
INTRODUCTION
Despite unprecedented successes in the field of medicine in the last fifty years, cancer remains a serious threat to human survival [1–4]. Chemotherapeutics, in combination with or in addition to surgery and radiotherapy, play an important role in increasing life expectancy of cancer patients [5–9]. Tumors are clones of rapidly dividing cells unregulated by normal mechanisms of growth suppression. Chemotherapy aims at interfering with this uncontrolled process of cell division [10, 11]. However, many cancer drugs typically lack specificity to transformed cells [12–14]. Consequently, they also kill healthy cells undergoing rapid proliferation resulting in toxic side effects. Another limitation of chemotherapy is the development of resistance by tumor cells [15–17]. Thus, a more effective alternative could be other classes of drugs with the property to specifically target cancer cells without toxicity to normal cells. One more requirement is a lower tendency for development of resistance against such drugs compared to conventional chemotherapeutics. AMPs are an untapped resource with low propensity to elicit development of resistance by its target and to display toxicity to healthy cells undergoing rapid proliferation [18–20]. AMPs are an essential component of the host innate immunity [21–23]. Although they display considerable diversity in both primary and secondary structures, [24–27] the cationic amphipathic motif is a typical structural feature of AMPs and an important determinant of antimicrobial functions [28–31]. While AMP investigations have been largely focused on antimicrobial properties, there is increasing evidence that AMPs display antitumor functions, perhaps in the context of a multifunctional host defense system of multicellular organisms. Alternatively, the antitumor property could be a “side-activity” not necessarily implicated in the natural selection of the common cationic amphipathic structure of AMPs. We reviewed herein the potential of AMPs for application to antitumor therapy as anticancer peptides (ACPs) with novel mechanisms. Broad structural and functional properties of AMPs not pertaining to selective action against cancer cells and antitumor efficacy are reviewed elsewhere [32–39].
Basis for antitumor property of AMPs: selective recognition of cancer cells via electrostatic interactions
Most AMPs are short (typically 10 to 50 residues long) cationic peptides with an amphipathic structure [40–48]. They are structurally diverse in both amino acid compositions and secondary structures (α-helix, β-sheets, extended helix, and loop) (Figure 1). There are multiple reviews of AMP structures, [49–52] which we do not discuss here. AMPs generally recognize their target via electrostatic interactions with negatively charged lipids on cell membranes [53–56]. Because these interactions are not mediated by specific receptors, conversion of L to D enantiomers does not necessarily disrupt the binding capacity of AMPs as shown by Papo and others [57–59]. AMPs display strong interactions with bacterial membranes due to high density of electronegative charges on the bacterial surface, such as lipopolysaccharide (LPS) on the outer membrane of gram-negative bacteria [60–63] or lipoteichoic acid (LTA) on gram-positive bacterial membranes [64–68]. Of note, there is no consensus sequence for binding activities, as cationic AMPs (typical charge of +2 or more) of all types of secondary structures (α-helix, β-sheets, loop alone or in combination; Figure 1) and diverse primary sequences with different positive charges are able to recognize their microbial targets [69–73]. It is, therefore, evident that the cationic amphipathic motif is the main requirement for activity whereas the primary sequence determines specificity or spectrum of activity. While AMPs may display toxicity to mammalian cells, toxic concentrations are commonly one log of magnitude higher compared to minimum inhibitory concentrations against bacteria [74, 75]. Hence, it is logical to predict that AMPs with promising pre-clinical data will display a minimum therapeutic index (maximum tolerated dose/minimum therapeutic dose) required for efficacy in specific applications.
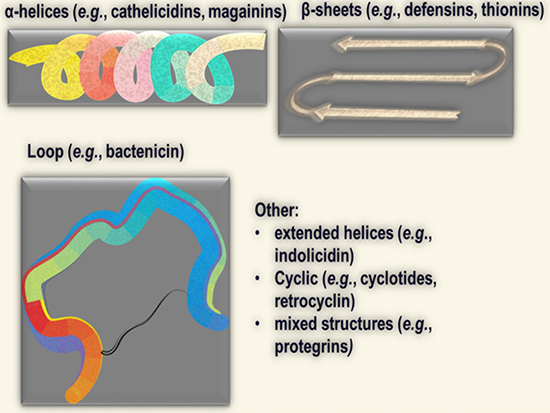
Figure 1: Main structural classes of cationic antimicrobial peptides (AMPs).
Some AMPs specifically target tumor or cancer cells because transformed cells generally incorporate phosphatidylserine (PS, 3–9% of the total amount of phospholipids) in the outer leaflet of the plasma membrane (Figure 2) [76–78]. PS is usually found on the inner leaflet of the cytoplasmic membrane of normal mammalian cells. However, it can be transferred to the outer leaflet of the plasma membrane of cells undergoing apoptosis, which disrupts the asymmetry observed for normal mammalian cell membranes [79, 80]. Hence, this change in asymmetry is typically shared by both apoptotic cells and several types of cancer cells and facilitates recognition and clearance of these cells by monocytes [81–83]. Other factors that may contribute to elevated negative charges on cancer cells include heparin sulfates, [84–86] and O-glycosylated mucins on the surface of tumor cells [87–90]. However, the density of electronegative charge (due to a single phosphate group on PS) on cancer cells is relatively lower compared to negative charges (due to multiple phosphate groups on LPS, LTA, in addition a phosphate group on PS) on bacterial cell membranes. As a result, AMP affinity for cancer cells is inherently weaker than the affinity for bacteria. Of note, electronegative charges do not always enhance activity. As shown by Fadnes et al. (2009), Cell surface heparin sulfate inhibits the bovine AMP lactoferricin by sequestering the peptide away from the lipid membrane [91]. Cancer cell membranes display other properties that may facilitate killing by AMPs compared to normal cell membranes. Some transformed cells may incorporate lower levels of cholesterol in their membranes, enhancing fluidity. For instance, cell membranes of human leukemia and lung cancers display increased fluidity due to a lower level of cholesterol in their membranes compared to membranes of normal leukocytes and pulmonary cells [92–94]. This increase in membrane fluidity may potentiate lytic effects of AMPs as in the case of cecropins and other peptides [95–97]. Conversely, some cancer cells incorporate elevated cholesterol levels as part of lipid rafts (e.g., prostate cancer) in their membranes compared to normal cell membranes [98, 99]. Therefore, the role of cholesterol content in cationic ACP activity against cancer cells remains unclear. For instance, some enveloped viruses are susceptible to AMPs (e.g., LL37 activity against herpes simplex and influenza viruses) although they incorporate high cholesterol content (lipid rafts) in their membranes [100, 101]. Another interesting property of cancer cells, which may enhance AMP binding, is the increase in surface area with increasing number of microvilli [102, 103]. Upon binding to cancer cells, AMPs may either disrupt the membrane or penetrate the cell and attack the mitochondria leading to apoptosis. The defensins (29–45 amino acids long), an important class of Cys-rich antimicrobial peptides (β-sheets, Figure 1), were among the first AMPs to be discovered and to demonstrate antitumor activity. Although these AMPs have been isolated from different species including plants, [104, 105] the α-(Cys1-Cys6, Cys2-Cys4 and Cys3-Cys5 bridges, with Cys residues numbered based on location from the N-terminus) and β-defensins (Cys1-Cys5, Cys2-Cys4 and Cys3-Cys6 bridges) synthesized in humans are the most studied defensins to date [104–109]. Antitumor activities of α-defensins, notably the human neutrophil peptides (HNP) 1–3, have been demonstrated via both membranolytic and apoptotic mechanisms as well as inhibition of neovascularization required for tumor growth [110, 111]. However, the HNPs also kill normal cells such as fibroblasts, epithelial cells, and leucocyte. Similarly, the plant defensins also display a lack of selectivity towards tumor cells [112–115]. As a result, the defensins are generally not efficient ACP therapeutics, or they require structural optimization to achieve antitumor selectivity. We examined hereafter the antitumor properties of AMPs derived from animals based on two sets of mechanisms, selective plasma membrane disruption or non-membranolytic cytotoxicity.
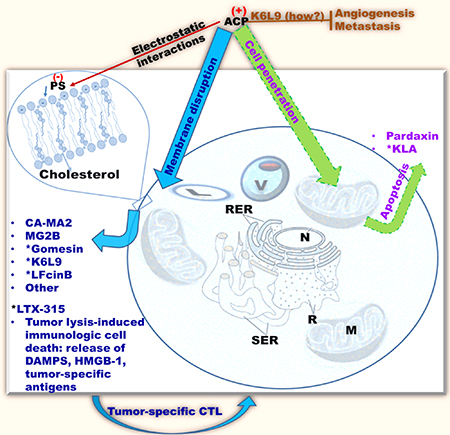
Figure 2: Common antitumor mechanisms of cationic AMPs classified as ACPs. ACPs (anticancer peptides), or cationic AMPs with anticancer properties, selectively recognize cancer cells by electrostatic interactions with negatively charged phospholipids on the surface of eukaryotic cells [e.g., PS (phosphatidylserine)]. Some ACPs demonstrate in vivo efficacy (e.g., *MGB2, Gomesin, K6L9, LTX-315); ACPs tend to kill cancer cells by membrane perturbation (blue/cyan), although some (e.g., KLA, Pardaxin) may penetrate the target cell and disrupt the mitochondrial membrane resulting in apoptosis (green/purple). (How?), mechanism unclear; brown perpendicular bar, inhibition; CTL, cytotoxic T-Lymphocytes; M, mitochondria, SER, smooth endoplasmic reticulum; RER, rough endoplasmic reticulum; R, ribosomes; N, nucleus; V, vacuole; L, lysosome; only cholesterol and other lipids are shown; membrane proteins are omitted for clarity.
