
INTRODUCTION
TIAM1 is a molecular conduit to a complex network of signaling pathways that control cell-fate decisions. TIAM1 N-terminal region binds to MYC box II, a highly conserved domain among MYC proteins, to inhibit MYC-mediated apoptosis [1]. This region also functions as an auto-inhibitory loop which can be released upon phosphorylation by PKCs [2, 3]. In addition to MYC signaling, TIAM1 can also associate with activated RAS oncoprotein [4]. TIAM1-deficient mice are resistant to the development of RAS-induced skin tumors, revealing that TIAM1 is a critical regulator of RAS-induced tumor formation [5]. Closer to its N-terminus, the tyrosine 384 is subjected to SRC-dependent phosphorylation, which in turn recruits GRB2/SOS complex and leads to RAS/MAPK activation [6]. Therefore, TIAM1 has a dual function, signaling as an effector or as an activator molecule for RAS.
TIAM1 also activates RAC to stimulate membrane ruffling, cell migration and invasion [7]. RAC activation is catalyzed by TIAM1 Dbl homology domain (DH) that works in tandem with a pleckstrin homology domain (PH) [8, 9]. TIAM1 has an additional PH domain closer to the N-terminal region which determines the activation of distinct RAS-mediated signaling pathways such as c-Jun NH2-terminal kinase (JNK) [10]. Altogether, cellular and molecular data reveal TIAM1 as a molecular conduit to MYC, RAS and RAC signaling and integrates these three pathways in a network to orchestrate pleiotropic responses in the cell.
Cellular polarity in neuroblastoma cells is controlled by TIAM1-dependent RAC activation mediated by the polarity complex PAR-6-PAR-3 [11]. Nonetheless, the role of TIAM1 in neuroblastoma cells has been mostly focused in neuritogenesis. TIAM1 is a specific regulator of RAC during neuritogenesis [12] and is required for ephrin-B1 and EphA2 signaling in neurite outgrowth [13]. More recently, a whole-genome sequence analysis of 87 neuroblastomas showed three mutations in TIAM1 as well as other mutations in regulators of the Rac/Rho pathway, suggesting that defects in neuritogenesis may contribute to neuroblastoma [14].
In the present study, we screened primary neuroblastomas by next generation sequencing and our results reveal that TIAM1 variants in the domains signaling to MYC, RAS and RAC significantly predict better prognosis in neuroblastoma patients. Moreover, three high-risk patients (age >1.5 years) with MYCN amplification also had a TIAM1 variant but did not relapse. Notably, two of these patients are already free of disease. These novel observations implicate TIAM1 as a key signaling molecule in neuroblastoma and as a potential target for therapeutic intervention.
RESULTS
Technical characteristics of the NB-panel
To identify altered druggable targets and outcome predictors in neuroblastoma, we analyzed a collection of 106 primary tumors by next generation sequencing (NGS). A customized amplicon-based gene panel (NB-panel) containing 26 genes with implications in neuroblastoma or other cancers was designed and implemented (Table 1, see Supplementary Table 1 for genomic coordinates). Mean values and ranges of relevant technical parameters are shown in Table 2. Interestingly, many of the MYCN amplified cases displayed a rather low uniformity (less than 50%), with a strong deviation of the readings towards MYCN amplicons. This sequestering of the readings always coincided with MYCN amplification, suggesting an additional approach to predict amplification status by NGS. However, since some MYCN amplified cases displayed a reasonably high uniformity and equal distribution of the readings throughout the amplicons of the panel, this approach is not fully reliable as a predictor as it may fail to detect some positives. The sequestering of the readings towards MYCN sequencing may rely on other factors such as the percentage of neuroblasts in the sample or the ratio of MYCN amplification.
Table 1: Genes and covered exons included in the customized NB-panel
Gene |
Locus |
Ref Seq |
Total exons |
Covered exons |
|---|---|---|---|---|
ABL1 |
9q |
NM_007313.2 |
11 |
6 |
AKT1 |
14q |
NM_001014431.1 |
14 |
1, 5-10 |
ALK |
2p |
NM_004304.4 |
29 |
20-29 |
ARID1A |
1p |
NM_006015.4 |
20 |
1-20 |
ATM |
11q |
NM_000051.3 |
63 |
9-12, 16-19, 21, 25-29, 31, 32, 37, 39-46, 54, 55, 58, 61-63 |
ATRX |
Xq |
NM_000489.4 |
35 |
8-10, 12-15, 17, 18, 21, 22, 26, 30-32, 35 |
BRAF |
7q |
NM_004333.4 |
18 |
15 |
CHEK2 |
22q |
NM_001005735.1 |
16 |
6, 8, 14-16 |
CKIT |
4q |
NM_000222.2 |
21 |
9, 11, 13, 17 |
EGFR |
7p |
