
INTRODUCTION
In the United States, colorectal cancer (CRC) is the third most common cancer in both women and men; in 2008, it caused about 50,000 deaths. Since approximately 50% of all CRC patients die of metastatic disease that is not apparent at surgery [1], efforts are underway to discover molecular determinants that identify patients who are at risk of developing recurrent CRC following surgical resection. These individuals could benefit from adjuvant chemotherapy.
The Smad4 (mothers against decapentaplegic homolog 4) gene is located at chromosome locus 18q21.1 [2, 3]. At some stages of CRC, there is loss of heterozygosity (LOH) at this locus, and such loss is associated with a poor prognosis for lymph node-negative (stage II) CRC patients [2, 4–6]. However, the prognostic value of LOH of Smad4, particularly in lymph node-positive (stage III) patients, remains controversial [5, 7–9]. There is a higher prevalence of Smad4 mutations in CRCs [10, 11], particularly in those with distant metastases (35%) than in locally advanced tumors (~10%) [11]. Moreover, animal studies show that Smad4 inactivation is involved in the malignant transformation of gastrointestinal (GI) adenomas [12], and, during tumor progression, there are reductions in Smad4 mRNA levels [13]. Similarly, CRC patients with tumors expressing low levels of SMAD4 mRNA or protein or immunophenotypic expression levels have worse survival outcomes than CRC patients with tumors expressing high levels of SMAD4 [14, 15]. Other investigations, however, have found Smad4 mutations in only a small proportion of CRCs [16–19].
Smad4, a member of the Smad family of proteins, is an intracellular transducer that mediates the transforming growth factor (TGF-β)-Smad-dependent signaling pathway and translocation of the TGF-β complex into the nucleus. In the nucleus, these signals inhibit the growth of colon epithelial cells by regulating genes related to cell proliferation, differentiation, and apoptosis [3, 11, 20–23]. In CRCs (all stages), low immunophenotypic expression of Smad4 correlates with poor patient survival [17, 24–28]. Some recent reports show a poor prognostic value for low phenotypic expression of Smad4 but no association for 18q21 allelic imbalance in CRCs [14]. These results, however, are contradictory to those reported for most of studies of CRCs [5, 8, 29–31].
The present study evaluated the role of Smad4 alterations (mutations, LOH expression, and sub-cellular localization) in CRC tissues from patients who did not receive adjuvant chemotherapy. The association between the abnormal variations in Smad4 and overall patient survival and by tumor stage was evaluated. The results provide evidence that SMAD4 is a prognostic marker for some patients with CRC.
RESULTS
Fifty-five cases (55/209, 26%) and 77 cases (77/209, 37%) exhibited low SMAD4 immunohistochemical protein (IHC) expression in the cytoplasm and nucleus, respectively (Table 1). Kaplan-Meier survival analyses were conducted for nuclear and cytoplasmic protein expression for the entire study sample and by tumor stage. For the overall sample, there was a marginally statistically significant association between low nuclear SMAD4 IHC levels and poorer survival of these patients (log-rank, P = 0.07) (data not shown). There was, however, no association for patients with low SMAD4 IHC expression in cytoplasm (log-rank, P = 0.28) (data not shown). The 74 Stage II and 64 Stage III cases were then analyzed separately. For stage III patients, a low nuclear SMAD4 IHC level was significantly associated with shorter survival (log-rank, P = 0.02; Figure 1E). However, for Stage II patients, high cytoplasmic IHC expression of SMAD4 was associated with an increased risk of cancer-specific mortality (log-rank, P = 0.047; Figure 1C). These results suggest that, for stage III patients, low nuclear IHC expression of SMAD4 is a prognostic marker for shorter survival and recurrence. In contrast, a high cytoplasmic IHC level was associated with worse survival of stage II patients. There was no significant association between IHC expression of SMAD4 and age, sex, ethnicity, location, or histological grade (Table 1). However, there was a trend towards statistical significance for association between nuclear (p=0.08) and cytoplasmic (p=0.07) protein expression with tumor stage (Table 1).