Evidence for selective membrane disruption
One of the earliest indications that AMPs may be a source of anti-tumor therapy is a study by Cruciani and colleagues [116]. Discovered by Michael Zasloff, the magainins are a family of AMPs with broad-spectrum antibacterial activity found in the African frog Xenopus larvae [117–119]. Cruciani et al. showed that magainin 2 and its analogues demonstrated activity selectively against both hematopoietic and solid tumor cells. The cytotoxic effects of the magainins were rapid (within 1 hour) at a concentration as low at 12 μg/mL and were not observed against normal lymphocytes even at up to 200 μg/mL. The peptides induced ion channels leading to leakage of Na+, K+, and Cl- ions. The cytotoxic effects of the peptides were abrogated when the electrical gradient was eliminated prior to peptide exposure, indicating the membrane potential is essential to the peptide activity. Exposure of the mitochondria to the peptide resulted in inhibition of respiration and leakage of glucose through the peptide-induced channels [120]. Subsequently, an interesting link was established between two independent discoveries [the Cecropins (insect) and magainins (frog)] by Boman and Zasloff, respectively [119, 121, 122]. In this study a hybrid between Cecropin A and magainin 2 (CA-MA-2, KWKLFKKI-P-KFLHSAKKF-NH2) was constructed with three more derivatives based on proline substitutions in the hinge region [123]. These investigators discovered that the activities against several tumor cell lines were enhanced compared to toxicity to erythrocytes and primary fibroblasts. The cationic peptide CA-MA-2 displayed no detectable hemolysis and cytotoxicity against the primary cell NIH-3T3 fibroblast at concentrations up to 100 μM. In contrast, the 50% inhibition concentration (IC50) against several tumor cell lines was as low as 20 μM. it is important to note that antimicrobial activity was always higher than the anti-tumor effects, suggesting that these AMPs could be further optimized specifically for enhanced antitumor properties. Disruption of large phosphatidylcholine (PC)/phosphatidylserine (PS)-based unilamellar (mixed PC-PS) vesicles by CA-MA-2 indicated a membrane perturbation mechanism, although it would have been informative for the investigators to include the parent peptides Cecropin A and Magainin 2 in this study. Another example of cell membrane disruption was more directly demonstrated by the AMP chrysophsin-1 (FFGWLIKGAIHAGKAIHGLI) [124]. Using fluorescent, scanning and transmission electron microscopy, combined with LDH release, the investigators showed convincingly that the cationic amphipathic peptide disrupted the plasma membrane of several cancer cell lines at much lower concentrations compared to the CA-MA-2 peptide. Cancer cell death by apoptosis was ruled out as caspase expression and activities were not affected by chrysophsin-1.
While these previous studies show great promise, they fell short of demonstrating anti-tumor efficacy in vivo. In addition, AMP activity against cancer cells is much lower than their antimicrobial activity. Hence, two main problems remained to be addressed: (1) the limited anti-tumor activity and (2) specificity toward tumor cells. Both limitations can be overcome if the specificity of AMPs towards tumor cells is enhanced. The strength of AMP interaction with cancer cells may affect both activity and specificity. Hence, in 2011 Liu and colleagues published an elegant study addressing this limitation. They reasoned that linking an AMP to a cancer-homing peptide would enhance specificity and activity against cancer cells. Isolated from frog skin, Bombesin appeared to be a good candidate as the 14-residue peptide recognizes a variety of human cancer cells. The question was which AMP would be the best choice for this experiment? Magainin II (MG2), also derived from frog skin, was one of the most studied AMPs at the time. In fact, the magainins are likely to become the first classical family of AMPs to be used clinically as the derivative pexiganan (cream, 0.08%) is currently in phase III clinical trials in patients with mild infections of diabetic foot ulcers [125–127]. Hence, MG2 was linked to Bombesin (MG2B, GIGKFLHSAKKFGKAFVGEIMNSGG-QRLGNQWAVGHLM). The MG2B peptide displayed higher cytolytic effects compared to MG2. In fact, the in vivo efficacy of MG2B was demonstrated in mice bearing MCF-7 tumor grafts. With a daily intratumoral injection of MG2B (20 mg/kg) for 5 days, there was a significant reduction of the tumor size in mice [128]. There are several other examples of membranolytic effects of ACPs, including the use of the ACP gomesin (ZCRRLCYKQRCVTYCRGR) in a cream formulation for successful topical treatment in mice [129, 130]. Importantly, one of the most significant anti-cancer effects of a membranolytic ACP was demonstrated by Papo and colleagues (Figures 2 and 3) [131–133]. The investigators constructed a D-enantiomer of an engineered ACP K6L9. With a daily dose of the peptide (9 mg/kg) injected systemically every other day for a total of nine doses, immunodeficient mice implanted with both breast and prostate metastatic cancers were protected against malignant disease. The tumors became necrotic, and the density of the tumor-induced neovascularization was significantly reduced. The selective binding of the ACP to the negatively charged PS and cytoplasmic membrane depolarization were also demonstrated. The ACP D-K6L9 (LKLLKKLLKKLLKLL) is the most successful demonstration of the systemic anticancer efficacy of an ACP to date [58]. Notably, D-K6L9 is made of only two amino acids. As natural AMPs work in the context of a multifunctional immune system, they are more efficient at protecting the host against disease than they are at curing established illnesses (more preventive than therapeutic). Hence, it is logical to engineer AMPs with more optimized structures for therapeutic applications as opposed to using AMPs as they are produced in nature. We predict that the success of AMP engineering will be facilitated by the establishment of a definitive framework for distinguishing the unique role of the cationic from that of the hydrophobic domain in selectivity toward the target versus host cells. Once a guideline for selective killing of different target cells is established, it will then be possible to design AMPs with enhanced therapeutic index with less trial and error. In the case of the peptide K6L9, the investigators use the principle of amphipathicity to design a 15-residue AMP (charge = +6) with an idealized amphipathic helix (Figure 3). In addition, the use of D-amino acids enhances stability, addressing a concern for lability of AMPs [58, 132–134]. This is an important example of how AMP engineering can lead to enhanced results. Yet, the most advanced (in terms of clinical trials) cationic ACP is not D-K6L9. Over the last two decades, there has been an increasing interest in bovine lactoferricin (LFcinB). LFcinB is derived from the natural milk protein bovine lactoferrin (LFB) by pepsin digestion [135–139]. In addition to its antimicrobial properties, [140–143] LFcinB demonstrates enhanced activities against cancer cells compared to normal mammalian cells [139, 144, 145]. Importantly, high-resolution imaging revealed a membranolytic mechanism [146]. These studies reached a turning point when Eliassen and colleagues (2006) began to examine the in vivo efficacy of LFcinB in mice, demonstrating the growth inhibition of neuroblastoma xenografts [147]. While the peptide could be localized in the mitochondria with caspase activation, the lytic effects on the cytoplasmic membrane represented the primary cytotoxic mechanism, as pan-inhibition of the caspase enzymatic cascade could not reverse the cytotoxic effects of the peptide. These studies led to further structural optimization using 3,3-diphenylalanine (Dip) substitutions for Trp at specific positions of the 9-mer template. These investigations resulted in enhanced selectivity (IC50 below 5–10 μM) against a variety of tumors compared to normal cells. More importantly, these studies revealed a cytolytic-immunogenic dual cell death mechanism [148–156]. Lysis of the tumor induced the release of danger-associated molecular patterns (DAMPs) including high mobility group box protein 1 (HMGB1, reviewed by Frank et al., 2015 [157]) in addition to antigens from the dead tumor cells. As potent stimulators of immune responses, HMGB1 potentiated A20 lymphoma-specific activation of immature dendritic cells with subsequent generation of tumor-specific cytotoxic T-lymphocytes and tumor cell lysis. The immunogenic cell death was further confirmed by the adoptive transfer of syngeneic A20 lymphoma-specific CTLs and resulting protection of immunodeficient mice from tumor implants. Further, LTX-315-treated mice in remission from the tumor were protected from a second challenge by the syngeneic tumor and not by a different tumor type, indicating the specificity of the immunogenic cell death. Hence, the potent derivative LTX-315 was selected for advanced preclinical studies. LTX-315 is now (2016) in phase 1 clinical trial as described later. The study of LTX-315 is yet another illustration of how natural AMPs can be optimized for specific clinical applications.
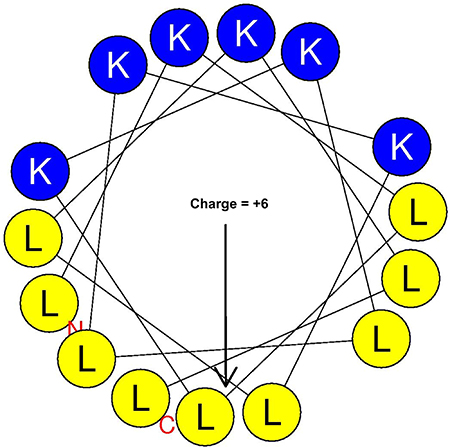
Figure 3: Helical wheel analysis of the engineered AMP K6L9 designed by Papo et al. [57]. The peptide was modeled to form an idealized amphipathic helix with only two amino acids; a structural optimization strategy that has been shown to enhance antimicrobial functions, now applied to antitumor properties as well. Arrow indicates direction of the hydrophobic moment. Structural motifs: yellow, hydrophobic; blue, cationic.
Non-membranolytic mechanisms of ACP
The ACP K6L9, while exerting a direct membranolytic effect on cancer cells, demonstrates anti-angiogenic and anti-metastatic effects, an anti-tumor mechanism that remains to be elucidated. Another common anti-cancer mechanism is the potentiation of apoptosis in cancer cells by ACP [158–160]. While ACPs selectively interact with cancer compared to normal cell membranes, some AMPs may both perturb the membrane and penetrate the cells while other non-lytic ACPs may simply traverse the membrane and access the intracellular compartment. In both cases, the ACP disrupts the mitochondria (as expected because of the bacterial origin of mitochondria) and induces programmed cell death. This apoptotic mechanism is demonstrated by swelling of the mitochondria, disruption of the mitochondrial membrane, translocation of PS to the surface of the cell, and stimulation of apoptotic markers (e.g., caspase enzymatic pathway). One of the earliest examples of an apoptotic ACP was reported by Mai et al., 2001 [161]. The authors used a KLA repeat AMP [(KLAKLAK)2] conjugated with a transduction peptide (RRQRRTSKLMKRGGKLAKLAKKLAKLAK) by a glycine pacer. The intratumoral injection of the chimeric ACP resulted in the translocation of the peptide into the cytoplasm of the tumor cells, disruption of the mitochondria, and stimulation of apoptotic enzymatic cascade by caspase 3 activation. Other ACPs have the inherent property to penetrate the cancer cells without the need for conjugation with a cell penetrating peptide. An example of such a peptide (GFFALIPKIISSPLFKTLLSAVGSALSSSGGQE) is pardaxin, which induces apoptosis of squamous cell carcinoma cells by caspase 3 activation [162]. In addition, some AMPs may be used as cell penetrating peptides to transfer an anticancer drug into cancer cells. This is the case of the AMP PR39 (RRRPRPPYLPRPRPPPFFPPRLPP RIPPGFPPRFPPRFP) used as a cell penetrating peptide to transfer Stat3 siRNA (the cargo) into breast cancer cells [163–165]. As previously mentioned, another interesting antitumor mechanism is the immunogenic cell death conferring long-lasting protection against future challenges from the same tumor type. Such a mechanism is displayed by the ACP LTX-315, [151] which was selected for advanced preclinical studies. LTX-315 is now in phase 1 clinical trial [151, 155, 156]. Noteworthy is an antitumor mechanism occurring via the lysosomal-mitochondrial death pathway by the defensin Brevenin-2R (in vitro activity) [166]. As the anticancer mechanisms of AMPs become increasingly clear, structural optimization for enhanced anticancer potency will be more achievable and, therefore, clinical development more likely to succeed.