NM_005228.3 |
28 |
18-21 |
ERBB2 |
17q |
NM_004448.3 |
27 |
19, 20 |
FLT3 |
13q |
NM_004119 |
24 |
11, 12, 14, 17 |
MYCN |
2p |
NM_005378.4 |
3 |
2, 3 |
NF1 |
17q |
NM_001042492.2 |
58 |
1-58 |
NOTCH1 |
9q |
NM_017617 |
34 |
6-8, 11-13, 26, 27, 34 |
NRAS |
1p |
NM_002524 |
7 |
2, 3 |
PDGFRA |
4q |
NM_006206.4 |
23 |
12, 14, 18 |
PIK3CA |
3q |
NM_006218.2 |
21 |
8, 10, 21 |
PTEN |
10q |
NM_000314.4 |
9 |
5-8 |
PTPN11 |
12q |
NM_002834.3 |
16 |
3, 7, 8, 12, 13 |
RB1 |
13q |
NM_000321.2 |
27 |
1-27 |
RET |
10q |
NM_020975.4 |
20 |
10-16 |
SRC |
20q |
NM_198291.2 |
14 |
12 |
STK11 |
19p |
NM_000455.4 |
10 |
1-8 |
TIAM1 |
21q |
NM_003253.2 |
29 |
5-29 |
TP53 |
17p |
NM_000546.5 |
11 |
5-9 |
Table 2: Technical performance of 106 primary neuroblastoma samples analyzed with the customized NB-Panel
|
Mean (Range) for all patients (n=106) |
|---|---|
Mapped reads |
1,623,834 (127,973 – 5,415,928) |
Reads on target |
93.69 % (74.30 – 99.41) |
Mean depth |
2,654 (154 – 9,653) |
Read uniformity |
78.21 % (7.58 – 94.34) |
Mean read length |
147 bp (109-177) |
Distribution of variants in primary neuroblastomas
Sequencing data analysis was performed based on low frequency allelic variants located on exons or splicing sites, except NF1 gene where both exon and intron low frequency allelic variants were considered. A combination of predictors was used to estimate malignancy. In this manner, the screening for rare variants revealed that only 62 of 106 neuroblastomas (58%) harbor at least one variant in any of the genes included in NB-panel (see Supplementary Table 2 for a detailed description of the identified variants and the predictors used in the determination). The genes most frequently mutated were ATM and NF1 (16 and 15 cases, respectively), followed by TIAM1 and ALK (12 cases each). Most tumors contained no variants or had just one or two, further supporting the simplicity of genetic alterations in primary tumors that gain in complexity later on upon relapse [15]. We also generated an additional list containing the silent variants outside of the splicing sites as well as deep intronic variants beyond 10 bases from the splicing site (Supplementary Table 3). Allelic variants localized 5’ and 3’ UTR were also included in this list. The fact that we failed to detect variants in 42% of the cases suggests that other genetic alterations should be taken into account and therefore, a second version of NB-panel may be required to identify other pathogenic changes in primary neuroblastomas.
TIAM1 variant associates with better outcome in primary neuroblastomas
Kaplan-Meier survival analysis revealed that variants in TIAM1 gene associated significantly with increased disease-free survival (Figure 1A) as well as with overall survival (Figure 1B). All but one of the 12 cases with TIAM1 variant reported no relapse. Interestingly, this one patient is currently free of disease. No other mutated gene showed a prognostic significance, perhaps owing to low incidence or the need of higher number of cases in the cohort.
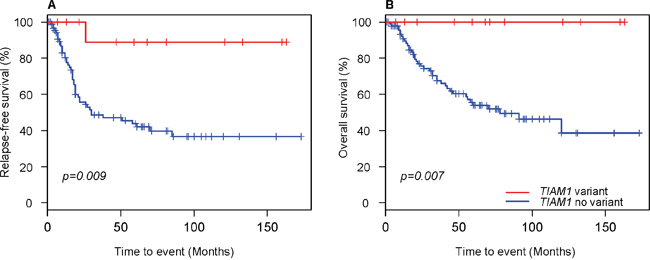
Figure 1: Kaplan–Meier curves for neuroblastoma patients with and without TIAM1 variants. (A) Relapse-free survival and (B) Overall survival. Censored cases are denoted as crosses along the plots. Log-rank p values were used to compare survival curves between patients with and without TIAM1 variants.
Heatmap representation of the clinical parameters and variants revealed that high-risk group clusters together with a defined division between survival and mortality (Figure 2). This distribution suggests that there are genetic and clinical factors in the high-risk group that may predispose for the outcome. MYCN amplification is a high-risk factor that can be subdivided into survivors and not survivors within the high-risk group in the hierarchical structure of the heatmap (Figure 2). This result also supports the existence of key determinants that define outcome in MYCN amplified patients. Among the 11 cases with 4S stage, a metastatic group in children under 12 months of age, 3 had also MYCN amplification but did not relapse, further supporting the better outcome in this group [16]. We also identified several variants in 4S stage: 2 cases with a variant in ATM gene, 1 case with concomitant ATM and RET variants, 2 cases in NF1 and 1 in ATRX, all alive and disease free. The only 4S case with a variant in ALK underwent progression and was the only one that did not survive, suggesting that ALK inhibitors should also be considered for treatment in 4S stage.