Table 1: Characteristics of the study sample (n = 209) according to 18q21 LOH and protein expression
Characteristics of study sample |
n (%) |
18q21 LOH |
P |
Nuclear Localization |
P |
Cytoplasmic Localization |
P |
|||
|---|---|---|---|---|---|---|---|---|---|---|
No n (%) 73 (38.9) |
Yes n (%) 118 (61.1) |
Low n(%) 77 (36.8) |
High n (%) 132 (63.2) |
Low n(%) 55 (26.3) |
High n (%) 154 (73.7) |
|||||
18q21 deletion |
|
|
|
|
|
|
|
|
|
0.74 |
No LOH |
73 (38.9) |
-- |
-- |
|
29 (37.7) |
62 (47.0) |
0.19 |
25 (45.5) |
66 (42.9) |
|
LOH |
118 (61.1) |
-- |
-- |
|
48 (62.3) |
70 (53.0) |
|
30 (55.5) |
88 (57.1) |
|
Cytoplasmic staining |
|
|
|
|
|
|
< 0.001 |
|
|
|
Low |
55 (26.3) |
-- |
-- |
|
45 (59.7) |
9 (6.8) |
|
-- |
-- |
|
High |
154 (73.7) |
-- |
-- |
|
31 (40.3) |
123 (93.2) |
|
-- |
-- |
|
Age |
|
|
|
0.79 |
|
|
0.37 |
|
|
0.23 |
< 65 |
92 (44.0) |
41 (45.1) |
51 (43.2) |
|
37 (48.0) |
55 (41.7) |
|
28 (50.9) |
64 (41.6) |
|
≥ 65 |
117 (56.0) |
50 (54.9) |
67 (56.8) |
|
40 (52.0) |
77 (58.3) |
|
27 (49.1) |
90 (58.4) |
|
Sex |
|
|
|
0.99 |
|
|
0.73 |
|
|
0.42 |
Male |
108 (51.7) |
47 (51.7) |
61 (51.7) |
|
41 (53.3) |
67 (50.8) |
|
31 (56.4) |
77 (50.0) |
|
Female |
101 (48.3) |
44 (48.4) |
57 (48.3) |
|
36 (46.8) |
65 (49.2) |
|
24 (43.6) |
77 (50.0) |
|
Race |
|
|
|
0.84 |
|
|
0.41 |
|
|
0.85 |
Caucasian |
127 (60.8) |
56 (61.5) |
71 (60.2) |
|
44 (57.1) |
83 (62.9) |
|
34 (61.8) |
93 (60.4) |
|
African American |
82 (39.2) |
35 (38.5) |
47 (39.8) |
|
33 (42.9) |
49 (37.1) |
|
21 (38.2) |
61 (39.6) |
|
Tumor Stage |
|
|
|
0.45 |
|
|
0.08 |
|
|
0.07 |
Stage I |
34 (16.3) |
16 (17.6) |
18 (15.3) |
|
14 (18.1) |
20 (15.1) |
|
8 (14.6) |
26 (16.9) |
|
Stage II |
70 (33.5) |
25 (27.5) |
45 (38.1) |
|
18 (23.4) |
52 (39.4) |
|
13 (23.6) |
57 (37.0) |
|
Stage III |
64 (30.6) |
30 (33.0) |
34 (28.8) |
|
25 (32.4) |
39 (29.6) |
|
17 (30.9) |
47 (30.5) |
|
Stage IV |
41 (19.6) |
20 (22.0) |
21 (17.8) |
|
20 (26.0) |
21 (15.9) |
|
17 (30.9) |
24 (15.6) |
|
Tumor size |
|
|
|
0.91 |
|
|
0.75 |
|
|
0.68 |
≤ 65 |
160 (78.1) |
69 (78.4) |
91 (77.8) |
|
61 (79.2) |
99 (77.3) |
|
44 (80.0) |
116 (77.3) |
|
> 65 |
45 (21.9) |
19 (21.6) |
26 (22.2) |
|
16 (20.8) |
29 (22.7) |
|
11 (20.0) |
34 (22.7) |
|
Tumor grade |
|
|
|
0.06 |
|
|
0.33 |
|
|
0.94 |
Low |
149 (71.3) |
71 (78.0) |
78 (66.1) |
|
58 (75.3) |
91 (68.9) |
|
39 (70.9) |
110 (71.4) |
|
High |
60 (28.7) |
20 (22.0) |
40 (33.9) |
|
19 (24.7) |
41 (31.1) |
|
16 (29.1) |
44 (28.6) |
|
Tumor location |
|
|
|
0.05 |
|
|
0.75 |
|
|
0.92 |
Rectum |
42 (20.1) |
19 (20.9) |
23 (19.5) |
|
15 (19.5) |
27 (20.4) |
|
11 (20.0) |
31 (20.1) |
|
Distal colon |
64 (30.6) |
21 (23.1) |
43 (36.4) |
|
26 (33.8) |
38 (28.8) |
|
18 (32.7) |
46 (29.9) |
|
Proximal colon |
103 (49.3) |
51 (56.0) |
52 (44.1) |
|
36 (46.7) |
67 (50.8) |
|
26 (47.3) |
77 (50.0) |
|
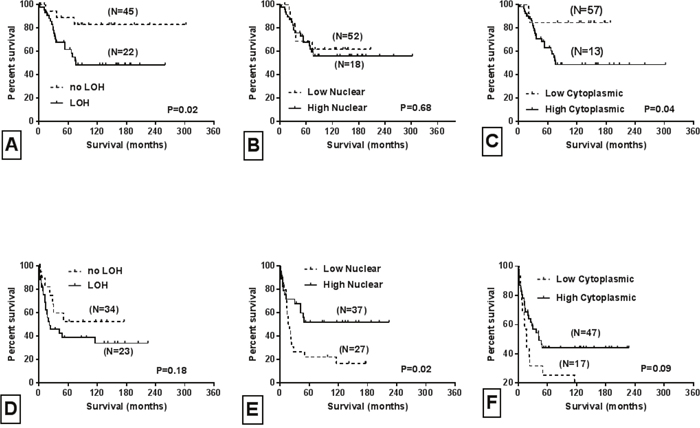
Figure 1: Univariate survival curves based on tumor stage, LOH and Smad4 sub-cellular localization. Kaplan-Meier survival curves for stage II patients according to: A. LOH status, B. nuclear Smad4 expression, C. cytoplasmic Smad4 expression. Kaplan-Meier survival curves for stage III patients according to: D. LOH status, E. nuclear Smad4 expression, F. cytoplasmic Smad4 expression. Log rank P values are provided.