Of note, AMPs synthesized in plants are another important source of peptide therapeutics that could be used as anticancer agents. However, for many of these AMPs, the anticancer mechanisms are unclear. AMPs derived from plants are beyond the scope of this review and are reviewed elsewhere [32]. A list of representative AMPs with selective antitumor mechanism is shown Table 1.
Table 1: Representative antimicrobial peptides with selective antitumor mechanisms
AMP name |
Amino acid sequence |
Source |
Tumor target |
Mechanism |
Reference |
|---|---|---|---|---|---|
Alpha-defensin-1 |
ACYCRIPACIAGERRYG TCIYQGRLWAFCC |
Human |
HTC/STC |
Apoptosis |
Xu et al., 2008 [109] |
Antiangiogenic |
|||||
BMAP-28 |
GGLRSLGRKILRAWK KYG |
Bovine |
HTC |
MP/Ca influx |
Risso et al., 2002 [158] |
Apoptosis |
|||||
Brevenin-2R |
KFALGKVNAKLQSLN AKSLKQSGCC |
Frog |
STC |
LDP |
Ghavami et al., 2008 [165] |
Buforin IIb |
RAGLQFPVG[RLLR]3 |
Frog |
HTC/STC |
Apoptosis |
Lee et al., 2008159 |
CA-MA-2 |
KWKLFKKI-P-KFLHS AKKF |
Hybrid |
STC |
MP |
Shin et al., 2000 [122] |
Cecropin A |
KWKLFKKIEKVGQNIR DGIIKAGPAVAVVGQA TQIAK |
Silk moth |
HTC |
MP |
Hui et al., 2002 [96] |
Cecropin B |
KWKVFKKIEKMGRNI RNGIVKAGPAIAVLGE AKAL |
Silk moth |
HTC/STC |
MP/Apoptosis |
Li et al., 2016 [94] |
chrysophsin-1 |
FFGWLIKGAIHAGKA IHGLI |
Red sea bream |
HTC/STC |
MP |
Hsu et al., 2011 [123] |
D-K6L9 |
LKLLKKLLKKLLKLL |
Engineered |
STC |
MP |
Papo et al., 2006 [57] |
Gomesin |
*ZCRRLCYKQRCVTY CRGR |
Spider |
STC |
MP |
Domingues et al., 2010 [128] |
KLA |
RRQRRTSKLMKRGGK LAKL-AKKLAKLAK- (KLAKLAK)2 |
Engineered |
STC |
MP |
Mai et al., 2001 [160] |
lactoferricin B |
FKC1RRWQWRMKKLG APSITC1VRRAF |
Bovine |
HTC/STC |
MP/Apoptosis |
Eliassen et al, 2002 [145] |
LL37 |
LLGDFFRKSKEKIGKEFKR IVQRIKDFLRNLVPRTES |
Human |
Ovarian CA |
MP |
Chuang et al. 2009 [210] |
LTX-315 |
K-K-W-W-K-K-W-Dip-K |
Engineered |
HTC/STC |
MP/ICD |
Haug et al., 2016148 |
Phase I/II trial |
|||||
Magainin 2 |
GIGKFLHSAKKFGKAF VGEIMNS |
Frog |
HTC/STC |
MP |
Cruciani et al., 1991115] |
Melittin |
GIGAVLKVLTTGLPALIS WIKRKRQQ |
Insect |
STC |
MP |
Wang et al., 2009 [56] |
MG2B |
GIGKFLHSAKKFGKAF VGEIMNSGG-QRLGNQ WAVGHLM |
Hyprid AMP |
MCF-7 tumor |
MP |
Liu et al., 2011 [128] |
Pardaxin |
GFFALIPKIISSPLFKTLL SAVGSALSSSGGQE |
Fish |
STC |
MP |
Han et al., 2016 [162] |
*pyroglutamic acid; HTC, hematological tumor cells; STC, solid tumor cells; MP, membrane permeabilization; LDP, lysosomal death pathway; ICD, immunological cell death..
Prospect for clinical use of AMPs as anticancer agents
The clinical development of AMPs as ACPs faces some of the same challenges to AMP clinical development as antimicrobial agents. AMPs are not traditional drugs. Because they have multifunctional properties, the adaptation from their natural environment to clinical applications without structural optimization is rather challenging. While AMPs work well in the context of a competent immune system, it is likely that in nature a particular AMP structure is not completely optimized for a single function (e.g., antibacterial, antiviral, or anticancer). The field also faces some unfounded criticisms that have hindered support for AMP development. Some of these criticisms are that (1) “AMPs are labile and likely to have poor pharmacokinetic properties”, an assumption based on the peptidic nature of AMPs; (2) “AMPs are expensive to make”; (3) “AMPs are not good drugs” because they do not recognize specific receptors.
Although stability is an important concern, the first criticism is based on the assumption that all peptides have similar stability and clearance mechanisms. One of the shortcomings of AMPs is the lack of correlation between in vitro susceptibility testing and efficacy in animal models, with some exceptions including some of the AMPs discussed in this review and elsewhere [58, 156, 167–169]. Assays establishing correlation between in vitro stability and bioavailability in animal models might address this concern. Currently, AMPs with optimized structures are amenable to parenteral administration including systemic, respiratory, intramuscular, intraperitoneal, or subcutaneous unless otherwise contraindicated [167, 168, 170–172]. Although the peptidic nature of AMPs precludes oral delivery, the development of specific delivery systems protecting AMPs from degradation by digestive enzymes may increase intestinal absorption and the feasibility of oral administration. Several strategies can be used to enhance PK properties of AMPs. One approach is the utilization of D-enantiomers to increase stability, [133] although such strategy is only indicated if decreasing peptide clearance enhances therapeutic efficacy and does not potentiate toxic side effects. Other strategies include end-to-end cyclization, C-terminus amidation, pegylation [attachment of polyethylene glycol (PEG) to a molecule], and liposomal delivery. Cyclisation and amidation can confer peptide stability by decreasing susceptibility to protease digestion, as demonstrated by the modification of gomesin and other AMPs [173, 174]. As a principle, all AMPs used in our laboratory are amidated [18, 20, 175, 176]. As shown by the Gumbleton group and others, pegylation of AMPs can result in lower host toxicity without affecting antimicrobial activity [177]. Pegylation enhances the pharmacological properties of a given drug in a number of ways [178–183]. Because it increases hydrophilicity, PEG serves as a shield that protects against protease digestion, prolongs circulation time, and reduces the glomerular filtration rate. An interesting example of AMP pegylation is a recent design by Kelly et al., 2016 as shown in Figure 4 [183]. A pro-peptide was designed using the AMP P18 covalently attached to a linker region that is sensitive to the cysteine protease cathepsin B, followed by a PEG region to confer thermodynamic stability to the peptide. In vitro studies show that selectivity index can be enhanced against an ovarian carcinoma cell line (A2780). While this strategy is still conceptual, as it requires extensive in vivo studies for proof of concept, it shows how a pro-peptide can be designed in combination with pegylation to enhance the PK properties of an ACP or AMP. Another strategy could be the liposomal formulation of AMPs [184–187]. However, one shortcoming is that AMPs are membrane active and could, therefore, bind and disrupt the liposome. Packaging the molecule as an inactive pro-peptide in the liposome could theoretically overcome this problem. One last potential caveat is whether we are able to produce liposomes that can discriminate between the target and normal cells. An AMP delivered inside a eukaryotic cell will probably interact with mitochondria as it would with a bacterial cell, based on similarities between a bacterial cell and mitochondrial membranes. One strategy that is already developed is to tag the lysosome with a cancer cell-specific ligand for targeted delivery of the peptide [188]. Finally, designing AMPs as part of nanoparticles for delivery is another strategy that may improve PK properties of AMPs and should be explored [189–191]. All of these strategies for enhancing PK properties should be considered only when indicated, as AMPs are highly diverse in structure, which affects PK properties.

Figure 4: Strategy to improve the PK properties of AMPs adapted from Kelly et al., 2016 [183]. The AMP P18 is amidated at the C-terminus. In addition, it is covalently bound to the protease cathepsin B-sensitive linker for the release of the cancer-active drug; this linker is also covalently attached to a polyethylene glycol (PEG) polymer, which is a hydrophilic moiety that serves as a protective shield from protease degradation and drug clearance.
The second criticism is based on an outdated notion of the highly impractical cost of peptide production. While cost remains a concern, peptide- and protein-based drugs have been used clinically for decades from anti-hypertensive (e.g. Lisinopril) [192–196] and anti-diabetic (e.g., insulin) [197, 198] to immune, antiviral (e.g., fuzeon), [199, 200] antibacterial, [201–203] and hormonal therapy [196]. Modern technology and larger scale synthesis have also significantly reduced the production cost of AMPs. The third concern is based on the fact that AMPs do not select their targets via specific receptors [204]. This nonreceptor-mediated recognition is one of the major reasons AMPs are less likely to invoke selection of resistance compared to current antibiotics/anticancer drugs and the basis for broad selectivity against diverse types of multidrug-resistant microbial pathogens and transformed cells. Lipid-mediated selectivity of AMPs should be considered as a major strength, not a weakness. These misconceptions against AMPs have considerably hampered the progress of their development, particularly AMP engineering for structural optimization in the United States. The success of the ACPs K6L9, LTX-315, and other engineered antibacterial AMPs [20, 167, 168, 175, 176] illustrates the need for AMP engineering and, thus, the establishment of a rational framework for predicting structural determinants of the selective killing of different target cells. Such a guideline for structural optimization, combined with PK-enhancing strategies, would enhance our ability to increase the therapeutic index of AMPs with significantly less trial and error.