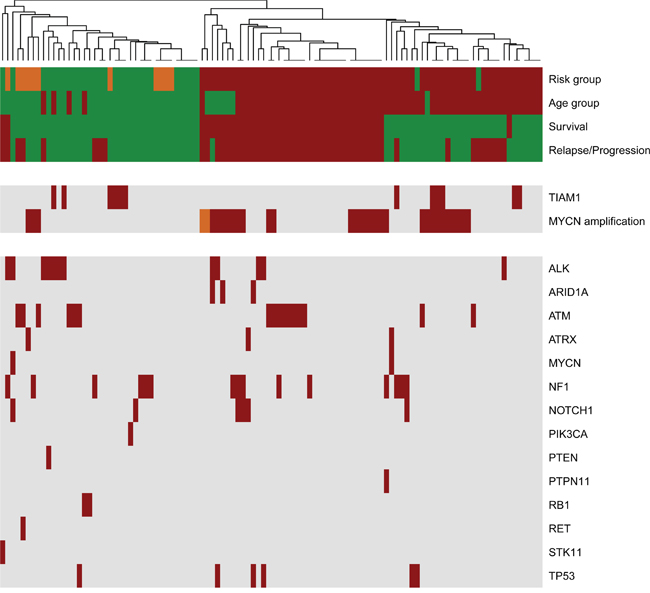
Figure 2: Landscape of clinical variations and identified genetic variants in primary neuroblastomas. The heatmap was clustered based on Manhattan distances between cases. Distances were estimated with clinical data and identified variants with NB-panel in 106 primary neuroblastomas and using the Average-Method. Each column summarizes one patient. Each row represents the indicated clinical variable (clustered on the upper rows) or genetic variations (clustered in two groups to highlight those cases with TIAM1 variants and/or MYCN amplification). Risk group (green, low and intermediate risk; red, high risk; orange, stage 4S); Age group (green, <18 months; red, >18 months); Survival (green, alive; red, death), Relapse/Progression (green, disease-free; red, event of progression or relapse); MYCN amplification (green, no amplification; red, amplification; orange, heterogenic amplification); Genes (red, variant found; grey, no variant detected).
TIAM1 variants are organized in two groups: the high-risk group and the low/intermediate group (Figure 2). Interestingly, the hierarchical classification for TIAM1 variants in the high risk clusters in the better outcome subtype (right side cases in Figure 2), suggesting that TIAM1 variants could be one of the genetic alterations that confer the high-risk group with a better outcome. The coexistence of TIAM1 variants with other alterations is frequent. In addition to the 3 cases with MYCN amplification, 2 cases have concomitant variants/mutations in ALK and 1 in NF1 (Figure 3). Moreover, TIAM1 variant status is of better prognosis regardless of MYCN amplification: out of 32 cases with MYCN amplification, 18 died and 14 survived (Table 3). Among the 14 survivors, 3 have a mutation in TIAM1 gene, one finished treatment and two of them are already free of disease (Table 3). These results strongly suggest that TIAM1 variant is a genetic alteration that is associated with better clinical outcome, even in the high-risk group with MYCN amplification.
Table 3: Frequency of TIAM1 variants in neuroblastomas based on MYCN amplification and overall survival
Variables |
MYCN |
Total |
|
|---|---|---|---|
Amplified |
Not amp |
||
|
|
No. with TIAM1 variant / total |
|
Survival |
3/14 |
9/53 |
12/67 |
Not survival |
0/18 |
0/21 |
0/39 |
Total |
3/32 |
9/74 |
12/106 |
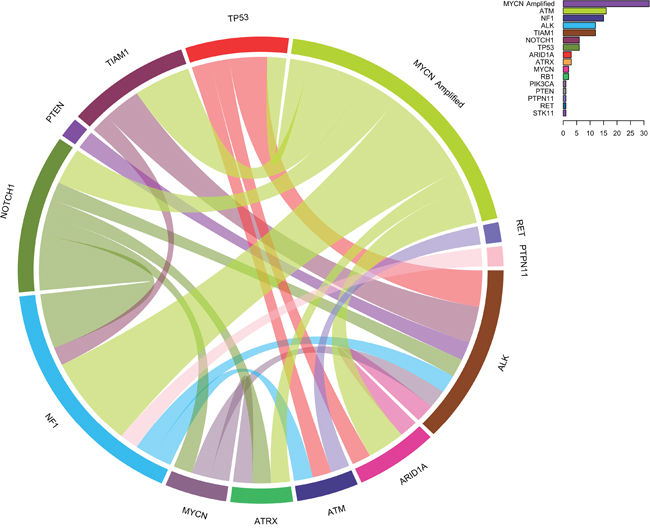
Figure 3: Circos diagram showing concomitantly mutated genes and/or MYCN amplification. The diagram represents paired concomitant genetic alterations found in 106 primary neuroblastomas using NB-panel. Frequencies of all variants are indicated in the chart on the upper right corner.
TIAM1 variant is an independent prognostic variable in primary neuroblastomas
To further explore the prognostic value of TIAM1 variant in neuroblastoma, we performed a multivariate Cox regression analysis. As expected, status of MYCN amplification and age were two independent variables that significantly associated with overall survival (Table 4). Noteworthy, TIAM1 variant was a clear independent variable to predict prognosis in primary neuroblastoma, further supporting its value as an outcome predictor in the disease.
