
INTRODUCTION
Cancer remains a leading cause of death worldwide. The complexity of tumor and the acquired or inherent resistance to treatments [1] contribute to less efficiency of traditional treatment of cancer, including surgery, chemotherapy and radiotherapy, which are not feasible for patients with advanced stage [2]. As a result, new treatments based on their unique mechanism of action are urgently needed. Cancer virotherapy, which employs a conditionally replicative virus, has attracted considerable attention, because it can kill cancer cells, but not normal cells, by selectively replicating in and transmitting among cancer cells [3]. Among oncolytic viruses currently explored to treat cancer, adenoviruses are the most widely used.
Oncolytic adenoviruses are engineered to selectively replicate in and kill tumor cells using different strategies. By virtue of its deleted E1B 55KDa gene, Onxy-015 was the first reported oncolytic adenovirus designed to replicate exclusively in p53-deleted or mutated cells [4]. The binding of EA1 to pRb protein through the CR2 region of E1A gene was required for adenovirus replication in normal cells rather than in tumor cells, and thus Ad5-Δ24 selectively replicate in tumor cells by deleting the CR2 region [5]. Oncolytic adenoviruses were also constructed by replacing viral promoters with tumor-specific promoters such as survivin promoter [6], PSA promoter [7], or telomerase promoter [8].
The most frequently used oncolytic adenoviruses are based on human adenovirus serotype 5, for its biology is well elucidated. However, preclinical and clinical trials have revealed several drawbacks of this serotype, one of which is widespread preexisting anti-AdHu5 immunity in humans due to prior infection in youth [9]. In addition, studies indicated that the hexon of AdHu5 was associated with blood coagulation factor X (FX), resulting in the direction of adenoviruses to FX receptors abundantly expressed in liver cells, which caused massive transduction to the liver [10, 11]. Recently, some low-seroprevalence human adenoviruses or adenoviruses originated from other species such as chimpanzee were modified into viral vectors, to circumvent preexisting anti-Adhu5 immunity [12, 13]. Importantly, some of these adenoviruses, such as AdHu26 and AdHu48, did not bind to FX [14], leading to diminished transduction to the liver. AdHu5 and chimpanzee adenoviruses are antigenically distinct; AdHu5-neutralizing antibodies could hardly influence the chimpanzee adenoviruses-derived vector, which enabled simian-derived adenoviral vectors to become promising vector prototypes [15].
AdC7, originated from chimpanzee, circulates rarely in human populations, and it is not subject to neutralization by preexisting anti-AdHu5 antibodies, as confirmed by previous studies [15, 16]. Compared to AdHu5-derived oncolytic adenoviruses, AdC7-originated oncolytic adenoviruses have a great potential to diminish liver transduction, because the FX-AdC7 complexes are unstable [17]. An oncolytic AdC7 platform therefore has some advantages over an AdHu5 platform. AdC7 was initially constructed into a replication-deficient vector, which was used to develop a great variety of vaccines including (but not limited to) HIV vaccines [18], Ebola vaccines [19] and influenza vaccines [16, 20]. To our knowledge, that chimpanzee adenoviruses are engineered into oncolytic viruses has not been previously reported. In this study, we constructed the oncolytic chimpanzee adenovirus AdC7-SP/E1A-ΔE3, in which the E1 promoter was replaced by the tumor-specific survivin promoter and the E3 region was deleted. These recombinant adenoviruses could replicate in many types of cancer cell lines, including NCI-H508, Huh7, A549, and SiHa. Moreover, AdC7-SP/E1A-ΔE3 could efficaciously kill NCI-H508 and Huh7 in vitro and in vivo, indicating that AdC7-SP/E1A-ΔE3 has the potential to treat such cancers.
RESULTS
E1A expression driven by survivin promoter in tumor cell lines
Survivin, a small member of the inhibitor of apoptosis family, is expressed in tumor cells and fetal tissues but not in terminally differentiated cells, and thus is considered a potential target in tumor treatment [21]. Previous studies indicated that survivin promoter is a tumor-specific promoter, and that a wide range of tumor cells express the survivin protein [6]. The present study used western blotting to assay the expression of survivin in four tumor cell lines and a normal cell line. As shown in Figure 1A, all four tumor cell lines (~1 × 106 cells) expressed the survivin protein, indicating that they each have survivin promoter activity. In addition, normal MRC-5 cells expressed the survivin protein.
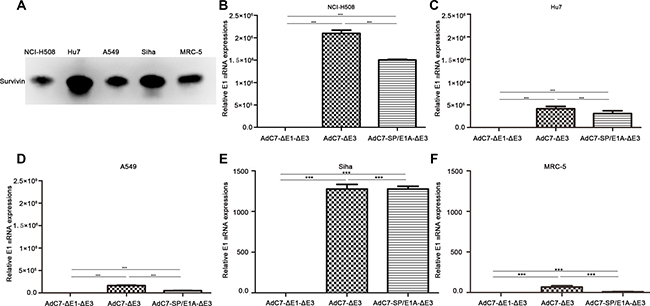
Figure 1: Expression of survivin and E1A genes in different cell types. (A) Western blotting was performed to detect survivin proteins in 1 × 106 cells. (B–F) A panel of NCI-H508 (B), Huh7 (C), A549 (D), SiHa (E), and MRC-5 (F) cells was infected with AdC7-ΔE1-ΔE3, AdC7-ΔE3, and AdC7-SP/E1A-ΔE3 at 10 MOI. E1A mRNA was quantitated by real-time PCR at 24 h post infection. Relative levels of E1A mRNA were shown in reference to GAPDH expression; relative E1 mRNAs in cells infected with AdC7-ΔE1-ΔE3 were defined as 1. Data shown are means ± standard errors of the means (SEM); statistical significance was determined by one-way ANOVA: ***P < 0.0001. Each experiment was performed three times.
AdC7-SP/E1A-ΔE3 is an oncolytic adenovirus in which E1A expression is driven by a survivin promoter and the E3 region is deleted; AdC7-ΔE3 is a modified adenovirus without the E3 region; and AdC7-ΔE1-ΔE3 is a non-replicating adenovirus lacking the E1 and E3 regions. Strong activity of survivin promoter in various types of tumor cell has been confirmed by previous studies [12, 22]. To explore the expression of E1A by AdC7-SP/E1A-ΔE3, we infected a panel of tumor cell lines with AdC7-SP/E1A-ΔE3. Figure 1B–1E show that tumor cell lines (NCI-H508, Huh7, A549, and SiHa) infected with AdC7-SP/E1A-ΔE3 exhibited significant increases in E1A expression relative to counterparts infected with AdC7-ΔE1-ΔE3 (p < 0.0001). As shown in Figure 1F, MRC-5 cells expressed considerably less E1A mRNA when infected with AdC7-SP/E1A-ΔE3 than when infected with AdC7-ΔE3 (p < 0.0001).