Despite the aforementioned challenges, there are a few AMPs being evaluated as anticancer (in addition to antimicrobial) agents in advanced phases of clinical development. The human cathelicidin LL37 is currently (2016) in Phase 2 clinical trial for melanoma (lesions at least 10 mm and not completely resectable) by intratumoral injections in patients with no known immune deficiency, a collaborative effort of M.D. Anderson Cancer Center and National Cancer Institute [205]. The first patients were enrolled in July 2015. LL37 is the single most studied human AMP [206–208]. It is normally found in human skin, reproductive, and respiratory systems and known to have multiple functional properties including (but not limited to) antibacterial, antiviral, antifungal, immunomodulatory, and anticancer activities [209–211]. Another AMP, LTX-315, is in phase 1 trial for PK and efficacy treatment of multiple types of transdermally accessible tumors. This is a lactoferrin-derived lytic peptide that binds and lyses tumor cells. The resulting tumor necrosis leads to enhanced presentation of tumor antigens and induced innate and adaptive immunity against the tumor as described above. This clinical trial started on October 28, 2013, and the last update (December 2016) indicates that LTX-315 is still in phase 1 “Open-label, multi-arm, multi-centre, multi-dose, dose Escalation Study” for exploration of efficacy as monotherapy or in combination with either ipilimumab or pembrolizumab in patients with transdermally accessible tumors” [212]. Noteworthy are other AMPs in clinical trial as anti-infective agents (e.g., OP-145 for otitis media and pexiganan for diabetic foot ulcer), but not as ACPs. Toward the goal for clinical applications, it is possible to improve the therapeutic index of current chemotherapeutic agents by considering combination therapy. Combining AMPs with a chemotherapeutic agent may help decrease dosage, which would result in lower toxicity. While the current clinical trial of LTX-315 including combination with immunotherapeutics (Ipilimumab or Pembrolizumab) is a step in the right direction, combination therapy using AMPs and chemotherapeutic agents needs to be explored for possible synergy [183].
Concluding remarks
Since the discovery of AMPs more than three decades ago, no other class of compounds has matched their versatility as multifunctional compounds. AMPs have the potential to become the only class of drugs that can be used against polymicrobial co-infections (e.g., bacterial and viral [213]) and cancer. However, the multifunctionality determined by a typical AMP structure suggests that no single property is completely optimized in natural AMPs in the context of maintaining multiple functions. Rational peptide engineering is essential to AMP development for clinical applications. An important task is to dissect the structural determinants of each property to uncouple each of AMP functions for “application-specific optimization”. Only when such studies are conducted in a systematic way will we begin to significantly explore the clinical potential of AMPs as a diverse class of therapeutics. AMP research has been largely occurring outside of the United States. Despite a vast literature in AMP research, this is still an area that is critically underfunded by the National Institute of Health (NIH). Because of the initial failure of AMPs to reach the Clinique, the resulting bias has largely hindered the advancement of AMP research in the United States. Hence, there is a pressing need for the NIH and pharmaceutical companies to support more AMP research to collect the evidence necessary to assess whether the promise of AMPs will ever come to fruition. As an essential component of the immune system, AMPs warrant such exploration.
ACKNOWLEDGMENTS AND FUNDING
This research is supported by NIH awards R01 HL091938, HL-125128, and a grant (CIA-123062) from Flight Attendant Medical Research Institute.
CONFLICTS OF INTEREST
None.
REFERENCES
1. Hashim D, Boffetta P, La Vecchia C, Rota M, Bertuccio P, Malvezzi M, Negri E. The global decrease in cancer mortality: trends and disparities. Ann Oncol. 2016; 27:926–933.
2. Torre LA, Siegel RL, Ward EM, Jemal A. Global Cancer Incidence and Mortality Rates and Trends–An Update. Cancer Epidemiol Biomarkers Prev. 2016; 25:16–27.
3. Saika K, Machii R. Cancer mortality attributable to tobacco by region based on the WHO Global Report. Jpn J Clin Oncol. 2012; 42:771–72.
4. Shibuya K, Mathers CD, Boschi-Pinto C, Lopez AD, Murray CJ. Global and regional estimates of cancer mortality and incidence by site: II. Results for the global burden of disease 2000. BMC Cancer. 2002; 2:37.
5. Daskivich TJ. Life Expectancy and Treatment Choice for Men with High-risk Prostate Cancer. Eur Urol. 2015; 68:59–60.
6. Arrington AK, Goldstein L, Kruper L, Vito C, Yim J, Chen SL. Life expectancy after curative-intent treatment of breast cancer: impact on long-term follow-up care. Am Surg. 2014; 80:604–09.
7. Delpierre C, Lamy S, Kelly-Irving M, Molinié F, Velten M, Tretarre B, Woronoff AS, Buemi A, Lapôtre-Ledoux B, Bara S, Guizard AV, Colonna M, Grosclaude P. Life expectancy estimates as a key factor in over-treatment: the case of prostate cancer. Cancer Epidemiol. 2013; 37:462–68.
8. Repetto L, Comandini D, Mammoliti S. Life expectancy, comorbidity and quality of life: the treatment equation in the older cancer patients. Crit Rev Oncol Hematol. 2001; 37:147–52.
9. Haybittle JL. Life expectancy as a measurement of the benefit shown by clinical trials of treatment for early breast cancer. Clin Oncol (R Coll Radiol). 1998; 10:92–94.
10. Kolberg HC, Villena-Heinsen C, Deml MM, Kraemer S, Diedrich K, Friedrich M. Relationship between chemotherapy with paclitaxel, cisplatin, vinorelbine and titanocene dichloride and expression of proliferation markers and tumour suppressor gene p53 in human ovarian cancer xenografts in nude mice. Eur J Gynaecol Oncol. 2005; 26:398–402.
11. Pierard GE, Focan C, Lapiere CM. Cell proliferation in a malignant angioendothelioma during sequential chemotherapy. J Cutan Pathol. 1979; 6:479–485.
12. Kato Y, Sato J, Kato R, Takata R, Obara W. [Side effect and supportive care to combination of gemcitabine and cisplatin chemotherapy for the advanced urothelial cancer]. [Article in Japanese]. Nihon rinsho. 2015; 73:609–613.
13. Larsen ME, Rowntree J, Young AM, Pearson S, Smith J, Gibson OJ, Weaver A, Tarassenko L. Chemotherapy side-effect management using mobile phones. Conf Proc IEEE Eng Med Biol Soc. 2008; 2008:5152–5155.
14. Saxena A. Cancer chemotherapy and its side effect management. Nurs J India. 2006; 97:109–10.
15. Chism DD, De Silva D, Whang YE. Mechanisms of acquired resistance to androgen receptor targeting drugs in castration-resistant prostate cancer. Expert Rev Anticancer Ther. 2014; 14:1369–78.
16. Chatterjee S, Damle SG, Sharma AK. Mechanisms of resistance against cancer therapeutic drugs. Curr Pharm Biotechnol. 2014; 15:1105–12.
17. Tamburrino A, Piro G, Carbone C, Tortora G, Melisi D. Mechanisms of resistance to chemotherapeutic and anti-angiogenic drugs as novel targets for pancreatic cancer therapy. Front Pharmacol. 2013; 4:56.
18. Deslouches B, Steckbeck JD, Craigo JK, Doi Y, Burns JL, Montelaro RC. Engineered cationic antimicrobial peptides to overcome multidrug resistance by ESKAPE pathogens. Antimicrob Agents Chemother. 2015; 59:1329–33.
19. Steckbeck JD, Deslouches B, Montelaro RC. Antimicrobial peptides: new drugs for bad bugs? Expert Opin Biol Ther. 2014; 14:11–14.
20. Deslouches B, Steckbeck JD, Craigo JK, Doi Y, Mietzner TA, Montelaro RC. Rational design of engineered cationic antimicrobial peptides consisting exclusively of arginine and tryptophan, and their activity against multidrug-resistant pathogens. Antimicrob Agents Chemother. 2013; 57:2511–21.
21. Dutta P, Das S. Mammalian Antimicrobial Peptides: Promising Therapeutic Targets Against Infection and Chronic Inflammation. Curr Top Med Chem. 2016; 16:99–129.
22. Hiemstra PS, Amatngalim GD, van der Does AM, Taube C. Antimicrobial Peptides and Innate Lung Defenses: Role in Infectious and Noninfectious Lung Diseases and Therapeutic Applications. Chest. 2016; 149:545–51.
23. Shagaghi N, Palombo EA, Clayton AH, Bhave M. Archetypal tryptophan-rich antimicrobial peptides: properties and applications. World J Microbiol Biotechnol. 2016; 32:31.
24. Schmitt P, Rosa RD, Destoumieux-Garzon D. An intimate link between antimicrobial peptide sequence diversity and binding to essential components of bacterial membranes. Biochim Biophys Acta. 2016; 1858:958–970.
25. Aguilera-Mendoza L, Marrero-Ponce Y, Tellez-Ibarra R, Llorente-Quesada MT, Salgado J, Barigye SJ, Liu J. Overlap and diversity in antimicrobial peptide databases: compiling a non-redundant set of sequences. Bioinformatics. 2015; 31:2553–59.
26. He X, Yang S, Wei L, Liu R, Lai R, Rong M. Antimicrobial peptide diversity in the skin of the torrent frog, Amolops jingdongensis. Amino Acids. 2013; 44:481–487.
27. Padhi A, Verghese B. Molecular diversity and evolution of myticin-C antimicrobial peptide variants in the Mediterranean mussel, Mytilus galloprovincialis. Peptides. 2008; 29:1094–101.
28. Dennison SR, Wallace J, Harris F, Phoenix DA. Amphiphilic alpha-helical antimicrobial peptides and their structure/function relationships. Protein Pept Lett. 2005; 12:31–39.
29. Bulet P, Hetru C, Dimarcq JL, Hoffmann D. Antimicrobial peptides in insects; structure and function. Dev Comp Immunol. 1999; 23:329–44.
30. Hwang PM, Vogel HJ. Structure-function relationships of antimicrobial peptides. Biochem Cell Biol. 1998; 76:235–246.
31. Sitaram N, Nagaraj R. Host-defense antimicrobial peptides: importance of structure for activity. Curr Pharm Des. 2002; 8:727–42.
32. Guzmán-Rodríguez JJ, Ochoa-Zarzosa A, Lopez-Gomez R, Lopez-Meza JE. Plant antimicrobial peptides as potential anticancer agents. Biomed Res Int. 2015; 2015:735087.
33. Farkas A, Maróti G, Kereszt A, Kondorosi É. Comparative Analysis of the Bacterial Membrane Disruption Effect of Two Natural Plant Antimicrobial Peptides. Front Microbiol. 2017; 8:51.
34. Gomarasca M, F C Martins T, Greune L, Hardwidge PR, Schmidt MA, Rüter C. Bacterial-derived cell-penetrating peptides deliver gentamicin to kill intracellular pathogens. Antimicrob Agents Chemother. 2017; 61:e02545–16.
35. Coelho ML, de Souza Duarte AF, do Carmo de Freire Bastos M. Bacterial Labionin-Containing Peptides and Sactibiotics: Unusual Types of Antimicrobial Peptides with Potential Use in Clinical Settings (A Review). Curr Top Med Chem. 2016; 17:1177–1198.
36. Jung HJ, Kim Y, Lee HB, Kwon HJ. Antiangiogenic activity of the lipophilic antimicrobial peptides from an endophytic bacterial strain isolated from red pepper leaf. Mol Cells. 2015; 38:273–78.
37. Gomes KM, Duarte RS, de Freire Bastos MD. Lantibiotics produced by Actinobacteria and their potential applications (a review). Microbiology. 2017; 163:109–21.