Table 4: Multivariate Cox analysis of relapse-free and overall survival for TIAM1 variant, MYCN amplification and age at diagnosis
Variables |
Multivariate Analysis |
|||||
|---|---|---|---|---|---|---|
Relapse-free survival |
Overall survival |
|||||
HR |
95% CI |
p |
HR |
95% CI |
p |
|
*Age |
1.017 |
1.008-1.025 |
0.0002 |
1.010 |
1.001-1.018 |
0.022 |
TIAM1 variant |
|
|
|
|
|
|
No |
1.000 |
0.025-0.833 |
0.021 |
1.000 |
0.028-0.206 |
0.006 |
Yes |
0.222 |
|
|
0.083 |
|
|
MYCN amplification |
|
|
|
|
|
|
No |
1.000 |
0.723-2.514 |
0.321 |
1.000 |
1.368-5.029 |
0.004 |
Yes |
1.375 |
|
|
2.623 |
|
|
Abbreviations: HR, Hazard Ratio; CI, confidence interval
*Age was consider as continuous variable measured in months
Distribution of variants in TIAM1 defines an integrated signaling network in primary neuroblastoma
To understand the role of the identified variants in neuroblastoma etiology and progression, we assessed the predictive effect of each variant in the signaling domains of TIAM1 protein and the clinical features of each patient. As outlined in Figure 4, the variants we identified in TIAM1 gene are distributed in the three major signaling regions: 1) the N-terminus region that enables the binding to MYC family members to promote transcriptional co-activation and inhibit cellular apoptosis, 2) the RAS-binding domain (RBD) to transduce its signaling to downstream effectors, and 3) the catalytic domain (DH-PH2) that enables RAC activation and thus, activates cellular migration and neuritogenesis. Sequencing of the tumor in parallel with peripheral blood revealed the presence of TIAM1 germline variants in at least 5 out of the 12 cases (Table 5). No other known pathogenic variants were found in the peripheral blood (Supplementary Table 4). Analysis of paired samples (peripheral blood plus tumor from same patient) revealed no increase of TIAM1 variant allele ratios in 4 tumors (Supplementary Tables 2 and 4). Notably, 2 tumors showed a TIAM1 variant allele ratio close to 100% (Supplementary Table 2, variants A163V and D424E), suggesting that they are homozygous in the cancer cells. In one of these patients, the variant was detected in blood cell DNA at a ratio close to 50% (Supplementary Table 4, A163V), supporting a recessive character for this germline variant and that it became the exclusive allele during neuroblastoma formation. These variants in TIAM1 appear to be rare and improve clinical outcome in neuroblastoma presumably by causing a partial or complete dysfunction of the protein.
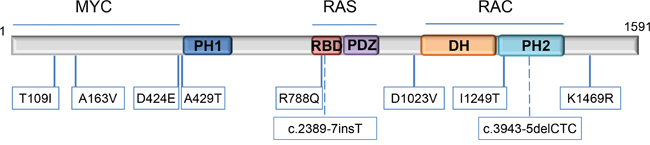
Figure 4: Location of TIAM1 variants. Variants are mapped to specific amino acids on the TIAM1 linearized protein model. The specific domains are indicated (from the N-terminus onwards according to amino acid sequence): PH1: N-terminal PH domain; RBD: RAS-binding domain; PDZ: PSD-95/DlgA/ZO-1 domain; DH: DBL homology domain/ RHOGEF domain and PH2: C-terminal PH domain. Locations of the 2 intronic variants are represented by dashed lines. Proteins reported to bind with certain regions of TIAM1 are indicated above the structure.
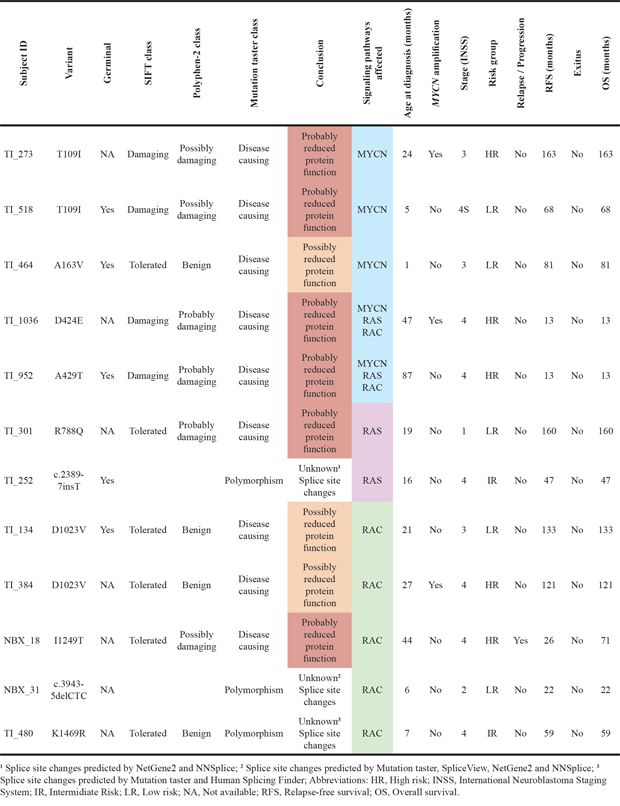
Table 5: Functional effect predictions and clinical data of patients with TIAM1 variants
TIAM1 variants were detected across risk groups, with 5 cases in the low risk, 5 cases in the high risk and 2 cases in intermediate risk (Table 5). TIAM1 variants are also evenly distributed among the stages, although with a higher tendency towards stage 4. In addition, TIAM1 variants T109I and D1023V were each found twice, although the distribution was equal in risk groups. These results suggest that TIAM1 variants do not associate with clinical stages and risk groups and therefore, TIAM1 variant is an independent factor that improves outcome probably by dysregulating signaling pathways important in neuroblastoma cells.