Efficient replication of AdC7-SP/E1A-ΔE3 in tumor cells
Having found that E1A was expressed in tumor cells infected with AdC7-SP/E1A-ΔE3, we speculated that AdC7-SP/E1A-ΔE3 was efficiently replicated in infected tumor cells, because E1A is required for adenovirus replication. To test this hypothesis, a panel of tumor cells was infected, at 10 MOI, with AdC7-ΔE1A-ΔE3, AdC7-ΔE3, or AdC7-SP/E1A-ΔE3, and relative viral genome copy numbers, which served as the readout for viral replication, were detected by RT-PCR. As shown in Figure 2A–2D, we confirmed that AdC7-SP/E1A-ΔE3 could replicate in a panel of tumor cell lines (NCI-H508, Huh7, A549, or SiHa); relative viral genome copy numbers were significantly higher in cells infected with AdC7-SP/E1A-ΔE3 than in cells infected with AdC7-ΔE1A-ΔE3 (p < 0.0001). Due to the deletion of the E1 region, AdC7-ΔE1A-ΔE3 is a non-replicating adenovirus. When infected with AdC7-SP/E1A-ΔE3, MCR-5 cells had dramatically lower viral copy numbers than when infected with AdC7-ΔE3, as shown in Figure 2E. To assay the replication competence of AdC7-SP/E1A-ΔE3 more accurately in tumor cells, progeny viruses produced in tumor cells were quantitated by TCID50 assay, after the infection of cells with adenoviruses at 10 MOI. As shown in Figure 2F, at 24 h after infection with AdC7-ΔE3 or AdC7-SP/E1A-ΔE3, adenoviruses were detectable in tumor cells but not in MRC-5 cells. In the NCI-H508 tumor cells, infected with AdC7-SP/E1A-ΔE3, 10 cells could yield 5 TCID50 of adenoviruses; the NCI-H508 cells produced 10-700 fold more progeny viruses than all other tumor cells. Figure 2G suggests that at 48 h post infection, tumor cell lines A549 and SiHa could produce more noticeable numbers of the progeny virus, and Huh7 cells produced the least virus among all tested tumor cell lines. While adenoviruses were detectable in MRC-5 cells infected with AdC7-SP/ E1A-ΔE3, only 0.006 TCID50 of progeny viruses were produced in ten of these infected MRC-5 cells. The dose of progeny viruses in MCR-5 was decreased three-fold when infected with AdC7-SP/ E1A-ΔE3, compared to when infected with AdC7-ΔE3.
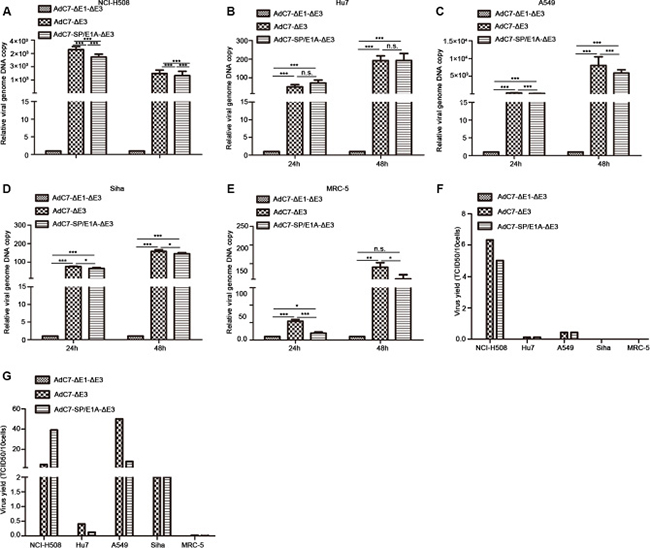
Figure 2: Replication of AdC7-ΔE1-ΔE3 in a panel of cells. (A–E) A panel of NCI-H508 (A), Huh7 (B), A549 (C), SiHa (D), and MRC-5 (E) cells was infected with AdC7-ΔE1-ΔE3, AdC7-ΔE3, and AdC7-SP/E1A-ΔE3 at 10 MOI, and sampled at different times post-infection. Relative viral genome copies were determined through real-time PCR. GAPDH was used as the reference and viral copies was defined as 1 in cells infected with AdC7-ΔE1-ΔE3. Data shown are means ± standard errors of the means (SEM); statistical significance was determined by one way ANOVA: ***P < 0.0001. Each experiment was performed three times. (F, G) A panel of NCI-H508, Huh7, A549, SiHa, and MRC-5 cells was infected with AdC7-ΔE1-ΔE3, AdC7-ΔE3, and AdC7-SP/E1A-ΔE3 at 10 MOI, and was twice collected and washed with PBS, at 24 h (F) and 48 h (G) post-infection. After three freeze-thaw cycles, progeny virus was quantitated with the TCID50 assay. Data shown are mean of two independent experiments.
Tumor cytotoxicity of AdC7-SP/E1A-ΔE3 in vitro
To assay the cytopathic effects (CPE) of AdC7-SP/E1A-ΔE3 on tumor cells, crystal violet cell staining assay was performed. A panel of tumor cells was stained with crystal violet 5 d after infection with AdC7-ΔE1A-ΔE3, AdC7-ΔE3, or AdC7-SP/E1A-ΔE3, at MOIs from 0.1–100. As shown in Figure 3A–3D, AdC7-SP/E1A-ΔE3 induced a prominent cytotoxic effect on NCI-H508 cells even when infection occurred at an MOI of 0.1; moreover, this virus decisively killed Huh7 cells at an MOI of 1. Although AdC7-SP/E1A-ΔE3 could replicate in A549 cells and SiHa cells (Figure 2), its cytotoxic effects on them were modest even at an MOI of 100. Figure 3E shows that AdC7-SP/E1A-ΔE3 had no cytotoxic effects on MRC-5 cells at 5 d post-infection. To further quantify the cytotoxic effects induced by AdC7-SP/E1A-ΔE3, cell viability was examined through MTT assay, which was carried out 3 d post-infection with three different adenoviruses at various MOIs. As shown in Figure 3F–3K, NCI-H508 cells and Huh7 cells were sensitive to adenoviral cytotoxicity at 10 MOI, while A549 and SiHa cell lines did not exhibit obvious cytotoxicity even at extremely high MOI (1000). Among all tested tumor cells, NCI-H508 cells were the most sensitive to AdC7-SP/E1A-ΔE3; AdC7-SP/E1A-ΔE3 killed these cells even at an MOI of 1. In addition, Figure 3K shows that AdC7-SP/E1A-ΔE3 was less cytotoxic than AdC7-ΔE3 in MRC-5 cell lines.
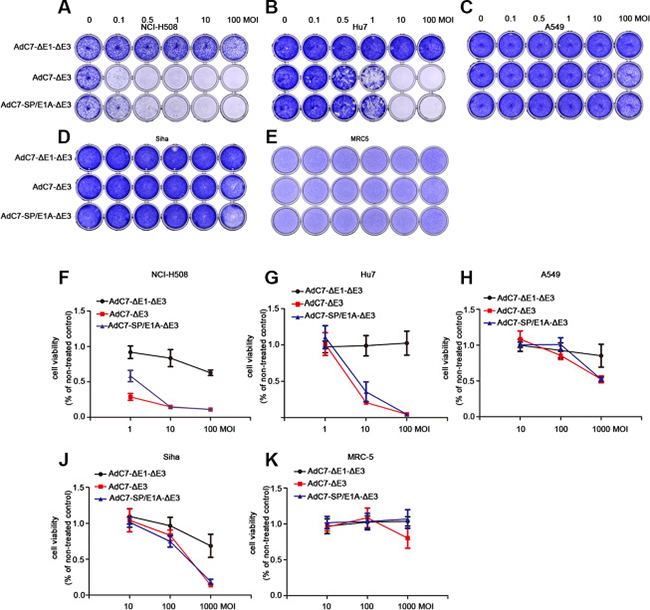
Figure 3: AdC7-ΔE1-ΔE3 induced cytotoxicity of tumor cells. A panel of NCI-H508, Huh7, A549, SiHa, and MRC-5 cells was infected with AdC7-ΔE1-ΔE3, AdC7-ΔE3, and AdC7-SP/E1A-ΔE3 at various MOIs. At 5 d post infection (A–E), cells were stained with crystal violet. Graph represents one of three different experiments. (F–K) Infected cells were subject to MTT assay kat 5 d post infection. Data are presented as means ± SEM of triplicate samples, and are representative of three independent experiments.