38. Barbosa AA, Mantovani HC, Jain S. Bacteriocins from lactic acid bacteria and their potential in the preservation of fruit products. Crit Rev Biotechnol. 2017; 3:1–13.
39. Ahmad V, Khan MS, Jamal QM, Alzohairy MA, Al Karaawi MA, Siddiqui MU. Antimicrobial potential of bacteriocins: in therapy, agriculture and food preservation. Int J Antimicrob Agents. 2017; 49:1–11.
40. Pirtskhalava M, Gabrielian A, Cruz P, Griggs HL, Squires RB, Hurt DE, Grigolava M, Chubinidze M, Gogoladze G, Vishnepolsky B, Alekseyev V, Rosenthal A, Tartakovsky M. DBAASP v.2: an enhanced database of structure and antimicrobial/cytotoxic activity of natural and synthetic peptides. Nucleic Acids Res. 2016; 44:6503.
41. Waghu FH, Barai RS, Gurung P, Idicula-Thomas S. CAMPR3: a database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res. 2016; 44:D1094–97.
42. Liu Y, Eichler J, Pischetsrieder M. Virtual screening of a milk peptide database for the identification of food-derived antimicrobial peptides. Mol Nutr Food Res. 2015; 59:2243–2254.
43. Di Luca M, Maccari G, Maisetta G, Batoni G. BaAMPs: the database of biofilm-active antimicrobial peptides. Biofouling. 2015; 31:193–99.
44. Niarchou A, Alexandridou A, Athanasiadis E, Spyrou G. C-PAmP: large scale analysis and database construction containing high scoring computationally predicted antimicrobial peptides for all the available plant species. PLoS One. 2013; 8:e79728.
45. Zhao X, Wu H, Lu H, Li G, Huang Q. LAMP: A Database Linking Antimicrobial Peptides. PLoS One. 2013; 8:e66557.
46. Piotto SP, Sessa L, Concilio S, Iannelli P. YADAMP: yet another database of antimicrobial peptides. Int J Antimicrob Agents. 2012; 39:346–51.
47. Li Y, Chen Z. RAPD: a database of recombinantly-produced antimicrobial peptides. FEMS Microbiol Lett. 2008; 289:126–29.
48. Fjell CD, Hancock RE, Cherkasov A. AMPer: a database and an automated discovery tool for antimicrobial peptides. Bioinformatics. 2007; 23:1148–55.
49. Hancock RE, Sahl HG. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. 2006; 24:1551–57.
50. Hilpert K, Elliott MR, Volkmer-Engert R, Henklein P, Donini O, Zhou Q, Winkler DF, Hancock RE. Sequence requirements and an optimization strategy for short antimicrobial peptides. Chem Biol. 2006; 13:1101–07.
51. Hancock RE, Brown KL, Mookherjee N. Host defence peptides from invertebrates—emerging antimicrobial strategies. Immunobiology. 2006; 211:315–22.
52. Brown KL, Hancock RE. Cationic host defense (antimicrobial) peptides. Curr Opin Immunol. 2006; 18:24–30.
53. Soblosky L, Ramamoorthy A, Chen Z. Membrane interaction of antimicrobial peptides using E. coli lipid extract as model bacterial cell membranes and SFG spectroscopy. Chem Phys Lipids. 2015; 187:20–33.
54. Kamimori H, Blazyk J, Aguilar MI. Lipid membrane-binding properties of tryptophan analogues of linear amphipathic beta-sheet cationic antimicrobial peptides using surface plasmon resonance. Biol Pharm Bull. 2005; 28:148–50.
55. Maget-Dana R. The monolayer technique: a potent tool for studying the interfacial properties of antimicrobial and membrane-lytic peptides and their interactions with lipid membranes. Biochim Biophys Acta. 1999; 1462:109–40.
56. Wang C, Chen T, Zhang N, Yang M, Li B, Lü X, Cao X, Ling C. Melittin, a major component of bee venom, sensitizes human hepatocellular carcinoma cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis by activating CaMKII-TAK1-JNK/p38 and inhibiting IkappaBalpha kinase-NFkappaB. J Biol Chem. 2009; 284:3804–13.
57. Mangoni ML, Papo N, Saugar JM, Barra D, Shai Y, Simmaco M, Rivas L. Effect of natural L- to D-amino acid conversion on the organization, membrane binding, and biological function of the antimicrobial peptides bombinins H. Biochemistry. 2006; 45:4266–76.
58. Papo N, Seger D, Makovitzki A, Kalchenko V, Eshhar Z, Degani H, Shai Y. Inhibition of tumor growth and elimination of multiple metastases in human prostate and breast xenografts by systemic inoculation of a host defense-like lytic peptide. Cancer Res. 2006; 66:5371–78.
59. Wakabayashi H, Matsumoto H, Hashimoto K, Teraguchi S, Takase M, Hayasawa H. N-Acylated and D enantiomer derivatives of a nonamer core peptide of lactoferricin B showing improved antimicrobial activity. Antimicrob Agents Chemother. 1999; 43:1267–69.
60. Shang D, Zhang Q, Dong W, Liang H, Bi X. The effects of LPS on the activity of Trp-containing antimicrobial peptides against Gram-negative bacteria and endotoxin neutralization. Acta Biomater. 2016; 33:153–65.
61. Nan YH, Bang JK, Jacob B, Park IS, Shin SY. Prokaryotic selectivity and LPS-neutralizing activity of short antimicrobial peptides designed from the human antimicrobial peptide LL-37. Peptides. 2012; 35:239–47.
62. Gustafsson A, Olin AI, Ljunggren L. LPS interactions with immobilized and soluble antimicrobial peptides. Scand J Clin Lab Invest. 2010; 70:194–200.
63. Giuliani A, Pirri G, Rinaldi AC. Antimicrobial peptides: the LPS connection. Methods Mol Biol. 2010; 618:137–154.
64. Malanovic N, Leber R, Schmuck M, Kriechbaum M, Cordfunke RA, Drijfhout JW, de Breij A, Nibbering PH, Kolb D, Lohner K. Phospholipid-driven differences determine the action of the synthetic antimicrobial peptide OP-145 on Gram-positive bacterial and mammalian membrane model systems. Biochim Biophys Acta. 2015; 1848:2437–2447.
65. Jasir A, Kasprzykowski F, Lindström V, Schalén C, Grubb A. New antimicrobial peptide active against Gram-positive pathogens. Indian J Med Res. 2004; 119:74–76.
66. Jasir A, Kasprzykowski F, Kasprzykowska R, Lindstrom V, Schalen C, Grubb A. New antimicrobial cystatin C-based peptide active against gram-positive bacterial pathogens, including methicillin-resistant Staphylococcus aureus and multiresistant coagulase-negative staphylococci. APMIS. 2003; 111:1004–1010.
67. Ruan Y, Shen T, Wang Y, Hou M, Li J, Sun T. Antimicrobial peptide LL-37 attenuates LTA induced inflammatory effect in macrophages. Int Immunopharmacol. 2013; 15:575–80.
68. Gustafsson A, Sigel S, Ljunggren L. The antimicrobial peptide LL37 and its truncated derivatives potentiates proinflammatory cytokine induction by lipoteichoic acid in whole blood. Scand J Clin Lab Invest. 2010; 70:512–518.
69. Ng TB, Cheung RC, Wong JH, Ye XJ. Antimicrobial activity of defensins and defensin-like peptides with special emphasis on those from fungi and invertebrate animals. Curr Protein Pept Sci. 2013; 14:515–531.
70. Crack LR, Jones L, Malavige GN, Patel V, Ogg GS. Human antimicrobial peptides LL-37 and human beta-defensin-2 reduce viral replication in keratinocytes infected with varicella zoster virus. Clin Exp Dermatol. 2012; 37:534–543.
71. Nava GM, Escorcia M, Castaneda MP. Molecular diversity of the antimicrobial domain of beta-defensin 3 and homologous peptides. Comp Funct Genomics. 2009:983636.
72. Nan YH, Bang JK, Shin SY. Design of novel indolicidin-derived antimicrobial peptides with enhanced cell specificity and potent anti-inflammatory activity. Peptides. 2009; 30:832–38.
73. Ladokhin AS, Selsted ME, White SH. CD spectra of indolicidin antimicrobial peptides suggest turns, not polyproline helix. Biochemistry. 1999; 38:12313–19.
74. Hollmann A, Martínez M, Noguera ME, Augusto MT, Disalvo A, Santos NC, Semorile L, Maffía PC. Role of amphipathicity and hydrophobicity in the balance between hemolysis and peptide-membrane interactions of three related antimicrobial peptides. Colloids Surf B Biointerfaces. 2016; 141:528–36.
75. Son M, Lee Y, Hwang H, Hyun S, Yu J. Disruption of interactions between hydrophobic residues on nonpolar faces is a key determinant in decreasing hemolysis and increasing antimicrobial activities of α-helical amphipathic peptides. ChemMedChem. 2013; 8:1638–42.
76. Utsugi T, Schroit AJ, Connor J, Bucana CD, Fidler IJ. Elevated expression of phosphatidylserine in the outer membrane leaflet of human tumor cells and recognition by activated human blood monocytes. Cancer Res. 1991; 51:3062–66.
77. Gerber DE, Hao G, Watkins L, Stafford JH, Anderson J, Holbein B, Oz OK, Mathews D, Thorpe PE, Hassan G, Kumar A, Brekken RA, Sun X. Tumor-specific targeting by Bavituximab, a phosphatidylserine-targeting monoclonal antibody with vascular targeting and immune modulating properties, in lung cancer xenografts. Am J Nucl Med Mol Imaging. 2015; 5:493–503.
78. Kenis H, Reutelingsperger C. Targeting phosphatidylserine in anti-cancer therapy. Curr Pharm Des. 2009; 15:2719–23.
79. Bevers EM, Williamson PL. Getting to the Outer Leaflet: Physiology of Phosphatidylserine Exposure at the Plasma Membrane. Physiol Rev. 2016; 96:605–45.
80. Connor J, Pak CC, Schroit AJ. Exposure of phosphatidylserine in the outer leaflet of human red blood cells. Relationship to cell density, cell age, and clearance by mononuclear cells. J Biol Chem. 1994; 269:2399–404.
81. Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 1994; 84:1415–20.
82. Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, Green DR. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995; 182:1545–1556.
83. Lee SH, Meng XW, Flatten KS, Loegering DA, Kaufmann SH. Phosphatidylserine exposure during apoptosis reflects bidirectional trafficking between plasma membrane and cytoplasm. Cell Death Differ. 2013; 20:64–76.