DISCUSSION
The implementation of our customized NB-panel in 106 primary neuroblastoma patients confirmed some of previously reported mutated genes in neuroblastoma with very similar frequencies, including ALK (11% of the patients) [17–21], NF1 (14%) [22], ATM (15%) [23], and others with lower frequency such as ATRX (3%) [20, 21], ARID1A (3%) [24] and STK11 (1%) [25]. Interestingly, we found mutated TP53 in 6% of the cases, a percentage higher than expected in primary neuroblastomas. Mutations in TP53 tumor suppressor have been reported to be rare at diagnosis but may be the cause of resistance acquired over chemotherapy [26]. In our cohort, the variant allele ratios for 6 cases with TP53 variants were found at frequencies close to 100% (2 cases) or close to 50% (4 cases) (Supplementary Table 2). Unfortunately, blood samples are not available to determine if these were germinal or somatic mutations. Nevertheless, it is very improbable that both alleles were inherited from each parent in the 2 cases with homozygous TP53 mutation. One possibility is that an event of uniparental disomy occurred, leading to a double copy of the inherited TP53 variant. Alternatively, the normal TP53 allele may have been deleted or exchanged by the mutated allele, suggesting that although rare, TP53 homozygous mutations also occur prior to chemotherapy in neuroblastomas. Additionally, we also found one patient with a variant in the receptor tyrosine kinase RET, in contrast to a previous report where no RET mutations were found in primary neuroblastomas [27]. Although the frequency was very low, our results may support screening for RET mutations in precision medicine panels since it is a druggable target that could be use in therapy, alone or in combination with inhibitors of its downstream effectors such as mTOR. Importantly, we noted some patients with concomitant MYCN amplification and NF1 variants (Figure 3) and the survival rate in this group is low (Figure 2). Based on the role of NF1 in RAS/RAF/MEK/ERK signaling, this data suggest that this high-risk group may require concomitant or alternative use of MEK inhibitors to improve outcome.
The role of TIAM1 in the invasion and metastasis of tumor cells has been well documented (reviewed in [28]). However, the oncogenic role of TIAM1 in neuroblastoma is still not clear. A recent study with 240 high-risk neuroblastoma patients did not identify any mutations in TIAM1 gene [21]. One plausible explanation is that screening for tumor-specific somatic mutations may have skipped TIAM1 protective germline variants. In contrast, another study with 87 neuroblastoma patients, investigators reported 3 mutations in TIAM1 gene, one with concomitant MYCN amplification and poor outcome [14]. Our results with 106 primary neuroblastomas reveal that TIAM1 has a higher incidence of variants (11%), and they significantly associate as an independent variable with better outcome, even in the concomitant presence of MYCN amplification or ALK mutation. One explanation for the higher percentage of TIAM1 variants found in our cohort vs. previous reports is that we focused strictly on primary neuroblastomas and we took into account both low and high risk groups. We report that TIAM1 variants improve clinical outcome and are evenly distributed in risk groups (Figure 1 and Figure 2). Hence, restricting the analysis to high risk group or inclusion of relapsed tumors as in published studies may have excluded or reduced the incidence of TIAM1 mutations. At least 5 out of 12 of these mutations are germline, suggesting that these rare variants could be inherited. Therefore, screening only for somatic mutations by terms of comparing sequences from blood and tumor would have censored these TIAM1 germline variants. Interestingly, A163V TIAM1 variant was heterozygous in blood and became homozygous in the tumor, suggesting a recessive phenotype. In addition, variant D424E was homozygous in the tumor and although no DNA from blood was sequenced, ExAC reported this variant in only 10 alleles out of 120256 sequenced, but none of them in homozygous (http://exac.broadinstitute.org/variant/21-32624197-G-T), also supporting a recessive phenotype for this variant. Collectively, our findings suggest that these variants promote a better outcome, presumably by impairing the function of TIAM1. One plausible explanation is that these variants disrupt or induce loss of function in oncogenic signaling networks mediated by TIAM1. All of these variants are localized in the interacting domains that signal to MYCN, RAS, and RAC, pathways that are fundamental for the oncogenic potential of neuroblastoma cells.
T109I and A163V variants are localized within TIAM1 N-terminal domain responsible for the binding to MYC and inhibition of its apoptotic effect [1]. Therefore, these variants may weaken or abolish the binding and facilitate MYC-mediated apoptosis (see Figure 4 and Table 5 for illustrative comprehension). D424E and A429T variants are predicted to inhibit the nearby PH1 domain, highly relevant for TIAM1 subcellular localization [10]. Thus, these variants may dampen or inhibit all TIAM1-dependent signaling pathways. R788Q variant is localized within the RAS binding domain and may inhibit RAS-dependent activation of TIAM1. On the other hand, D1023V is immediately upstream of DH domain, the TIAM1 catalytic domain involved in the nucleotide exchange of RAC. This variant may inhibit RAC signaling by reducing the efficiency of nucleotide exchange. I1249T maps to the PH2 domain which works in tandem with DH to facilitate TIAM1 translocation to the membrane and RAC activation. Therefore, this variant is also predicted to reduce or inhibit RAC signaling.
Turnover of TIAM1 is regulated by the ubiquitin-proteasome pathway: two lysine residues which are sensitive to ubiquitylation (K1404 and K1420) have been localized immediately after the tandem DH-PH2 domain at the C-terminus [29]. K1469 is localized close to the two ubiquitylated lysines and thus, variant K1469R found in one patient may alter TIAM1 localization/turnover or reduce its capacity as a MYCN transcriptional coactivator, as it occurs with other transcriptional coactivators such as NCOA3 and the estrogen receptor [30]. Alternatively, variant K1469R is predicted to generate an alternative splice-site change that may result in reduced mRNA/protein stability (Table 5). The proteasome inhibitor bortezomib with retinoic acid has been suggested as an alternative co-adjuvant therapy in neuroblastoma patients to prevent relapse [31]. Patient with TIAM1 variant K1469R also harbors an ALK variant R1275Q, suggesting that ALK inhibitors could also be an alternative in case of relapse. However, neither this patient nor patient with concomitant TIAM1 variant R788Q and ALK variant F1174L did relapse, further supporting the protective phenotype of TIAM1 variants in the outcome of the disease. Collectively, TIAM1 variants associated with better outcome in neuroblastoma patients and therefore, suggest that inhibiting TIAM1 in neuroblastoma could be used in conjunction with conventional therapy to improve outcome.