AdC7-SP/E1A-ΔE3 kills tumor cells through p53-independent mitochondrial apoptosis pathway
To explore the mechanism by which AdC7-SP/E1A-ΔE3 killed NCI-H508 and Huh7 cells, we assayed apoptosis of NCI-H508 and Huh7 through flow cytometry after AdC7-SP/E1A-ΔE3 infection. As shown in Figure 4A, AdC7-SP/E1A-ΔE3 could trigger NCI-H508 and Huh7 apoptosis because they were labeled by annexin V alone (early apoptosis), or both annexin V and PI (late apoptosis). To confirm the apoptosis that occurred in NCI-H508 and Huh7 cells, we used western blotting to examine the apoptosis inducers, cleaved caspase 3 and PARP. The data shown in Figure 4B indicated that cleaved caspase 3 and PARP were augmented in NCI-H508 and Huh7 cells infected with AdC7-SP/E1A-ΔE3. Since Adhu5 killed cells via autophagic cell death [23], AdC7-SP/E1A-ΔE3 is also likely to have induced autophagic cell death in NCI-H508 and Huh7 cells. Contrary to this hypothesis, autophagic cell death was not detectable in NCI-H508 and Huh7 cells infected with AdC7-SP/E1A-ΔE3; levels of p62 did not decrease, and levels of LC3 II did not increase (Figure 4B).
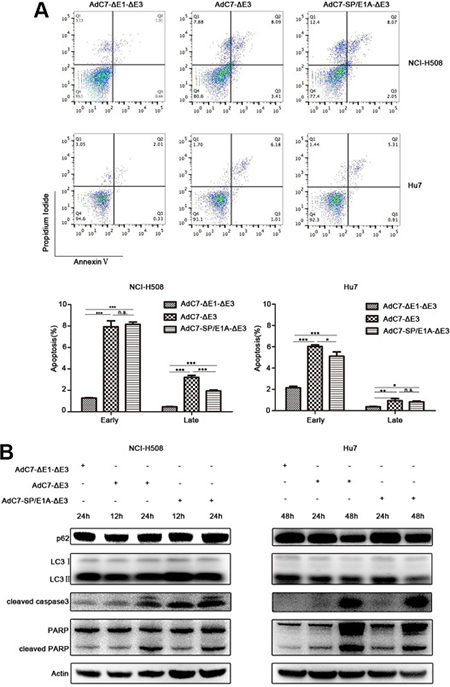
Figure 4: AdC7-ΔE1-ΔE3 trigger tumor cell apoptosis. (A) NCI-H508 and Huh7 cells were infected at 100 MOI with AdC7-ΔE1-ΔE3, AdC7-ΔE3, and AdC7-SP/E1A-ΔE3. Two cells were collected 24 h and 48 h post infection respectively. Flow cytometry was performed to evaluate tumor cell apoptosis after staining NCI-H508 and Huh7 with propidium iodide (PI)/annexin V. Early apoptosis: AV+/PI- (%); later stage apoptosis: AV+/PI+ (%). Data are presented as means ± SEM of triplicate samples, and are representative of three independent experiments. Statistical significance was determined by one way ANOVA: ***P < 0.0001, **< 0.001, *< 0.05. (B) NCI-H508 and Huh7 cells were infected at 100 MOI with AdC7-ΔE1-ΔE3, AdC7-ΔE3, and AdC7-SP/E1A-ΔE3. NCI-H508 cells were collected at 12 h and 24 h post infection, and Huh7 cells were collected 24 h and 48 h post infection. Western blotting was carried out to detect levels of p62, LC3, cleaved caspase3, and cleaved PARP in NCI-H508 and Huh7 cells. β-actin was used as a loading control.
Previous studies [24], in which the E1B-55K and E4-25K proteins of AdHu5 were associated with other cellular proteins to form a ubiquitin ligase complex that polyubiquitinated and degraded p53, implied that AdC7-SP/E1A-ΔE3 also degraded p53. This hypothesis was validated by the data in Figure 5, where p53 levels were markedly lowered in cells infected with AdC7-SP/E1A-ΔE3. These diminished p53 levels indicate that AdC7-SP/E1A-ΔE3 induced apoptosis in a p53-independent manner. It was further validated by the fact that the phosphorylation of p53 at Ser46, which induces the expression of relevant apoptosis genes, was significantly decreased in cells infected with AdC7-SP/E1A-ΔE3 (Figure 5). NCI-H508 infected with AdC7-SP/E1A-ΔE3 exhibited marked drops in levels of pro-survival proteins MCL-1and Bcl-2, while Huh7 exhibited marked drops in levels of pro-survival proteins MCL-1and Bcl-xl (Figure 5). We also note that both NCI-H508 and Huh7 cells have noticeably diminished levels of Bad phosphorylation at serine 112 and have increased expression of Bik (Figure 5).
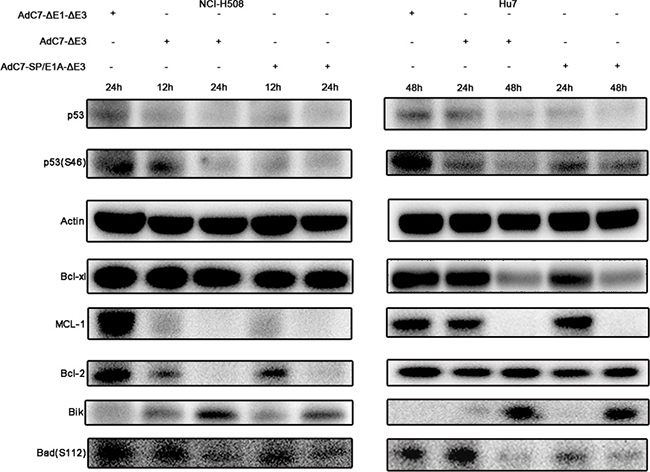
Figure 5: Tumor cell apoptosis is induced by AdC7-SP/E1A-ΔE3 via a p53-independent mitochondrial pathway. NCI-H508 and Huh7 cells were infected at 100 MOI with AdC7-ΔE1-ΔE3, AdC7-ΔE3, and AdC7-SP/E1A-ΔE3. NCI-H508 cells were collected at 12 h and 24 h post infection, and Huh7 cells were collected 24 h and 48 h post-infection. Western blotting was carried out to detect levels of p53, phosphorylation of p53, MCL-1, Bcl-2, Bcl-xl, Bik and phosphorylation of Bad in NCI-H508 and Huh7 cells. β-actin was used as a loading control.
AdC7-SP/E1A-ΔE3 inhibits tumor growth in xenograft tumor models
To examine the antitumor activity of AdC7-SP/E1A-ΔE3 in vivo, xenograft mouse tumor models were established. We inoculated matrigel mixtures of NCI-H508 or Huh7 tumor cells under the skin; when the tumor volume reached 100-150 mm3, we treated them with PBS, AdC7-ΔE1A-ΔE3, AdC7-ΔE3, or AdC7-SP/E1A-ΔE3 through intratumoral injection. As shown in Figure 6A, in NCI-H508 xenograft models, AdC7-ΔE3 and AdC7-SP/E1A-ΔE3 groups induced dramatic declines in tumor volume relative to control groups (PBS group, AdC7-ΔE1A-ΔE3), while AdC7-ΔE3 and AdC7-SP/E1A-ΔE3 groups had no significant difference in tumor volume. In Figure 6B, the number of TUNEL-positive cells was considerably more in AdC7-ΔE3 and AdC7-SP/E1A-ΔE3 groups than in the control groups (p < 0.0001). Similarly, the Huh7 xenograft experiments showed that AdC7-ΔE3 and AdC7-SP/E1A-ΔE3 significantly inhibited tumor growth (Figure 7A) by triggering tumor cell apoptosis, which was validated by TUNEL staining assay (Figure 7B).