84. Cole CL, Rushton G, Jayson GC, Avizienyte E. Ovarian cancer cell heparan sulfate 6-O-sulfotransferases regulate an angiogenic program induced by heparin-binding epidermal growth factor (EGF)-like growth factor/EGF receptor signaling. J Biol Chem. 2014; 289:10488–10501.
85. Kontro H, Joenväärä S, Haglund C, Renkonen R. Comparison of sialylated N-glycopeptide levels in serum of pancreatic cancer patients, acute pancreatitis patients, and healthy controls. Proteomics. 2014; 14:1713–23.
86. Tian Y, Esteva FJ, Song J, Zhang H. Altered expression of sialylated glycoproteins in breast cancer using hydrazide chemistry and mass spectrometry. Mol Cell Proteomics. 2012; 11:M111 011403.
87. Lee CS, Taib NA, Ashrafzadeh A, Fadzli F, Harun F, Rahmat K, Hoong SM, Abdul-Rahman PS, Hashim OH. Unmasking Heavily O-Glycosylated Serum Proteins Using Perchloric Acid: Identification of Serum Proteoglycan 4 and Protease C1 Inhibitor as Molecular Indicators for Screening of Breast Cancer. PLoS One. 2016; 11:e0149551.
88. Mu AK, Lim BK, Hashim OH, Shuib AS. Identification of O-glycosylated proteins that are aberrantly excreted in the urine of patients with early stage ovarian cancer. Int J Mol Sci. 2013; 14:7923–7931.
89. Osinaga E. Expression of cancer-associated simple mucin-type O-glycosylated antigens in parasites. IUBMB Life. 2007; 59:269–73.
90. Yoon WH, Park HD, Lim K, Hwang BD. Effect of O-glycosylated mucin on invasion and metastasis of HM7 human colon cancer cells. Biochem Biophys Res Commun. 1996; 222:694–99.
91. Fadnes B, Rekdal O, Uhlin-Hansen L. The anticancer activity of lytic peptides is inhibited by heparan sulfate on the surface of the tumor cells. BMC Cancer. 2009; 9:183.
92. Sok M, Sentjurc M, Schara M, Stare J, Rott T. Cell membrane fluidity and prognosis of lung cancer. Ann Thorac Surg. 2002; 73:1567–1571.
93. Sok M, Sentjurc M, Schara M. Membrane fluidity characteristics of human lung cancer. Cancer Lett. 1999; 139:215–20.
94. Berger M, Motta C, Boiret N, Aublet-Cuvelier B, Bonhomme J, Travade P. Membrane fluidity and adherence to extracellular matrix components are related to blast cell count in acute myeloid leukemia. Leukem & lymphom. 1994; 15:297–302.
95. Li X, Shen B, Chen Q, Zhang X, Ye Y, Wang F, Zhang X. Antitumor effects of cecropin B-LHRH’ on drug-resistant ovarian and endometrial cancer cells. BMC Cancer. 2016; 16:251.
96. Suttmann H, Retz M, Paulsen F, Harder J, Zwergel U, Kamradt J, Wullich B, Unteregger G, Stockle M, Lehmann J. Antimicrobial peptides of the Cecropin-family show potent antitumor activity against bladder cancer cells. BMC Urol. 2008; 8:5.
97. Hui L, Leung K, Chen HM. The combined effects of antibacterial peptide cecropin A and anti-cancer agents on leukemia cells. Anticancer Res. 2002; 22:2811–16.
98. Liu Y, Chen L, Gong Z, Shen L, Kao C, Hock JM, Sun L, Li X. Lovastatin enhances adenovirus-mediated TRAIL induced apoptosis by depleting cholesterol of lipid rafts and affecting CAR and death receptor expression of prostate cancer cells. Oncotarget. 2015; 6:3055–70. doi: 10.18632/oncotarget.3073.
99. Zhuang L, Lin J, Lu ML, Solomon KR, Freeman MR. Cholesterol-rich lipid rafts mediate akt-regulated survival in prostate cancer cells. Cancer Res. 2002; 62:2227–31.
100. Mulder KC, Lima LA, Miranda VJ, Dias SC, Franco OL. Current scenario of peptide-based drugs: the key roles of cationic antitumor and antiviral peptides. Front Microbiol. 2013; 4:321.
101. Rothan HA, Mohamed Z, Suhaeb AM, Rahman NA, Yusof R. Antiviral cationic peptides as a strategy for innovation in global health therapeutics for dengue virus: high yield production of the biologically active recombinant plectasin peptide. Omics. 2013; 17:560–567.
102. Chan SC, Hui L, Chen HM. Enhancement of the cytolytic effect of anti-bacterial cecropin by the microvilli of cancer cells. Anticancer Res. 1998; 18:4467–74.
103. Kolata GB. Microvilli: a major difference between normal and cancer cells? Science. 1975; 188:819–20.
104. Lacerda AF, Vasconcelos EA, Pelegrini PB, Grossi de Sa MF. Antifungal defensins and their role in plant defense. Front Microbiol. 2014; 5:116.
105. Vriens K, Cammue BP, Thevissen K. Antifungal plant defensins: mechanisms of action and production. Molecules. 2014; 19:12280–303.
106. Lehrer RI, Barton A, Daher KA, Harwig SS, Ganz T, Selsted ME. Interaction of human defensins with Escherichia coli. Mechanism of bactericidal activity. J Clin Invest. 1989; 84:553–61.
107. Panyutich AV, Voitenok NN, Lehrer RI, Ganz T. An enzyme immunoassay for human defensins. J Immunol Methods. 1991; 141:149–55.
108. Venkataraman N, Cole AL, Ruchala P, Waring AJ, Lehrer RI, Stuchlik O, Pohl J, Cole AM. Reawakening retrocyclins: ancestral human defensins active against HIV-1. PLoS Biol. 2009; 7:e95.
109. Muñoz A, Chu M, Marris PI, Sagaram US, Kaur J, Shah DM, Read ND. Specific domains of plant defensins differentially disrupt colony initiation, cell fusion and calcium homeostasis in Neurospora crassa. Mol Microbiol. 2014; 92:1357–74.
110. Xu N, Wang YS, Pan WB, Xiao B, Wen YJ, Chen XC, Chen LJ, Deng HX, You J, Kan B, Fu AF, Li D, Zhao X, Wei YQ. Human alpha-defensin-1 inhibits growth of human lung adenocarcinoma xenograft in nude mice. Mol Cancer Ther. 2008; 7:1588–97.
111. Ren XY, Li WY, Xu M. [Expression of human beta-defensin-2 in laryngeal squamous cell carcinoma and its correlation with CD1a(+) dendritic cells infiltrating in tumor.]. [Article in Chinese]. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 2009; 44:1029–33.
112. Ganz T, Oren A, Lehrer RI. Defensins: microbicidal and cytotoxic peptides of mammalian host defense cells. Med Microbiol Immunol (Berl). 1992; 181:99–105.
113. Kagan BL, Ganz T, Lehrer RI. Defensins: a family of antimicrobial and cytotoxic peptides. Toxicology. 1994; 87:131–49.
114. Lehrer RI, Lichtenstein AK, Ganz T. Defensins: antimicrobial and cytotoxic peptides of mammalian cells. Annu Rev Immunol. 1993; 11:105–28.
115. Lichtenstein AK, Ganz T, Nguyen TM, Selsted ME, Lehrer RI. Mechanism of target cytolysis by peptide defensins. Target cell metabolic activities, possibly involving endocytosis, are crucial for expression of cytotoxicity. J Immunol. 1988; 140:2686–94.
116. Cruciani RA, Barker JL, Zasloff M, Chen HC, Colamonici O. Antibiotic magainins exert cytolytic activity against transformed cell lines through channel formation. Proc Natl Acad Sci USA. 1991; 88:3792–96.
117. Moore KS, Bevins CL, Brasseur MM, Tomassini N, Turner K, Eck H, Zasloff M. Antimicrobial peptides in the stomach of Xenopus laevis. J Biol Chem. 1991; 266:19851–57.
118. Soravia E, Martini G, Zasloff M. Antimicrobial properties of peptides from Xenopus granular gland secretions. FEBS Lett. 1988; 228:337–40.
119. Zasloff M. Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc Natl Acad Sci USA. 1987; 84:5449–53.
120. Westerhoff HV, Hendler RW, Zasloff M, Juretić D. Interactions between a new class of eukaryotic antimicrobial agents and isolated rat liver mitochondria. Biochim Biophys Acta. 1989; 975:361–69.
121. Merrifield RB, Vizioli LD, Boman HG. Synthesis of the antibacterial peptide cecropin A (1–33). Biochemistry. 1982; 21:5020–31.
122. Hultmark D, Engstrom A, Bennich H, Kapur R, Boman HG. Insect immunity: isolation and structure of cecropin D and four minor antibacterial components from Cecropia pupae. Eur J Biochem. 1982; 127:207–217.
123. Shin SY, Kang JH, Jang SY, Kim Y, Kim KL, Hahm KS. Effects of the hinge region of cecropin A(1–8)-magainin 2(1–12), a synthetic antimicrobial peptide, on liposomes, bacterial and tumor cells. Biochim Biophys Acta. 2000; 1463:209–18.
124. Hsu JC, Lin LC, Tzen JT, Chen JY. Characteristics of the antitumor activities in tumor cells and modulation of the inflammatory response in RAW264.7 cells of a novel antimicrobial peptide, chrysophsin-1, from the red sea bream (Chrysophrys major). Peptides. 2011; 32:900–10.
125. Cirioni O, Simonetti O, Pierpaoli E, Barucca A, Ghiselli R, Orlando F, Pelloni M, Minardi D, Trombettoni MM, Guerrieri M, Offidani A, Giacometti A, Provinciali M. Enhanced Efficacy of Combinations of Pexiganan with Colistin Versus Acinetobacter Baumannii in Experimental Sepsis. Shock. 2016; 46:219–25.
126. Zhang XL, Jiang AM, Ma ZY, Li XB, Xiong YY, Dou JF, Wang JF. The synthetic antimicrobial peptide pexiganan and its nanoparticles (PNPs) exhibit the anti-helicobacter pylori activity
127. Dipexium Pharmaceuticals I. Dipexium Pharmaceuticals Initiates Pivotal Phase 3 Clinical Trial Of Locilex® In Patients With Mild Infections Of Diabetic Foot Ulcers. http://contentequisolvenet/dipexiumpharmaceuticals/news/2014–07-17_Dipexium_Pharmaceuticals_Initiates_Pivotal_Phase_54pdf, PRNewswire. 2014
128. Liu S, Yang H, Wan L, Cai HW, Li SF, Li YP, Cheng JQ, Lu XF. Enhancement of cytotoxicity of antimicrobial peptide magainin II in tumor cells by bombesin-targeted delivery. Acta Pharmacol Sin. 2011; 32:79–88.