MATERIALS AND METHODS
Patients and samples
All neuroblastoma samples (106 in total) were derived from primary tumours of untreated patients diagnosed with neuroblastoma between years 1997 and 2015 in Spanish cooperative hospitals. Patients were included in different national and European studies (LNESG I and II, INES, EUNS, LINES, N-AR-99, N-II-92, and HR-NBL1) and carefully selected in order to have all neuroblastoma subtypes represented. Patient’s follow-up was 43 months (range 1-173 months). The distribution of patients by stages was 9% stage 1; 7% stage 2; 24% stage 3; 51 stage 4 and 10% stage 4S. Median age of the cohort at diagnosis was 26.35 months (first quartile 10.1 months, third quartile 46.63 months). Median overall survival in the cohort was 43 months (first quartile 16 months, third quartile 76.75 months). MYCN was amplified in 30 patients (28%), heterogeneous amplified in 2 patiens (2%) and not amplified in 74 patients (70%). Tumor samples were centrally reviewed and histologic type was classified according to the International Neuroblastoma Pathology Classificarion (INPC) criteria [32, 33]. Staging and risk stratification were established according to International Neuroblastoma Staging System (INSS) and International Neuroblastoma Risk Group (INRG) [34]. Biological studies included status of MYCN, 1p and 11q, studied by FISH according to ENQUA guidelines [35, 36]. Patient’s follow-up was obtained from clinical records. The study was conducted in accordance with the Declaration of Helsinki and La Fe Research Ethics Committee approved this project. Parents or legal guardians signed an informed consent statement for sample and data management.
Genomic DNA was extracted from frozen tumor tissues by a standard proteinase K and phenol-chloroform extraction protocol. NanoDrop spectrophotometer (Thermo Scientific, Waltham, MA, USA) was used to measure the quality and quantity of the extracted DNA by A260 spectrophotometric absorbance.
Gene panel design
The customized NB-panel comprises 483 amplicons from 26 genes which are frequently mutated in cancer and/or potential genes to targeted-therapies (see Table 1 and Supplementary Table 1). Design of the panel primers were performed with the Ion Ampliseq Designer (pipeline version 4.2). The panel consists of 2 primer pools; each requires a minimum of 10 ng DNA input material. Panel size is 87.64 kb and amplicon lengths are between 125 and 275 bp.
Next generation sequencing
Sequencing analysis was performed on the Genomic Unit of La Fe Hospital Research Institute with the Ion PGM™ / Ion PROTON™ system (Life Technologies). Amplicon library was prepared using the Ion Ampliseq Library kit 2.0 (Life Technologies). Briefly, multiplex primer pool was added to 10 ng of genomic DNA and amplified with the following PCR cycles: 99°C for 2 min, followed by 99°C for 15 s and at 60°C for 4 min (17 cycles), and holding on at 10°C. Primers are partially digested using a FuPa reagent, and then sequencing adapters were ligated to the amplicons. The final libraries were purified with Agentcourt AMPure XP (Beckman Coulter) and quantified with Qubit DS HS assay Kit (Life Technologies). DNA template preparation was performed with the semiautomated Ion OneTouch™ 2 instrument and Ion One Touch enrichment system (ES) (Life Technologies). Finally DNA high-throughput sequencing was performed on PGM or Proton instrument. Ion PGM™ HiQ Sequencing kit on the Ion 318 sequencing chip (Life Technologies) was used for PGM instrument. In case of PROTON instrument, we used Ion PI™ HiQ Sequencing Kit on the Ion PI sequencing chip (Life Technologies).
Data from the Ion Torrent runs were analysed using the platform-specific pipeline software Torrent Suite v5.0 for base calling, trim adapter and primer sequences, filtering out poor quality reads and no call reads according to the barcode sequences. The sequence variants in each sample were identified using the Torrent Suite Variant Caller (TSVC; v4.0‑r76860) plug-in and browser extensible data (BED) files (chromosome coordinates) that specify the coding regions of the target genes within the human reference genome (hg19) retrieved from the NCBI database (build 37) as a reference. Finally, Ion Reporter software (https://ionreporter.thermofisher.com) was used to further annotate variant caller format (VCF) files and Integrated Genomic Viewer (IGV) software [37] was then used to complete the visualization and to discriminate false-positive variants. We considered only variants with balanced numbers of forward and reverse reads. Based on the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [38], variants with minor allele frequency (MAF) of > 1% in 1000G were considered polymorphisms and were excluded from further analyses. Investigation about the potential pathogenic role was done using databases (COSMIC, dbSNP, ClinVar (NCBI)), prediction algorithms (SIFT, PolyPhen-2 or Mutation Taster) or splice prediction tools (SpliceView, NetGene2, Human Splicing Finder, NNSplice).
Statistical analysis
Statistics were performed using R software (version 3.2.3). The heatmap diagram was performed to represent the most relevant clinical data and potentially pathogenic variants detected. The distance measure used for heatmap clustering was Manhattan and the clustering method was Average. Circos diagram was generated with R for visualizing data of concomitantly mutated genes and/or MYCN amplification. The diagram does not include patients with only one alteration. The survival curves were plotted according to the Kaplan–Meier method and comparisons were performed by log-rank test. A Cox regression analysis was used for multivariate analysis. A p value ≤0.05 was considered statistically significant. Relapse-free survival (RFS) was defined as the time from diagnosis to the time of first occurrence of relapse, progression or last follow-up; and overall survival (OS) was defined as the time from diagnosis until death or until last follow-up if the patient was alive.