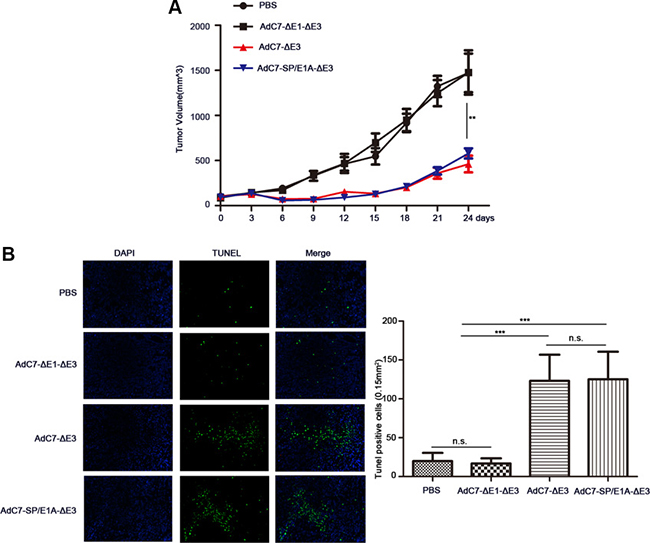
Figure 6: AdC7-SP/E1A-ΔE3 inhibit tumor growth in nude mouse NCI-H508 cell xenografts. NCI-H508 cells (107 ) were inoculated into the right flanks of nude mice, and tumor was injected with 5 × 108 PFU adenovirus (AdC7-ΔE1-ΔE3, AdC7-ΔE3, or AdC7-SP/E1A-ΔE3) suspended in 50 μL of PBS or 50 μL PBS alone when reaching 100–150 mm3. (A) Tumor volume (n = 6) was measured every three days. (B) In left images, TUNEL staining of tumor cells, and in right images, TUNEL positive cells (n = 5) were quantitated. Data are presented as means ± SEM. Statistical significance was determined by one way ANOVA: ***< 0.0001, **< 0.001. Data are representative of two independent experiments.
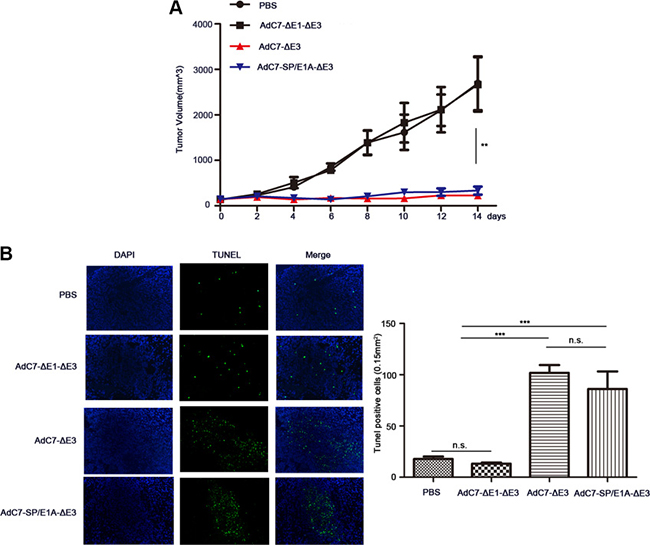
Figure 7: AdC7-SP/E1A-ΔE3 inhibit tumor growth in nude mouse Huh7 cell xenografts. Huh7 cells (5 × 106) were inoculated into the right flanks of nude mice, and tumor was injected with 5 × 108 PFU adenovirus (AdC7-ΔE1-ΔE3, AdC7-ΔE3, or AdC7-SP/E1A-ΔE3) suspended in 50 μL of PBS or 50 μL PBS alone when reaching 100-150 mm3. (A) Tumor volume (n = 6) was measured every two days. (B) In left images, TUNEL staining of tumor cells, and in right images, TUNEL positive cells (n = 5) were quantitated. Data are presented as means ± SEM. Statistical significance was determined by one way ANOVA: ***< 0.0001, **< 0.001. Data are representative of two independent experiments.
Antitumor efficacy of AdC7-SP/E1A-ΔE3 via systemic administration
To investigate the antitumor activity induced by AdC7-SP/E1A-ΔE3 via intravenous injection, we inoculate the mixture of Huh7 cells with matrigel under the skins in the xenograft mouse tumor models; when tumors reached 100–150 mm3, mice were intravenously injected with 1 × 109 PFU of AdC7-ΔE1A-ΔE3 or AdC7-SP/E1A-ΔE3. As shown in Figure 8A, the tumor volume of mice treated with AdC7-SP/E1A-ΔE3 was 1.8 fold smaller than that of mice injected with AdC7-ΔE1A-ΔE3 (p < 0.05). Furthermore, immunohistofluorescence indicated that the number of TUNEL positive tumor cells in the injected group with AdC7-SP/E1A-ΔE3 was 7.0 fold more than in the group treated with AdC7-ΔE1A-ΔE3(p < 0.0001) (Figure 8B).
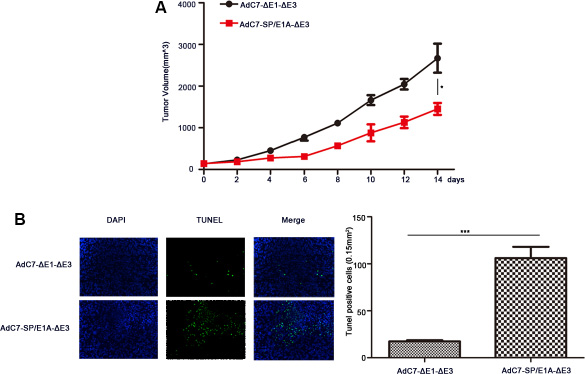
Figure 8: The antitumor efficacy of AdC7-SP/E1A-ΔE3 via systemic administration. Huh7 cells (5 × 106) were inoculated into the right flanks of nude mice, and tumor was injected with 1 × 109 PFU adenovirus (AdC7-ΔE1-ΔE3 or AdC7-SP/E1A-ΔE3) suspended in 50 μL of PBS three times at the interval of one day when reaching 100–150 mm3. (A) Tumor volume (n = 6) was measured every two days. (B) In left images, TUNEL staining of tumor cells, and in right images, TUNEL positive cells (n = 5) were quantitated. Data are presented as means ± SEM. Statistical significance was determined by student’s t test: ***< 0.0001, *< 0.05. Data are representative of two independent experiments.
DISCUSSION
To circumvent preexisting anti-AdHu5 immunity in populations, chimpanzee adenoviruses have been engineered into replication-deficient vaccine vectors [25]. These replication-deficient vectors have been used to evaluate a wide range of vaccines, such as HIV, Ebola, and rabies virus vaccines in preclinical and clinical trials [26–29]. Like other chimpanzee adenoviruses, AdC7 is not neutralized by anti-Adhu5 antibodies and rarely circulates in human populations [15, 16]. Unlike the hexon protein of AdHu5, AdC7 hexon is not associated with FX [17], indicating that AdC7 does not bind FX receptors expressed abundantly in liver cells, thus avoiding extensive liver sequestration of adenoviruses. Therefore, in this study, conditionally replicating AdC7-SP/E1A-ΔE3, in which the replication of AdC7-SP/E1A-ΔE3 in cells was dependent on survivin promoter activity, was constructed to tackle the drawbacks of AdHu5-based oncolytic adenoviruses: preexisting anti-AdHu5 immunity in most populations, and entry of AdHu5 into liver cells via FX factor. Although AdC7 has some advantages over AdHu5 as oncolytic viruses, AdC7 without modification could not address the AdHu5 drawback of nonselectivity in infecting target cells. This problem might be overcame by attempts to genetically alter the fiber protein [30] or to insert some peptides into the hexon region [31].
The traditional method for generating oncolytic adenoviruses is based on homologous recombination using shuttle vector in bacterial, yeast, and mammalian cells [32]. These systems require a laborious multistep procedure. The present study demonstrates a new oncolytic adenovirus-generating system based on a direct cloning method, which was initially used to produce replication-deficient vector for vaccines [33]. As shown in Figure 9, the system consisted of pshoncoE1 and ponco3 vectors. The vector pshoncoE1 was directly inserted into ponco3 to form oncolytic adenoviruses after manipulation of some viral genome in pshoncoE1. Based on our system, various oncolytic adenoviruses were easily constructed within a short period of time, and the potential of contaminating with unidentified pathogens from chimpanzee could be avoided.
