014; 10:977–83.
41. Kariolis MS, Miao YR, Diep A, Nash SE, Olcina MM, Jiang D, Ii DSJ, Kapur S, Mathews II, Koong AC, Rankin EB, Cochran JR, Giaccia AJ. Inhibition of the GAS6 / AXL pathway augments the efficacy of chemotherapies. J Clin Invest. 2016; 127:
42. Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, Moraga I, Raeber ME, Bowman GR, Novick P, Pande VS, Fathman CG, Boyman O, et al. Exploiting a natural conformational switch to engineer an interleukin-2 “superkine.” Nature. 2012; 484:529–33.
43. Deng P, Wang YL, Pattengale PK, Rettenmier CW. The role of individual cysteine residues in the processing, structure, and function of human macrophage colony-stimulating factor. Biochem Biophys Res Commun. 1996; 228:557–66.
44. Boesen TP, Soni B, Schwartz TW, Halkier T. Single-chain vascular endothelial growth factor variant with antagonistic activity. J Biol Chem. 2002; 277:40335–41.
45. Cho CH, Kammerer RA, Lee HJ, Steinmetz MO, Ryu YS, Lee SH, Yasunaga K, Kim KT, Kim I, Choi HH, Kim W, Kim SH, Park SK, et al. COMP-Ang1: a designed angiopoietin-1 variant with nonleaky angiogenic activity. Proc Natl Acad Sci U S A. 2004; 101:5547–52.
46. Gai SA, Wittrup KD. Yeast surface display for protein engineering and characterization. Curr Opin Struct Biol. 2007; 17:467–73.
47. Carter PJ. Potent antibody therapeutics by design. Nat Rev Immunol. 2006; 6:343–57.
48. Weiner GJ. Building better monoclonal antibody-based therapeutics. Nat Rev Cancer. 2015; 15:361–70.
49. Fuh G, Li B, Crowley C, Cunningham BC, Wells JA. Requirements for binding and signaling of the kinase domain receptor for vascular endothelial growth factor. J Biol Chem. 1998; 273:11197–204.
50. Tolbert WD, Daugherty J, Gao C, Xie Q, Miranti C, Gherardi E, Vande Woude G, Xu HE. A mechanistic basis for converting a receptor tyrosine kinase agonist to an antagonist. Proc Natl Acad Sci U S A. 2007; 104:14592–7.
51. Lokker NA, Mark MR, Luis EA, Bennett GL, Robbins KA, Baker JB, Godowski PJ. Structure-function analysis of hepatocyte growth factor: identification of variants that lack mitogenic activity yet retain high affinity receptor binding. EMBO J. 1992; 11:2503–10.
52. Jenkins N, Meleady P, Tyther R, Murphy L. Strategies for analysing and improving the expression and quality of recombinant proteins made in mammalian cells. Biotechnol Appl Biochem. 2009; 53:73–83.
53. Zhu J. Mammalian cell protein expression for biopharmaceutical production. Biotechnol Adv. 2012; 30:1158–70.
54. Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, Wittrup KD. Isolating and engineering human antibodies using yeast surface display. Nat Protoc. 2006; 1:755–68.
55. Moore SJ, Leung CL, Norton HK, Cochran JR. Engineering agatoxin, a cystine-knot peptide from spider venom, as a molecular probe for in vivo tumor imaging. PLoS One. 2013; 8:e60498