LOH in chromosome site 18q21 at the Smad4 locus
Genomic DNA was retrieved from 209 FFPE tissue blocks of CRCs and their corresponding normal tissues. Three microsatellite biomarkers (D18S474, D18S46 and D18S363) that surround the Smad4 gene were used to analyze LOH status of 18q21. In at least one of the three markers, LOH was detected in 118 of the 193 (61%) informative cases (Table 1). Forty (40/118, 34%) cases exhibited LOH at two or three microsatellite markers. Excluding those with microsatellite instability and the ‘not detectable’ cases, the highest percentage (68/118, 58%) of informative cases was found at the D18S363 locus. The other two biomarkers had values of 40% (49/121) for the D18S46 locus and 41% (48//118) for the D18S474 locus (data not shown). For all 193 CRC cases, there was no significant difference in survival between the cases with and without Smad4 LOH (log-rank, P = 0.14) (data not shown). Survival, however, was significantly associated with Smad4 LOH for 67 stage II patients (log-rank, P = 0.02; Figure 1A), but not for 57 stage III patients (log-rank, P = 0.18; Figure 1D). There was no significant difference between Smad4 LOH status according to demographics and most clinical features. There was, however, a borderline statistically significant association for tumor location (p = 0.05) (Table 1). LOH-positive tumors were more likely to be in the proximal (44.1%) and distal (36.4%) colon than in the rectum (19.5%, Table 1).
Analysis of Smad4 mutations
A set of primers that covered the entire Smad4 coding region (exon 1 through 11; codons 1 through 552) was used to screen for Smad4 mutations by direct sequencing after PCR amplification. Among 53 samples, only 4 Smad4 mutations were found (stage I, 5; II, 18; III, 21; IV, 9) (4/53, 7.5%). Genomic DNA was extracted from the corresponding FFPE tissues that exhibited Smad4 mutations that had been confirmed by exon (2, 8 10)-specific primers and direct DNA sequencing. Of these four mutations, there were two missense mutations (codon 356 and 474) leading to changes in the amino acid residues and two silent mutations (codon 118 and codon 464) (Figure 2). The four mutations were all in DNA from Caucasian patients; three of these were male. The 53 cases (21 LOH positive, 28 LOH negative, and 4 not informative) were analyzed for their mutational status; there were no Smad4 mutations in LOH-positive cases (2 were LOH-negative, 2 were not informative). Due to the small number of mutations, associations according to tumor stage could not be determined. These findings indicate that, in CRCs, Smad4 may not be a target for mutational inactivation.