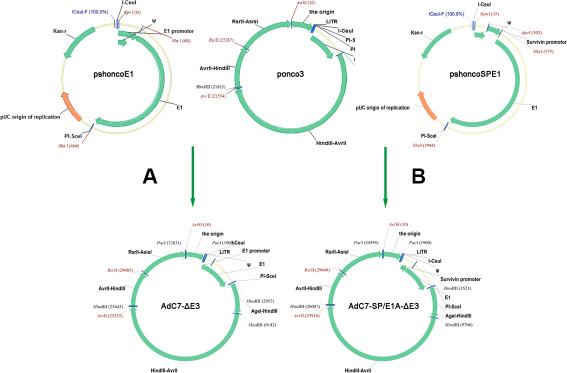
Figure 9: Flowchart of cloning AdC7-ΔE3 and AdC7-SP/E1A-ΔE3. (A, B) Construction of AdC7-ΔE3 and AdC7-SP/E1A-ΔE3. A fragment containing the E1 region was cut from pshoncoE1 and pshoncoSPE1, and inserted into the ponco3 region between I-Ceu / and PI-Sce /. See Methods for more detail.
Although the data in Figure 2 suggest that AdC7-SP/E1A-ΔE3 could replicate in tumor cells A549 and SiHa, implying that this virus has the potential to treat such tumors, it could not efficiently kill these tumor cells (Figure 3). In contrast, AdC7-SP/E1A-ΔE3 could not only replicate in but also efficiently kill tumor cells NCI-H508 and Huh7 (Figures 2–3). Different types of tumor cells vary in proteome, which may determine the differences in the sensitivities of different tumor cells to the same therapy [34, 35]. This could explain the phenomenon that some types of tumor cells such as A549 and SiHa are insensitive to AdC7-SP/E1A-ΔE3 while other types of tumor cells such as Huh7 and NCI-H508 are sensitive. The antagonism to apoptosis induced by AdC7-SP/E1A-ΔE3 in A549 and SiHa could promote the replication of AdC7-SP/E1A-ΔE3, for cells can limit viral replication through death of the infected cell, which is an important cellular defense mechanism against viral invasion [36]. However, NCI-H508 and HuH7 cells infected with AdC7-SP/E1A-ΔE3 could be labeled with Annexin V (Figure 4A), had increasing levels of apoptosis markers (cleaved caspase 3 and PARP) (Figure 4B), and did not experience declining levels of p62 and increasing levels of LC3 II (Figure 4B), suggesting that AdC7-SP/E1A-ΔE3 could kill NCI-H508 and Huh7 cell through apoptosis rather than autophagic cell death, for autophagic cell death is accompanied by the declining of p62 and the increases of LC3 II [37]. Our observations reflect that AdC7-SP/E1A-ΔE3 is distinct in anti-tumor mechanism from AdHu5, which induced autophagic cell death in tumor cells [23]. From the findings on AdHu5 [24] that E1-55k and E4-25k proteins interacted with other cellular proteins to form a ubiquitin ligase complex that degraded the p53 protein, it was speculated that AdC7-SP/E1A-ΔE3 might enable levels of p53 to decline in the infected cells; this hypothesis was validated by data in Figure 5. Moreover, phosphorylation of p53 at serine 46, which is thought to regulate the transcription of apoptosis-inducing genes [38], is absent in cells infected with AdC7-SP/E1A-ΔE3 (Figure 5). These results suggested that apoptosis in infected cells triggered by AdC7-SP/E1A-ΔE3 occurred in a p53-independent manner. The Bcl-2 family proteins, which included antiapoptotic proteins (Bcl-2, Bcl-xl, MCL-1, et al.), proapoptotic proteins (Bim, Bid, PUMA, Bik, Bad, et al.), and the effector proteins Bax and Bak, are well known to interact with each other to regulate apoptosis [39]. In the present study, after infection with AdC7-SP/E1A-ΔE3, NCI-H508 cells had declining levels of antiapoptotic protein MCL-1 and Bcl-2, whereas Huh7 cells had decreased expression of MCL-1and Bcl-xl (Figure 5). E1B 19k protein, the adenovirus Bcl-2 homolog, is required to inhibit human cell apoptosis induced by the infection of adenoviruses [40]. Bik, a potent pro-apoptotic protein, antagonizes anti-apoptotic E1B 19k protein [41]. Our results show that NCI-H508 and Huh7 Cells have increased expressions of Bik after infection with AdC7-SP/E1A-ΔE3. Bad, one of the BH3-only proapoptotic proteins, coupled the death signals to the mitochondrial-mediated pathway, and phosphorylation of Bad at serine 112 was thought to inactivate its proapoptotic function by binding to 14-3-3 scaffold proteins to dissociate Bad from antiapoptotic proteins [42]. Our results show that AdC7-SP/E1A-ΔE3 could enable levels of phosphorylation of Bad at serine 112 to dramatically decline in infected cells, indicating that AdC7-SP/E1A-ΔE3 induced the apoptosis of the infected cells via the Bad-mediated mitochondrial pathway.
In conclusion, we investigated the use of chimpanzee adenoviruses origin- AdC7-SP/E1A-ΔE3 as oncolytic virus for cancer treatment to address the disadvantages of oncolytic adenoviruses based on AdHu5. Importantly, compared to AdHu5, chimpanzee adenoviruses exhibit the lower transduction of hepatocytes [43], which could be explained by the observation that the hexon proteins derived from chimpanzee adenoviruses could not bind to blood coagulation factor X (FX) [17]. Moreover, systemic administration of AdC7-SP/E1A-ΔE3 could also inhibit tumor growth (Figure 8). Oncolytic adenoviruses based on AdC7 are therefore promising candidates for treatment of a variety of tumors, including those of the liver and colon.
MATERIALS AND METHODS
Cell lines and cell culture. Human embryonic kidney cell line 293, human colorectal cancer cell line NCI-H508, human hepatoma cell line Huh7, human lung cancer cell line A549, cervical carcinoma cell line SiHa, and human embryo lung fibroblastic MRC-5 cells were purchased from the American Type Culture Collection (Manassas, VA). All cell lines were cultured at 37°C in Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, NY), supplemented with 10% fetal bovine serum (Gibco) and antibiotics.
Adenoviral vectors. A fragment spanning from 809bp to 2691bp of pNEB193 (New England Biolabs, cat. no. N3051S), a fragment spanning from 1 bp to 132 bp of AdC7 (Simian Ad24/Pan7; ATCC, cat no. VR-593) genome, and a fragment spanning from 3027 bp to 4026 bp of AdC7 genome were amplified using following primers, respectively: (sense: actagtcagcctaggacatgcgatcgcaatcaggg gataacgcaggaaagaacatgt, antisense: ttattgatgatgttaattaac tatttttataggttaatgtcatgataataatggtttc); (sense: cctataaaaata gttaattaacatcatcaataatatacctcaaacttttggtgc, antisense: ctcc gcacccgacatagatgcgagctcgctaccttaggaccgttatagttattaaaaacg tcacccgccccgcccctaac); and (sense: agcgagctcgcatctatgtcgg gtgcggagaaagaggtaatgaaatggaaaatgaatcaataaataaacggagac ggttg, antisense: gagaccggtggtccagggcctaccgcgcgcgaaa). These fragments were then fused into new fragments through overlap PCR as previously described [44]. The fused fragment was digested with HindIII and AvrII, and was ligated with a fragment spanning nt 7152–23367 of AdC7 genome with a previously described method [33], forming plasmid ponco1. A fragment spanning nt 31800–36535 of AdC7 genome was amplified (sense: gtggacggctacgattgacggtccgtcacccccttatccagtgaaataaatatc; antisense: cagtgcgatcgcttaattaacatcatcaataatatacctcaaact tttggtgc), digested with AvrII and RsrII, and inserted into plasmid ponco1, forming ponco2. A fragment spanning nt 21533–25346 of AdC7 genome was amplified (sense: gctcaccacgactatttcttctccttg; antisense: tt atttcactggataagggggtgacggaccgtcaatcgtagccgtccaccgactcg c), digested with RsrII and AsisII, and inserted into plasmid ponco2, forming ponco3.