129. Domingues TM, Riske KA, Miranda A. Revealing the lytic mechanism of the antimicrobial peptide gomesin by observing giant unilamellar vesicles. Langmuir. 2010; 26:11077–11084.
130. Rodrigues EG, Dobroff AS, Cavarsan CF, Paschoalin T, Nimrichter L, Mortara RA, Santos EL, Fázio MA, Miranda A, Daffre S, Travassos LR. Effective topical treatment of subcutaneous murine B16F10-Nex2 melanoma by the antimicrobial peptide gomesin. Neoplasia. 2008; 10:61–68.
131. Tomasinsig L, Skerlavaj B, Papo N, Giabbai B, Shai Y, Zanetti M. Mechanistic and functional studies of the interaction of a proline-rich antimicrobial peptide with mammalian cells. J Biol Chem. 2006; 281:383–91.
132. Papo N, Braunstein A, Eshhar Z, Shai Y. Suppression of human prostate tumor growth in mice by a cytolytic D-, L-amino Acid Peptide: membrane lysis, increased necrosis, and inhibition of prostate-specific antigen secretion. Cancer Res. 2004; 64:5779–86.
133. Braunstein A, Papo N, Shai Y.
134. Cichoń T, Smolarczyk R, Matuszczak S, Barczyk M, Jarosz M, Szala S. D-K6L 9 peptide combination with IL-12 inhibits the recurrence of tumors in mice. Arch Immunol Ther Exp (Warsz). 2014; 62:341–51.
135. Jing W, Svendsen JS, Vogel HJ. Comparison of NMR structures and model-membrane interactions of 15-residue antimicrobial peptides derived from bovine lactoferricin. Biochem Cell Biol. 2006; 84:312–26.
136. Umeyama M, Kira A, Nishimura K, Naito A. Interactions of bovine lactoferricin with acidic phospholipid bilayers and its antimicrobial activity as studied by solid-state NMR. Biochim Biophys Acta. 2006; 1758:1523–1528.
137. Longhi C, Conte MP, Ranaldi S, Penta M, Valenti P, Tinari A, Superti F, Seganti L. Apoptotic death of Listeria monocytogenes-infected human macrophages induced by lactoferricin B, a bovine lactoferrin-derived peptide. Int J Immunopathol Pharmacol. 2005; 18:317–325.
138. Tomita M, Wakabayashi H, Yamauchi K, Teraguchi S, Hayasawa H. Bovine lactoferrin and lactoferricin derived from milk: production and applications. Biochem Cell Biol. 2002; 80:109–12.
139. Yoo YC, Watanabe S, Watanabe R, Hata K, Shimazaki K, Azuma I. Bovine lactoferrin and lactoferricin, a peptide derived from bovine lactoferrin, inhibit tumor metastasis in mice. Jpn J Cancer Res. 1997; 88:184–90.
140. Haug BE, Svendsen JS. The role of tryptophan in the antibacterial activity of a 15-residue bovine lactoferricin peptide. J Pept Sci. 2001; 7:190–196.
141. Tanaka T, Omata Y, Saito A, Shimazaki K, Yamauchi K, Takase M, Kawase K, Igarashi K, Suzuki N. Toxoplasma gondii: parasiticidal effects of bovine lactoferricin against parasites. Exp Parasitol. 1995; 81:614–617.
142. Bellamy W, Wakabayashi H, Takase M, Kawase K, Shimamura S, Tomita M. Killing of Candida albicans by lactoferricin B, a potent antimicrobial peptide derived from the N-terminal region of bovine lactoferrin. Med Microbiol Immunol (Berl). 1993; 182:97–105.
143. Bellamy W, Takase M, Wakabayashi H, Kawase K, Tomita M. Antibacterial spectrum of lactoferricin B, a potent bactericidal peptide derived from the N-terminal region of bovine lactoferrin. J Appl Bacteriol. 1992; 73:472–79.
144. Yoo YC, Watanabe S, Watanabe R, Hata K, Shimazaki K, Azuma I. Bovine lactoferrin and Lactoferricin inhibit tumor metastasis in mice. Adv Exp Med Biol. 1998; 443:285–91.
145. Yoo YC, Watanabe R, Koike Y, Mitobe M, Shimazaki K, Watanabe S, Azuma I. Apoptosis in human leukemic cells induced by lactoferricin, a bovine milk protein-derived peptide: involvement of reactive oxygen species. Biochem Biophys Res Commun. 1997; 237:624–28.
146. Eliassen LT, Berge G, Sveinbjørnsson B, Svendsen JS, Vorland LH, Rekdal Ø. Evidence for a direct antitumor mechanism of action of bovine lactoferricin. Anticancer Res. 2002; 22:2703–10.
147. Eliassen LT, Haug BE, Berge G, Rekdal O. Enhanced antitumour activity of 15-residue bovine lactoferricin derivatives containing bulky aromatic amino acids and lipophilic N-terminal modifications. J Pept Sci. 2003; 9:510–517.
148. Haug BE, Camilio KA, Eliassen LT, Stensen W, Svendsen JS, Berg K, Mortensen B, Serin G, Mirjolet JF, Bichat F, Rekdal Ø. Discovery of a 9-mer Cationic Peptide (LTX-315) as a Potential First in Class Oncolytic Peptide. J Med Chem. 2016; 59:2918–27.
149. Yamazaki T, Pitt JM, Vetizou M, Marabelle A, Flores C, Rekdal O, Kroemer G, Zitvogel L. The oncolytic peptide LTX-315 overcomes resistance of cancers to immunotherapy with CTLA4 checkpoint blockade. Cell Death Differ. 2016; 23:1004–1015.
150. Sistigu A, Manic G, Vitale I. LTX-315, CAPtivating immunity with necrosis. Cell Cycle. 2016; 15:1176–1177.
151. Zhou H, Forveille S, Sauvat A, Yamazaki T, Senovilla L, Ma Y, Liu P, Yang H, Bezu L, Müller K, Zitvogel L, Rekdal Ø, Kepp O, Kroemer G. The oncolytic peptide LTX-315 triggers immunogenic cell death. Cell Death Dis. 2016; 7:e2134.
152. Forveille S, Zhou H, Sauvat A, Bezu L, Muller K, Liu P, Zitvogel L, Pierron G, Rekdal O, Kepp O, Kroemer G. The oncolytic peptide LTX-315 triggers necrotic cell death. Cell Cycle. 2015; 14:3506–3512.
153. Eike LM, Yang N, Rekdal Ø, Sveinbjørnsson B. The oncolytic peptide LTX-315 induces cell death and DAMP release by mitochondria distortion in human melanoma cells. Oncotarget. 2015; 6:34910–23. doi: 10.18632/oncotarget.5308.
154. Zhou H, Forveille S, Sauvat A, Sica V, Izzo V, Durand S, Müller K, Liu P, Zitvogel L, Rekdal Ø, Kepp O, Kroemer G. The oncolytic peptide LTX-315 kills cancer cells through Bax/Bak-regulated mitochondrial membrane permeabilization. Oncotarget. 2015; 6:26599–614. doi: 10.18632/oncotarget.5613.
155. Camilio KA, Rekdal O, Sveinbjörnsson B. LTX-315 (Oncopore™): A short synthetic anticancer peptide and novel immunotherapeutic agent. OncoImmunology. 2014; 3:e29181.
156. Camilio KA, Berge G, Ravuri CS, Rekdal O, Sveinbjørnsson B. Complete regression and systemic protective immune responses obtained in B16 melanomas after treatment with LTX-315. Cancer Immunol Immunother. 2014; 63:601–13.
157. Frank MG, Weber MD, Watkins LR, Maier SF. Stress sounds the alarmin: The role of the danger-associated molecular pattern HMGB1 in stress-induced neuroinflammatory priming. Brain Behav Immun. 2015; 48:1–7.
158. Theansungnoen T, Maijaroen S, Jangpromma N, Yaraksa N, Daduang S, Temsiripong T, Daduang J, Klaynongsruang S. Cationic Antimicrobial Peptides Derived from Crocodylus siamensis Leukocyte Extract, Revealing Anticancer Activity and Apoptotic Induction on Human Cervical Cancer Cells. Protein J. 2016; 35:202–11.
159. Risso A, Braidot E, Sordano MC, Vianello A, Macrì F, Skerlavaj B, Zanetti M, Gennaro R, Bernardi P. BMAP-28, an antibiotic peptide of innate immunity, induces cell death through opening of the mitochondrial permeability transition pore. Mol Cell Biol. 2002; 22:1926–35.
160. Lee HS, Park CB, Kim JM, Jang SA, Park IY, Kim MS, Cho JH, Kim SC. Mechanism of anticancer activity of buforin IIb, a histone H2A-derived peptide. Cancer Lett. 2008; 271:47–55.
161. Mai JC, Mi Z, Kim SH, Ng B, Robbins PD. A proapoptotic peptide for the treatment of solid tumors. Cancer Res. 2001; 61:7709–7712.
162. Han Y, Cui Z, Li YH, Hsu WH, Lee BH.
163. Tian W, Li B, Zhang X, Dang W, Wang X, Tang H, Wang L, Cao H, Chen T. Suppression of tumor invasion and migration in breast cancer cells following delivery of siRNA against Stat3 with the antimicrobial peptide PR39. Oncol Rep. 2012; 28:1362–68.
164. Wu J, Parungo C, Wu G, Kang PM, Laham RJ, Sellke FW, Simons M, Li J. PR39 inhibits apoptosis in hypoxic endothelial cells: role of inhibitor apoptosis protein-2. Circulation. 2004; 109:1660–67.
165. Li J, Post M, Volk R, Gao Y, Li M, Metais C, Sato K, Tsai J, Aird W, Rosenberg RD, Hampton TG, Sellke F, Carmeliet P, Simons M. PR39, a peptide regulator of angiogenesis. Nat Med. 2000; 6:49–55.
166. Ghavami S, Asoodeh A, Klonisch T, Halayko AJ, Kadkhoda K, Kroczak TJ, Gibson SB, Booy EP, Naderi-Manesh H, Los M. Brevinin-2R(1) semi-selectively kills cancer cells by a distinct mechanism, which involves the lysosomal-mitochondrial death pathway. J Cell Mol Med. 2008; 12:1005–22.
167. Deslouches B, Gonzalez IA, DeAlmeida D, Islam K, Steele C, Montelaro RC, Mietzner TA. De novo-derived cationic antimicrobial peptide activity in a murine model of Pseudomonas aeruginosa bacteraemia. J Antimicrob Chemother. 2007; 60:669–72.
168. Deslouches B, Islam K, Craigo JK, Paranjape SM, Montelaro RC, Mietzner TA. Activity of the de novo engineered antimicrobial peptide WLBU2 against Pseudomonas aeruginosa in human serum and whole blood: implications for systemic applications. Antimicrob Agents Chemother. 2005; 49:3208–16.