ACKNOWLEDGMENTS
We are grateful to José María Millán for help in genetic analysis and to María José Aparisi and Laia Pedrola (Genomics Unit at IIS La Fe) for help with the analysis of sequences and with the use of predictors for the identified variants. We are also thankful to Lisandra Muñoz for valuable help with the initial analysis.
CONFLICTS OF INTEREST
Authors declare that this study has no financial and personal relationships with other people or organizations that could inappropriately influence this work.
GRANT SUPPORT
This study was financially supported by grant PIE13/0046 from the Spanish Health Institute Carlos III and by grant from Fundación Científica de la Asociación Española Contra el Cáncer (AECC 2016).
REFERENCES
1. Otsuki Y, Tanaka M, Kamo T, Kitanaka C, Kuchino Y, Sugimura H. Guanine nucleotide exchange factor, Tiam1, directly binds to c-Myc and interferes with c-Myc-mediated apoptosis in rat-1 fibroblasts. J Biol Chem. 2003; 278: 5132-5140.
2. Matsuzawa K, Akita H, Watanabe T, Kakeno M, Matsui T, Wang S, Kaibuchi K. PAR3-aPKC regulates Tiam1 by modulating suppressive internal interactions. Mol Biol Cell. 2016; 27: 1511-1523.
3. Wang S, Watanabe T, Matsuzawa K, Katsumi A, Kakeno M, Matsui T, Ye F, Sato K, Murase K, Sugiyama I, Kimura K, Mizoguchi A, Ginsberg MH, et al. Tiam1 interaction with the PAR complex promotes talin-mediated Rac1 activation during polarized cell migration. J Cell Biol. 2012; 199: 331-345.
4. Lambert JM, Lambert QT, Reuther GW, Malliri A, Siderovski DP, Sondek J, Collard JG, Der CJ. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat Cell Biol. 2002; 4: 621-625.
5. Malliri A, van der Kammen RA, Clark K, van der Valk M, Michiels F, Collard JG. Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature. 2002; 417: 867-871.
6. Woodcock SA, Rooney C, Liontos M, Connolly Y, Zoumpourlis V, Whetton AD, Gorgoulis VG, Malliri A. SRC-induced disassembly of adherens junctions requires localized phosphorylation and degradation of the rac activator tiam1. Mol Cell. 2009; 33: 639-653.
7. Michiels F, Habets GG, Stam JC, van der Kammen RA, Collard JG. A role for Rac in Tiam1-induced membrane ruffling and invasion. Nature. 1995; 375: 338-340.
8. Karnoub AE, Worthylake DK, Rossman KL, Pruitt WM, Campbell SL, Sondek J, Der CJ. Molecular basis for Rac1 recognition by guanine nucleotide exchange factors. Nat Struct Biol. 2001; 8: 1037-1041.
9. Worthylake DK, Rossman KL, Sondek J. Crystal structure of Rac1 in complex with the guanine nucleotide exchange region of Tiam1. Nature. 2000; 408: 682-688.
10. Michiels F, Stam JC, Hordijk PL, van der Kammen RA, Ruuls-Van Stalle L, Feltkamp CA, Collard JG. Regulated membrane localization of Tiam1, mediated by the NH2-terminal pleckstrin homology domain, is required for Rac-dependent membrane ruffling and C-Jun NH2-terminal kinase activation. J Cell Biol. 1997; 137: 387-398.
11. Nishimura T, Yamaguchi T, Kato K, Yoshizawa M, Nabeshima Y, Ohno S, Hoshino M, Kaibuchi K. PAR-6-PAR-3 mediates Cdc42-induced Rac activation through the Rac GEFs STEF/Tiam1. Nat Cell Biol. 2005; 7: 270-277.
12. Leeuwen FN, Kain HE, Kammen RA, Michiels F, Kranenburg OW, Collard JG. The guanine nucleotide exchange factor Tiam1 affects neuronal morphology; opposing roles for the small GTPases Rac and Rho. J Cell Biol. 1997; 139: 797-807.
13. Tanaka M, Ohashi R, Nakamura R, Shinmura K, Kamo T, Sakai R, Sugimura H. Tiam1 mediates neurite outgrowth induced by ephrin-B1 and EphA2. EMBO J. 2004; 23: 1075-1088.
14. Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J, Westerman BA, van Arkel J, Ebus ME, Haneveld F, Lakeman A, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012; 483: 589-593.
15. Schramm A, Koster J, Assenov Y, Althoff K, Peifer M, Mahlow E, Odersky A, Beisser D, Ernst C, Henssen AG, Stephan H, Schroder C, Heukamp L, et al. Mutational dynamics between primary and relapse neuroblastomas. Nat Genet. 2015; 47: 872-877.
16. Canete A, Gerrard M, Rubie H, Castel V, Di Cataldo A, Munzer C, Ladenstein R, Brichard B, Bermudez JD, Couturier J, de Bernardi B, Pearson AJ, Michon J. Poor survival for infants with MYCN-amplified metastatic neuroblastoma despite intensified treatment: the International Society of Paediatric Oncology European Neuroblastoma Experience. J Clin Oncol. 2009; 27: 1014-1019.
17. Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, Nakagawara A, Hayashi Y, Mano H, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008; 455: 971-974.
18. George RE, Sanda T, Hanna M, Frohling S, Luther W 2nd, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, Xue L, Zozulya S, Gregor VE, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008; 455: 975-978.
19. Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, Valteau-Couanet D, Frebourg T, Michon J, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008; 455: 967-970.
20. Lasorsa VA, Formicola D, Pignataro P, Cimmino F, Calabrese FM, Mora J, Esposito MR, Pantile M, Zanon C, De Mariano M, Longo L, Hogarty MD, de Torres C, et al. Exome and deep sequencing of clinically aggressive neuroblastoma reveal somatic mutations that affect key pathways involved in cancer progression. Oncotarget. 2016; 7: 21840-21852. doi: 10.18632/oncotarget.8187.
21. Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, Kim J, Lawrence MS, Lichenstein L, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013; 45: 279-284.
22. Holzel M, Huang S, Koster J, Ora I, Lakeman A, Caron H, Nijkamp W, Xie J, Callens T, Asgharzadeh S, Seeger RC, Messiaen L, Versteeg R, et al. NF1 is a tumor suppressor in neuroblastoma that determines retinoic acid response and disease outcome. Cell. 2010; 142: 218-229.
23. Mandriota SJ, Valentijn LJ, Lesne L, Betts DR, Marino D, Boudal-Khoshbeen M, London WB, Rougemont AL, Attiyeh EF, Maris JM, Hogarty MD, Koster J, Molenaar JJ, et al. Ataxia-telangiectasia mutated (ATM) silencing promotes neuroblastoma progression through a MYCN independent mechanism. Oncotarget. 2015; 6: 18558-18576. doi: 10.18632/oncotarget.4061.
24. Sausen M, Leary RJ, Jones S, Wu J, Reynolds CP, Liu X, Blackford A, Parmigiani G, Diaz LA Jr, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE, et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet. 2013; 45: 12-17.
25. Buryk MA, Picarsic JL, Creary SE, Shaw PH, Simons JP, Deutsch M, Monaco SE, Nikiforov YE, Witchel SF. Identification of Unique, Heterozygous Germline Mutation, STK11 (p.F354L), in a Child with an Encapsulated Follicular Variant of Papillary Thyroid Carcinoma within Six Months of Completing Treatment for Neuroblastoma. Pediatr Dev Pathol. 2015; 18: 318-323.
26. Tweddle DA, Malcolm AJ, Bown N, Pearson AD, Lunec J. Evidence for the development of p53 mutations after cytotoxic therapy in a neuroblastoma cell line. Cancer Res. 2001; 61: 8-13.
27. Peaston AE, Camacho ML, Norris MD, Haber M, Marsh DJ, Robinson BG, Hyland VJ, Marshall GM. Absence of MEN2A- or 2B-type RET mutations in primary neuroblastoma tumour tissue. Mol Cell Probes. 1998; 12: 239-242.
28. Boissier P, Huynh-Do U. The guanine nucleotide exchange factor Tiam1: a Janus-faced molecule in cellular signaling. Cell Signal. 2014; 26: 483-491.
29. Genau HM, Huber J, Baschieri F, Akutsu M, Dotsch V, Farhan H, Rogov V, Behrends C. CUL3-KBTBD6/KBTBD7 ubiquitin ligase cooperates with GABARAP proteins to spatially restrict TIAM1-RAC1 signaling. Mol Cell. 2015; 57: 995-1010.
30. Ferrero M, Avivar A, Garcia-Macias MC, Font de Mora J. Phosphoinositide 3-kinase/AKT signaling can promote AIB1 stability independently of GSK3 phosphorylation. Cancer Res. 2008; 68: 5450-5459.
31. Hammerle B, Yanez Y, Palanca S, Canete A, Burks DJ, Castel V, Font de Mora J. Targeting neuroblastoma stem cells with retinoic acid and proteasome inhibitor. PLoS One. 2013; 8: e76761.
32. Burgues O, Navarro S, Noguera R, Pellin A, Ruiz A, Castel V, Llombart-Bosch A. Prognostic value of the International Neuroblastoma Pathology Classification in Neuroblastoma (Schwannian stroma-poor) and comparison with other prognostic factors: a study of 182 cases from the Spanish Neuroblastoma Registry. Virchows Arch. 2006; 449: 410-420.
33. Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B, Stram DO, Gerbing RB, Lukens JN, Matthay KK, Castleberry RP. The International Neuroblastoma Pathology Classification (the Shimada system). Cancer. 1999; 86: 364-372.
34. Cohn SL, Pearson AD, London WB, Monclair T, Ambros PF, Brodeur GM, Faldum A, Hero B, Iehara T, Machin D, Mosseri V, Simon T, Garaventa A, et al. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol. 2009; 27: 289-297.
35. Ambros IM, Benard J, Boavida M, Bown N, Caron H, Combaret V, Couturier J, Darnfors C, Delattre O, Freeman-Edward J, Gambini C, Gross N, Hattinger CM, et al. Quality assessment of genetic markers used for therapy stratification. J Clin Oncol. 2003; 21: 2077-2084.
36. Noguera R, Canete A, Pellin A, Ruiz A, Tasso M, Navarro S, Castel V, Llombart-Bosch A. MYCN gain and MYCN amplification in a stage 4S neuroblastoma. Cancer Genet Cytogenet. 2003; 140: 157-161.
37. Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011; 29: 24-26.
38. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17: 405-424.