A fragment spanning nt 133–575 of AdC7 genome and a fragment spanning from nt 576–3932 of AdC7 genome were amplified using the following primers respectively: (sense: cgtaactat aacggtcctaaggtagcgaaggtacctacgtggccgtgaggcggagccggttt gca; antisense: ctttcagctagcctttcaaagtgtagatctgactcgc ggcgcg) and (sense: gaaaggctagctgaaagatgaggcacctgaga; antisense cataatgccatttcattacctctttctccgcacccgacatagattcta gattatttggttttcaccgtggcaaccgcggcc), digested with I-CeuI and NheI and with NheI and PI-SceI, respectively, and cloned into digested plasmid pshuttle (Clontech, cat. no. K1650-1) with I-CeuI and PI-SceI, forming pshoncoE1. The survivin promoter was synthesized with recursive PCR [45], and was fused with a fragment from nt 30–290 of pshoncoE1 into the new fragment. The fused fragment was digested with KpnI and NheI, and was cloned into digested pshoncoE1 with KpnI and NheI, forming pshoncoSPE1.
To create the E3-deleted adenovirus AdC7-ΔE3, the fragment containing the E1 region (which was produced by digesting pshoncoE1 with I-CeuI and PI-SceI) was inserted into ponco3, forming the recombinant plasmid pAdC7-ΔE3. Similarly, to create the E3-deleted adenovirus AdC7-SP/E1A-ΔE3, the fragment containing the E1 region (which was produced by digesting pshoncoSPE1 with I-CeuI and PI-SceI) was inserted into ponco3, forming the recombinant plasmid pAdC7-SP/E1A-ΔE3. The recombinant adenoviruses AdC7-ΔE3 and AdC7-SP/E1A-ΔE3 were rescued, propagated in HEK 293 cells, and purified as previously described [33]. AdC7-ΔE1A-ΔE3 of which E1 and E3 region was deleted was constructed as before [16].
E1A mRNA expression assay. Different cell types (~1 × 106 cells) were infected with 10 MOI of the adenoviruses AdC7-ΔE1A-ΔE3, AdC7-ΔE3, and AdC7-SP/E1A-ΔE3 in Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, NY) supplemented with 5% fetal bovine serum (Gibco) and antibiotics. Twenty-four hours later, cells were collected, and total cellular RNA was extracted using Trizol Reagent (Invitrogen). RNA (1 μg) from each sample was reverse transcribed to synthesize cDNA using a high-capacity RNA-to-cDNA synthesis kit (Applied Biosystems, Foster City, CA), according to the manufacturer’s protocol. E1A mRNA copy number was determined in triplicate experiments by quantitative real-time PCR using the 7900HT Real Time PCR system (Applied Biosystems) as previously described [46]. The E1A primers were sense, gctgctatgaggaatgcttg and antisense, ctttcactccctggttcgat; and the GAPDH primers were sense, ggtttacatgttccaatatgattcca and antisense, atgggatttccattgatgacaag. Data were analyzed using the 7900HT System SDS software (Applied Biosystems).
Adenoviral genome copy number assay. For analysis of adenoviral genome copy numbers, different cell types (~1 × 106 cells) were infected with 10 MOI of the adenoviruses AdC7-ΔE1A-ΔE3, AdC7-ΔE3, and AdC7-SP/E1A-ΔE3 in Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, NY) supplemented with 5% fetal bovine serum (Gibco) and antibiotics. Twenty-four hours and 48 h later, cells were collected and, after cells were washed twice with PBS, DNA was purified using the QIAamp Blood Mini kit (Qiagen), following the manufacturer’s instructions. Viral genome copy numbers were determined by quantitative real-time PCR, as previously described [47], using the 7900HT Real Time PCR system (Applied Biosystems). The fiber primers were sense, ggtaatggcatagctgcaaa and antisense, aaggtaaggccagtacccaa; and the GAPDH primers were sense, ggtttacatgttccaatatgattcca and antisense, atgggatttccattgatgacaag. Data were analyzed using the 7900HT System SDS software (Applied Biosystems).
Infectious Progeny Production. To determine virus progeny production, 2 × 105 cells were seeded in 6-well plates. Cells were infected with 10 MOI of the adenoviruses AdC7-ΔE1A-ΔE3, AdC7-ΔE3, and AdC7-SP/E1A-ΔE3. After 24 h and 48 h, cells were collected and washed twice with PBS. After three freeze/thaw cycles, the production was determined by TCID50 assay on 293 cells.
Cytopathic effect assay. Four tumor cell lines (NCI-H508, Huh7, A549, and SiHa) and a normal cell line (MRC-5) were grown to sub-confluence. These cultures were infected (or not) with AdC7-ΔE1A-ΔE3, AdC7-ΔE3, or AdC7-SP/E1A-ΔE3, at various MOIs. Five days after infection, cells were stained with 2% crystal violet in 20% methanol for 15 min, washed with distilled water, and photographed.
To quantify the cytopathic effect, cells (8 × 103/well) were seeded in 96-well plates and infected (or not) with AdC7-ΔE1A-ΔE3, AdC7-ΔE3, or AdC7-SP/E1A-ΔE3, at various MOIs. Cell survival was assayed at 5 d after infection by an MTT assay, as previously described [48].
Western blot analysis. For detection of the expression of survivin protein in different cell lines, ~1 × 106 cells from each group were lysed with RIPA buffer containing protease inhibitor cocktail (Roche), and the phosphatase inhibitor cocktail (Roche). For apoptosis assay, NCI-H508 and Huh7 cells (~1 × 106 cells) were infected with AdC7-ΔE1A-ΔE3, AdC7-ΔE3, or AdC7-SP/E1A-ΔE3, at various MOIs, collected at different time points, and lysed with the above buffers. Equal amounts of total extracted proteins were added to each lane on SDS-PAGE gels (Thermo Scientific, NP0336BOX). Protein expression levels were detected following transfer to PVDF membranes (Millipore, Bedford, MA); incubation with primary antibodies for Survivin, Cleaved Caspase-3, Cleaved PARP, p62, LC3A/B, p53, Phospho-p53 (Ser46), Mcl-1, Bcl-2, Bcl-xl, Bik or Phospho-Bad (Ser112) (Cell Signaling Technology, Danvers, MA); and subsequent incubation with secondary horseradish peroxidase-conjugated antibodies (Sigma-Aldrich, St Louis, MO). The expression of β-actin (Sigma-Aldrich) was measured as a normalization control for protein loading. The signal was detected using a chemiluminescence detection system (GE Health Life Sciences, Pittsburgh, PA).
Flow cytometry assay. Trypsinized cancer cells were washed twice with ice-cold PBS, and incubated with annexin-V-FITC/PI (Sigma-Aldrich) in binding buffer [49]; flow cytometry (FACS) was performed using the BD Fortessa flow cytometer (BD Biosciences, CA, USA); and FACS data were analyzed using the FlowJo software (TreeStar, Ashland, OR, USA).
Xenograft cancer model. All animal experiments were approved by the Institutional Animal Care and Use Committee of Institut Pasteur of Shanghai. Six to eight-week-old female BALB/c nude mice were purchased from Shanghai Laboratory Animal Center (Shanghai, China), and housed under specific pathogen free conditions. Mice were injected subcutaneously (s.c.) on the right dorsal flank with 107 NCI-H508 cells, or 5 × 106 human Huh7 cells mixed with a matrix gel (1:1, BD Biosciences, San Jose, CA). Tumor volume (V) was calculated using the formula: V (mm3) = length × width2/2. When the tumors reached a volume of 100–150 mm3, animals were randomly divided into PBS, AdC7-ΔE1A-ΔE3, AdC7-ΔE3, and AdC7-SP/E1A-ΔE3 groups. A dose of 5 × 108 PFU adenovirus suspended in 50 μL of PBS or 50 μL PBS alone was administrated intratumorally for three times at an interval of 1 d. Tumor volumes were measured using digital calipers every 3 d for NCI-H508 cells, and every 2 d for Huh7 cells.