169. Makobongo MO, Gancz H, Carpenter BM, McDaniel DP, Merrell DS. The oligo-acyl lysyl antimicrobial peptide C12K-2β12 exhibits a dual mechanism of action and demonstrates strong in vivo efficacy against Helicobacter pylori. Antimicrob Agents Chemother. 2012; 56:378–90.
170. Aranha C, Gupta S, Reddy KV. Contraceptive efficacy of antimicrobial peptide Nisin:
171. Desbois AP, Gemmell CG, Coote PJ.
172. Narayana JL, Huang HN, Wu CJ, Chen JY. Efficacy of the antimicrobial peptide TP4 against Helicobacter pylori infection:
173. Chan LY, Zhang VM, Huang YH, Waters NC, Bansal PS, Craik DJ, Daly NL. Cyclization of the antimicrobial peptide gomesin with native chemical ligation: influences on stability and bioactivity. ChemBioChem. 2013; 14:617–24.
174. Dennison SR, Morton LH, Phoenix DA. Effect of amidation on the antimicrobial peptide aurein 2.5 from Australian southern bell frogs. Protein Pept Lett. 2012; 19:586–591.
175. Deslouches B, Hasek ML, Craigo JK, Steckbeck JD, Montelaro RC. Comparative functional properties of engineered cationic antimicrobial peptides consisting exclusively of tryptophan and either lysine or arginine. J Med Microbiol. 2016; 65:554–65.
176. Deslouches B, Phadke SM, Lazarevic V, Cascio M, Islam K, Montelaro RC, Mietzner TA. De novo generation of cationic antimicrobial peptides: influence of length and tryptophan substitution on antimicrobial activity. Antimicrob Agents Chemother. 2005; 49:316–22.
177. Morris CJ, Beck K, Fox MA, Ulaeto D, Clark GC, Gumbleton M. Pegylation of antimicrobial peptides maintains the active peptide conformation, model membrane interactions, and antimicrobial activity while improving lung tissue biocompatibility following airway delivery. Antimicrob Agents Chemother. 2012; 56:3298–308.
178. André M, Besse S, Chezal JM, Mounetou E. PEGylation enhances the tumor selectivity of melanoma-targeted conjugates. Org Biomol Chem. 2015; 13:388–97.
179. Da Pieve C, Blackshaw E, Missailidis S, Perkins AC. PEGylation and biodistribution of an anti-MUC1 aptamer in MCF-7 tumor-bearing mice. Bioconjug Chem. 2012; 23:1377–81.
180. Vine KL, Lobov S, Indira Chandran V, Harris NL, Ranson M. Improved pharmacokinetic and biodistribution properties of the selective urokinase inhibitor PAI-2 (SerpinB2) by site-specific PEGylation: implications for drug delivery. Pharm Res. 2015; 32:1045–54.
181. Mattheolabakis G, Wong CC, Sun Y, Amella CA, Richards R, Constantinides PP, Rigas B. Pegylation improves the pharmacokinetics and bioavailability of small-molecule drugs hydrolyzable by esterases: a study of phospho-Ibuprofen. J Pharmacol Exp Ther. 2014; 351:61–66.
182. Yang Z, Wang J, Lu Q, Xu J, Kobayashi Y, Takakura T, Takimoto A, Yoshioka T, Lian C, Chen C, Zhang D, Zhang Y, Li S, et al. PEGylation confers greatly extended half-life and attenuated immunogenicity to recombinant methioninase in primates. Cancer Res. 2004; 64:6673–6678.
183. Kelly GJ, Kia AF, Hassan F, O’Grady S, Morgan MP, Creaven BS, McClean S, Harmey JH, Devocelle M. Polymeric prodrug combination to exploit the therapeutic potential of antimicrobial peptides against cancer cells. Org Biomol Chem. 2016; 14:9278–86.
184. Spinks CB, Zidan AS, Khan MA, Habib MJ, Faustino PJ. Pharmaceutical characterization of novel tenofovir liposomal formulations for enhanced oral drug delivery:
185. Dawidczyk CM, Russell LM, Hultz M, Searson PC. Tumor accumulation of liposomal doxorubicin in three murine models: Optimizing delivery efficiency. Nanomedicine. 2017 Feb 28. [Epub ahead of print].
186. Cern A, Marcus D, Tropsha A, Barenholz Y, Goldblum A. New drug candidates for liposomal delivery identified by computer modeling of liposomes’ remote loading and leakage. J Control Release. 2017; 252:18–27.
187. Abu Lila AS, Ishida T. Liposomal Delivery Systems: Design Optimization and Current Applications. Biol Pharm Bull. 2017; 40:1–10.
188. Li H, Yuan D, Sun M, Ping Q. Effect of ligand density and PEG modification on octreotide-targeted liposome via somatostatin receptor
189. Pazos E, Sleep E, Rubert Pérez CM, Lee SS, Tantakitti F, Stupp SI. Nucleation and Growth of Ordered Arrays of Silver Nanoparticles on Peptide Nanofibers: Hybrid Nanostructures with Antimicrobial Properties. J Am Chem Soc. 2016; 138:5507–10.
190. Sandreschi S, Piras AM, Batoni G, Chiellini F. Perspectives on polymeric nanostructures for the therapeutic application of antimicrobial peptides. Nanomedicine (Lond). 2016; 11:1729–44.
191. Brandelli A. Nanostructures as promising tools for delivery of antimicrobial peptides. Mini Rev Med Chem. 2012; 12:731–41.
192. Coca A, Sobrino J, Módol J, Soler J, Mínguez A, Plana J, De la Sierra A. A multicenter, parallel comparative study of the antihypertensive efficacy of once-daily lisinopril vs enalapril with 24-h ambulatory blood pressure monitoring in essential hypertension. J Hum Hypertens. 1996; 10:837–41.
193. Rengo F, Canonico V. [Quinapril versus lisinopril in the treatment of essential hypertension in elderly patients with low blood renin]. [Article in Italian]. Minerva Cardioangiol. 1996; 44:631–35.
194. Bellissant E, Nguyen PC, Giudicelli JF. Pharmacokinetic-pharmacodynamic model relating lisinopril plasma concentrations to regional hemodynamic effects in healthy volunteers. J Cardiovasc Pharmacol. 1996; 28:470–478.
195. Zornoff LA, Matsubara BB, Matsubara LS, Paiva SA, Tornero MT, Spadaro J. Effects of lisinopril on experimental ischemia in rats. Influence of infarct size. Arq Bras Cardiol. 1999; 73:359–372.
196. Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov Today. 2015; 20:122–28.
197. Velemínský J. Plasma insulin response to glucose infusion in obese non-diabetic and diabetic subjects. Horm Metab Res. 1980; 12:651–55.
198. Farrar D, Tuffnell DJ, West J, West HM. Continuous subcutaneous insulin infusion versus multiple daily injections of insulin for pregnant women with diabetes. Cochrane Database Syst Rev. 2016; 6:CD005542.
199. Ashkenazi A, Wexler-Cohen Y, Shai Y. Multifaceted action of Fuzeon as virus-cell membrane fusion inhibitor. Biochim Biophys Acta. 2011; 1808:2352–2358.
200. Formal approval for Fuzeon. AIDS Patient Care STDS. 2004; 18:736.
201. Hancock RE, Chapple DS. Peptide antibiotics. Antimicrob Agents Chemother. 1999; 43:1317–23.
202. Khmel IA, Metlitskaia AZ, Fomenko DE, Katrukha GS, Basiuk EI, Kurepina NE, Lipasova VA, Bezrukov VM. [Microcins--new peptide antibiotics from enterobacteria and genetic control of their synthesis]. [Article in Russian]. 1999; 33:113–19.
203. Shi WL, Chen XL, Wang LX, Gong ZT, Li S, Li CL, Xie BB, Zhang W, Shi M, Li C, Zhang YZ, Song XY. Cellular and molecular insight into the inhibition of primary root growth of Arabidopsis induced by peptaibols, a class of linear peptide antibiotics mainly produced by Trichoderma spp. J Exp Bot. 2016; 67:2191–205.
204. Lee TH, Hall KN, Aguilar MI. Antimicrobial Peptide Structure and Mechanism of Action: A Focus on the Role of Membrane Structure. Curr Top Med Chem. 2016; 16:25–39.
205. Center MD. Induction of Antitumor Response in Melanoma Patients Using the Antimicrobial Peptide LL37. http://appswhoint/trialsearch/Trial2aspx?TrialID=NCT02225366. July 2015, updated on January 9, 2017; ClinicalTrials.gov.
206. Kościuczuk EM, Lisowski P, Jarczak J, Strzałkowska N, Jóźwik A, Horbańczuk J, Krzyżewski J, Zwierzchowski L, Bagnicka E. Cathelicidins: family of antimicrobial peptides. A review. Mol Biol Rep. 2012; 39:10957–70.
207. Tomasinsig L, Zanetti M. The cathelicidins—structure, function and evolution. Curr Protein Pept Sci. 2005; 6:23–34.
208. Zanetti M. Cathelicidins, multifunctional peptides of the innate immunity. J Leukoc Biol. 2004; 75:39–48.
209. Ordonez SR, Amarullah IH, Wubbolts RW, Veldhuizen EJ, Haagsman HP. Fungicidal mechanisms of cathelicidins LL-37 and CATH-2 revealed by live-cell imaging. Antimicrob Agents Chemother. 2014; 58:2240–48.
210. Tomasinsig L, De Conti G, Skerlavaj B, Piccinini R, Mazzilli M, D’Este F, Tossi A, Zanetti M. Broad-spectrum activity against bacterial mastitis pathogens and activation of mammary epithelial cells support a protective role of neutrophil cathelicidins in bovine mastitis. Infect Immun. 2010; 78:1781–88.
211. Chuang CM, Monie A, Wu A, Mao CP, Hung CF. Treatment with LL-37 peptide enhances antitumor effects induced by CpG oligodeoxynucleotides against ovarian cancer. Hum Gene Ther. 2009; 20:303–13.
212. plc LBAaTI. A Phase I, Open-label, Multi-arm, Multi-centre, Multi-dose, Dose Escalation Study of LTX-315 as Monotherapy or in Combination With Either Ipilimumab or Pembrolizumab in Patients With Transdermally Accessible Tumours. https://clinicaltrials.gov/ct2/show/NCT01986426?term=anticancerpeptide&rank=10. 2016.
213. Melvin JA, Lashua LP, Kiedrowski MR, Yang G, Deslouches B, Montelaro RC, Bomberger JM. Simultaneous Antibiofilm and Antiviral Activities of an Engineered Antimicrobial Peptide during Virus-Bacterium Coinfection. mSph. 2016; 1.