To examine the antitumor activity induced by AdC7-SP/E1A-ΔE3 via systemic administration, mice were injected subcutaneously (s.c.) on the right dorsal flank with the mixture of 5 × 106 human Huh7 cells with a matrix gel (1:1, BD Biosciences, San Jose, CA). Tumor volume (V) was calculated as above. When the tumors reached a volume of 100–150 mm3, animals were randomly divided into AdC7-ΔE1A-ΔE3 and AdC7-SP/E1A-ΔE3 groups. A dose of 1× 109 PFU adenovirus suspended in 50 μL of PBS was administrated intravenous for three times at an interval of one day. Tumor volumes were measured using digital calipers every two day.
Immunohistochemistry. Tumors were fixed with 1 mL 4% PFA overnight, dehydrated in ethanol, paraffin embedded, and cut into 5 μm sections. DNA fragmentation was determined by TdT-mediated dUTP nick end labeling (TUNEL) as described by the manufacturer (Roche Applied Science, Mannheim, Germany). Fluorescent images were obtained using an EVOS fluorescent microscope (AMG, Bothell, WA).
Statistical analysis. The Prism 5 Software package (GraphPad, La Jolla, CA) was used to draw the graphs and perform statistical analysis. Differences between groups were analyzed by one-way analysis of variance (ANOVA) with Tukey adjustment. For all tests, P < 0.05 was considered significant.
Abbreviations
AdHu5, human adenovirus serotype 5; ChAd, chimpanzee adenoviruses; FX, blood coagulation factor X; SP, survivin promoter; PSA, Prostate-specific antigen.
ACKNOWLEDGMENTS
We thank the members of the anti-infection immunity and vaccine research unit of Institut Pasteur of Shanghai, for discussions and support throughout this study.
CONFLICTS OF INTEREST
The Authors do not have any conflicts of interest.
GRANT SUPPORT
This project was supported by grants from National Natural Science Foundation of China (31670946), “Knowledge Innovation Program” and “100 Talent Program” from Chinese Academy of Sciences to DZ, and Natural Science Foundation of Shanghai (17ZR1433500) to TC.
REFERENCES
1. Rosa DD, Ismael G, Lago LD, Awada A. Molecular-targeted therapies: lessons from years of clinical development. Cancer treatment reviews. 2008; 34:61–80.
2. Wang Y, Liu T, Huang P, Zhao H, Zhang R, Ma B, Chen K, Huang F, Zhou X, Cui C, Liu X. A novel Golgi protein (GOLPH2)-regulated oncolytic adenovirus exhibits potent antitumor efficacy in hepatocellular carcinoma. Oncotarget. 2015; 6:13564–13578. doi: 10.18632/oncotarget.3769.
3. Uusi-Kerttula H, Hulin-Curtis S, Davies J, Parker AL. Oncolytic Adenovirus: Strategies and Insights for Vector Design and Immuno-Oncolytic Applications. Viruses. 2015; 7:6009–6042.
4. Bischoff JR, Kim DH, Williams A, Heise C, Horn S, Muna M, Ng L, Nye JA, SampsonJohannes A, Fattaey A, McCormick F. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996; 274:373–376.
5. Fueyo J, Gomez-Manzano C, Alemany R, Lee PS, McDonnell TJ, Mitlianga P, Shi YX, Levin VA, Yung WK, Kyritsis, AP. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000; 19:2–12.
6. Kamizono J, Nagano S, Murofushi Y, Komiya S, Fujiwara H, Matsuishi T, Kosai, K. Survivin-responsive conditionally replicating adenovirus exhibits cancer-specific and efficient viral replication. Cancer research. 2005; 65:5284–5291.
7. Yu DC, Sakamoto GT, Henderson DR. Identification of the transcriptional regulatory sequences of human kallikrein 2 and their use in the construction of Calydon virus 764, an attenuated replication competent adenovirus for prostate cancer therapy. Cancer research. 1999; 59:1498–1504.
8. Huang TG, Savontaus MJ, Shinozaki K, Sauter BV, Woo SLC. Telomerase-dependent oncolytic adenovirus for cancer treatment. Gene Ther. 2003; 10:1241–1247.
9. Chirmule N, Propert KJ, Magosin SA, Qian Y, Qian R, Wilson JM. Immune responses to adenovirus and adeno-associated virus in humans. Gene Ther. 1999; 6:1574–1583.
10. Kalyuzhniy O, Di Paolo NC, Silvestry M, Hofherr SE, Barry MA, Stewart PL, Shayakhmetov, DM. Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105:5483–5488.
11. Waddington SN, McVey JH, Bhella D, Parker AL, Barker K, Atoda H, Pink R, Buckley SM, Greig JA, Denby L, Custers J, Morita T, Francischetti IM, et al. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell. 2008; 132:397–409.
12. Weaver EA, Barry MA. Low seroprevalent species D adenovirus vectors as influenza vaccines. Plos One. 2013; 8:e73313.
13. Cheng C, Wang L, Ko SY, Kong WP, Schmidt SD, Gall JG, Colloca S, Seder RA, Mascola JR, Nabel, GJ. Combination recombinant simian or chimpanzee adenoviral vectors for vaccine development. Vaccine. 2015; 33:7344–7351.
14. Xu W, Zhang Z, Yang Y, Hu Z, Wang CH, Morgan M, Wu Y, Hutten R, Xiao X, Stock S, Guise T, Prabhakar BS, Brendler C, Seth P. Ad5/48 hexon oncolytic virus expressing sTGFbetaRIIFc produces reduced hepatic and systemic toxicities and inhibits prostate cancer bone metastases. Molecular therapy. 2014; 22:1504–1517.
15. Farina SF, Gao GP, Xiang ZQ, Rux JJ, Burnett RM, Alvira MR, Marsh J, Ertl HC, Wilson JM. Replication-defective vector based on a chimpanzee adenovirus. J Virol. 2001; 75:11603–11613.
16. Cheng T, Wang X, Song Y, Tang X, Zhang C, Zhang H, Jin X, Zhou D. Chimpanzee adenovirus vector-based avian influenza vaccine completely protects mice against lethal challenge of H5N1. Vaccine. 2016; 34:4875–83.
17. Belousova N, Mikheeva G, Xiong C, Soghomonian S, Young D, Le Roux L, Naff K, Bidaut L, Wei W, Li C, Gelovani J, Krasnykh V. Development of a targeted gene vector platform based on simian adenovirus serotype 24. J Virol. 2010; 84:10087–10101.
18. Lorin C, Vanloubbeeck Y, Baudart S, Ska M, Bayat B, Brauers G, Clarinval G, Donner MN, Marchand M, Koutsoukos M, Mettens P, Cohen J, Voss G. Heterologous prime-boost regimens with a recombinant chimpanzee adenoviral vector and adjuvanted F4 protein elicit polyfunctional HIV-1-specific T-Cell responses in macaques. Plos One 2015; 10:e0122835.
19. Kobinger GP, Feldmann H, Zhi Y, Schumer G, Gao G, Feldmann F, Jones S, Wilson JM. Chimpanzee adenovirus vaccine protects against Zaire Ebola virus. Virology. 2006; 346:394–401.
20. Roy S, Kobinger GP, Lin J, Figueredo J, Calcedo R, Kobasa D, Wilson, JM. Partial protection against H5N1 influenza in mice with a single dose of a chimpanzee adenovirus vector expressing nucleoprotein. Vaccine. 2007; 25:6845–6851.
21. Garg H, Suri P, Gupta JC, Talwar GP, Dubey, S. Survivin: a unique target for tumor therapy. Cancer cell international. 2016; 16:49.
22. Li B, Liu X, Fan J, Qi R, Bo L, Gu J, Qian Q, Qian C, Liu, X. A survivin-mediated oncolytic adenovirus induces non-apoptotic cell death in lung cancer cells and shows antitumoral potential in vivo. J Gene Med. 2006; 8:1232–1242.
23. Ito H, Aoki H, Kuhnel F, Kondo Y, Kubicka S, Wirth T, Iwado E, Iwamaru A, Fujiwara K, Hess KR, Lang FF, Sawaya R, Kondo S. Autophagic cell death of malignant glioma cells induced by a conditionally replicating adenovirus. Journal of the National Cancer Institute. 2006; 98:625–636.
24. Sarnow P, Hearing P, Anderson CW, Halbert DN, Shenk T, Levine AJ. Adenovirus Early Region-1b 58,000-Dalton Tumor-Antigen Is Physically Associated with an Early Region-4 25,000-Dalton Protein in Productively Infected-Cells. J Virol. 1984; 49:692–700.
25. Tatsis N, Tesema L, Robinson ER, Giles-Davis W, McCoy K, Gao GP, Wilson JM, Ertl HCJ. Chimpanzee-origin adenovirus vectors as vaccine carriers. Gene Ther. 2006; 13:421–429.
26. Bliss CM, Ewer K, Anagnostou N, Havelock T, Poulton I, Lawrie A, Choudhary P, Hill, A. A phase I study to assess safety and immunogenicity of novel schedules for vaccination with candidate malaria vaccines ChAd63 ME-TRAP and MVA ME-TRAP. Immunology. 2011; 135:77–77.
27. Sheehy SH, Duncan, CJA, Elias SC, Collins KA, Ewer KJ, Spencer AJ, Williams AR, Halstead FD, Moretz SE, Miura K, Epp C, Dicks MD, Poulton ID, et al. Phase Ia Clinical Evaluation of the Plasmodium falciparum Blood-stage Antigen MSP1 in ChAd63 and MVA Vaccine Vectors. Molecular Therapy. 2011; 19:2269–2276.
28. de Barra E, Hodgson SH, Ewer KJ, Bliss CM, Hennigan K, Collins A, Berrie E, Lawrie AM, Gilbert SC, Nicosia A, McConkey SJ, Hill AV. A Phase Ia Study to Assess the Safety and Immunogenicity of New Malaria Vaccine Candidates ChAd63 CS Administered Alone and with MVA CS. Plos One. 2014; 9:e115161.
29. Xiang Z, Gao G, Reyes-Sandoval A, Cohen CJ, LY, Bergelson JM, Wilson JM, Ertl, H.C. Novel, chimpanzee serotype 68-based adenoviral vaccine carrier for induction of antibodies to a transgene product. J Virol. 2002; 76:2667–2675.
30. Behr M, Kaufmann JK, Ketzer P, Engelhardt S, Muck-Hausl M, Okun PM, Petersen G, Neipel F, Hassel JC, Ehrhardt A, Enk AH, Nettelbeck DM. Adenoviruses Using the Cancer Marker EphA2 as a Receptor In Vitro and In Vivo by Genetic Ligand Insertion into Different Capsid Scaffolds. Plos One. 2014; 9.
31. Jin C, Yu D, Cancer M, Nilsson B, Leja J, Essand, M. Tat-PTD-modified oncolytic adenovirus driven by the SCG3 promoter and ASH1 enhancer for neuroblastoma therapy. Human gene therapy. 2013; 24:766–775.
32. Yuan M, Webb E, Lemoine NR, Wang, YH. CRISPR-Cas9 as a Powerful Tool for Efficient Creation of Oncolytic Viruses. Viruses-Basel. 2016; 8.
33. Zhou D, Zhou X, Bian A, Li H, Chen H, Small JCLY, Giles-Davis W, Xiang Z, Ertl, HC. An efficient method of directly cloning chimpanzee adenovirus as a vaccine vector. Nature protocols. 2010; 5:1775–1785.
34. Dukhanin AS, Romanova EA, Dukhanina, EA. Molecular mechanisms of different sensitivity of tumor cells to dexamethasone. Bulletin of experimental biology and medicine. 2001; 131:73–76.
35. Wang XB, Liu QH, Wang P, Wang ZZ, Tong WY, Zhu B, Wang Y, Li CD. Comparisons among sensitivities of different tumor cells to focused ultrasound in vitro. Ultrasonics. 2009; 49:558–564.
36. Banerjee S, Uppal T, Strahan R, Dabral P, Verma SC. The Modulation of Apoptotic Pathways by Gammaherpesviruses. Frontiers in microbiology 7:2016.
37. Ganesan R, Hos NJ, Gutierrez S, Fischer J, Stepek JM, Daglidu E, Kronke M, Robinson, N. Salmonella Typhimurium disrupts Sirt1/AMPK checkpoint control of mTOR to impair autophagy. PLoS pathogens. 2017; 13:e1006227.
38. Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y, Taya Y. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell. 2000; 102:849–862.
39. Zheng JH, Viacava Follis A, Kriwacki RW, Moldoveanu T. Discoveries and controversies in BCL-2 protein-mediated apoptosis. The FEBS journal. 2016; 283:2690–2700.
40. Tarodi B, Subramanian T, Chinnadurai G. Functional Similarity between Adenovirus E1b 19k Gene and Bcl2 Oncogene - Mutant Complementation and Suppression of Cell-Death Induced by DNA-Damaging Agents. International journal of oncology. 1993; 3:467–472.
41. Han J, Sabbatini P, White E. Induction of apoptosis by human Nbk/Bik, a BH3-containing protein that interacts with E1B 19K. Molecular and cellular biology. 1996; 16:5857–5864.
42. Hayakawa J, Ohmichi M, Kurachi H, Kanda Y, Hisamoto K, Nishio Y, Adachi K, Tasaka K, Kanzaki T, Murata, Y. Inhibition of BAD phosphorylation either at serine 112 via extracellular signal-regulated protein kinase cascade or at serine 136 via Akt cascade sensitizes human ovarian cancer cells to cisplatin. Cancer research. 2000; 60:5988–5994.
43. Roy S, Gao G, Lu Y, Zhou X, Lock M, Calcedo R, Wilson JM. Characterization of a family of chimpanzee adenoviruses and development of molecular clones for gene transfer vectors. Human gene therapy. 2004; 15:519–530.
44. Shevchuk NA, Bryksin AV, Nusinovich YA, Cabello FC, Sutherland M, Ladisch, S. Construction of long DNA molecules using long PCR-based fusion of several fragments simultaneously. Nucleic Acids Res. 2004; 32.
45. Prodromou C, Pearl LH. Recursive PCR: a novel technique for total gene synthesis. Protein engineering. 1992; 5:827–829.
46. Chi YD, Wang X, Yang Y, Zhang C, Ertl HCJ, Zhou, DM. Survivin-targeting Artificial MicroRNAs Mediated by Adenovirus Suppress Tumor Activity in Cancer Cells and Xenograft Models. Mol Ther-Nucl Acids. 2014; 3.
47. Ketzer P, Kaufmann JK, Engelhardt S, Bossow S, von Kalle C, Hartig JS, Ungerechts G, Nettelbeck DM. Artificial riboswitches for gene expression and replication control of DNA and RNA viruses. Proceedings of the National Academy of Sciences of the United States of America. 2014; 111:E554–562.
48. Chi Y, Wang X, Yang Y, Zhang C, Ertl HC, Zhou, D. Survivin-targeting Artificial MicroRNAs Mediated by Adenovirus Suppress Tumor Activity in Cancer Cells and Xenograft Models. Molecular therapy. Nucleic acids. 2014; 3:e208.
49. Pigault C, Follenius-Wund A, Schmutz M, Freyssinet JM, Brisson A. Formation of two-dimensional arrays of annexin V on phosphatidylserine-containing liposomes. J Mol Biol. 1994; 236:199–208.