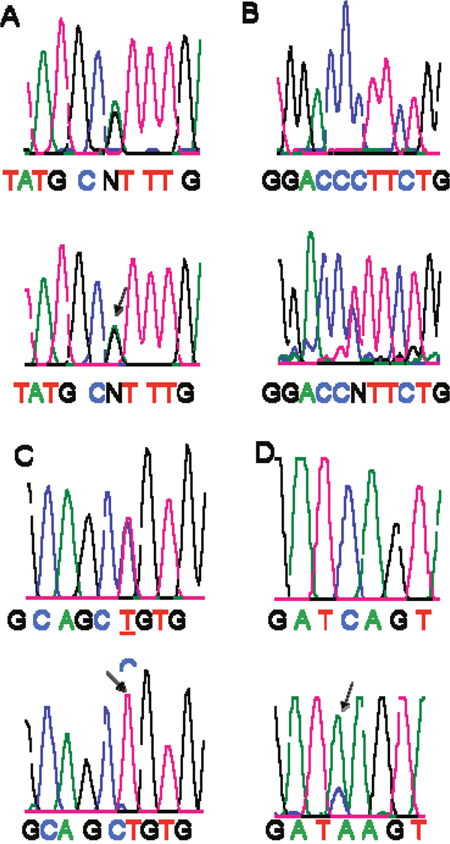
Figure 2: Detection of Smad4 gene mutations. A silent mutation, GCG (Ala) to GCA (Ala) at codon118 Panel-A. A missense mutation, CCT (Pro) to CTT (Leu) at codon 356 Panel-B. A silent mutation, GCC (Ala) to GCT (Ala) at codon 464 Panel-C. A missense mutation, TCA (Ser) to TAA (stop) at codon 474 Panel-D. Arrows show the mutations. Tumor tissues are below, and corresponding normal tissues are above.
Bivariate associations
The bivariate associations of SMAD4 characteristics, demographic variables, and tumor-related measures are shown in Table 2. For all study participants, LOH of Smad4 was associated with increased hazard of death, however, this association is statistically not significant (HR = 1.15; 95% CI, 0.78-1.71). Low nuclear expression of SMAD4 protein was also associated with an increased hazard (HR = 1.44; 95% CI, 0.97-2.12) of marginal statistical significance. For all stages, cytoplasmic expression of SMAD4 protein was not associated with mortality.
Table 2: Bivariate associations with colon cancer-specific death
Study characteristics |
Unadjusted HR (95% CI) |
|---|---|
18q21 deletion |
|
no LOH |
Ref |
LOH |
1.15 (0.78, 1.71) |
Cytoplasmic expression |
|
Low |
Ref |
High |
0.78 (0.52, 1.19) |
Nuclear expression |
|
High |
Ref |
Low |
1.44 (0.97, 2.12) |
Age |
|
< 65 |
Ref |
≥ 65 |
0.91 (0.62, 1.35) |
Sex |
|
Male |
Ref |
Female |
1.57 (1.06, 2.32) |
Race |
|
Caucasian |
Ref |
African American |
1.21 (0.82, 1.79) |
Tumor stage |
|
Stage I |
Ref |
Stage II |
1.71 (0.73, 3.96) |
Stage III |
3.88 (1.73, 8.72) |
Stage IV |
10.95 (4.80, 24.99) |
Tumor size |
|
≤ 65 |
Ref |
> 65 |
0.93 (0.58, 1.49) |
Tumor grade |
|
Low |
Ref |
High |
1.71 (1.14, 2.58) |
Tumor location |
|
Rectum |
Ref |
Distal colon |
1.84 (1.02, 3.32) |
Proximal colon |
1.70 (0.97, 2.99) |
HR, hazard ratio.
Multivariate results
The associations for Smad4 LOH and immunophenotypic expression of this protein with death due to CRC are shown in Table 3. This table depicts the overall association of LOH and phenotypic expression of the protein as well as stage-specific associations after adjustment for Smad4 status, tumor stage, tumor grade, and location. Overall, LOH was not associated with CRC-specific death (HR = 1.45; 95% CI, 0.91-2.31). However, it was associated with a 3.3-fold increased hazard of death for stage II patients that was of borderline statistical significance (HR = 3.30; 95% CI, 0.93-11.69). Although there was an association for stage I patients, this relationship failed to reach statistical significance due to only seven events (deaths due to CRC) among these patients. Cytoplasmic protein expression was not associated with mortality for all stages combined. However, among stage II patients, a high cytoplasmic protein level was associated with a 4.7-fold (HR = 4.71; 95% CI, 0.98-22.65) increased hazard of cancer-specific death that was borderline statistically significant. Overall, low nuclear protein expression of SMAD4 was associated with a 1.7-fold (HR = 1.70; 95% CI, 0.96-3.00) increased hazard of death that was of marginal statistical significance. The lack of nuclear protein expression was primarily confined to stage III patients, who showed a 2.7-fold difference (HR = 2.67; 95% CI, 0.98-7.32) that was also borderline statistically significant (Table 3).
Table 3: Association of Smad4 allelic status and phenotypic expression with overall mortality and by tumor stage
Variable |
Overall HR1 (95% CI) |
Stage I HR1 (95% CI) |
Stage II HR1 (95% CI) |
Stage III HR1 (95% CI) |
Stage IV HR1 (95% CI) |
|---|---|---|---|---|---|
18q21 deletion |
|
|
|
|
|
no LOH |
Ref |
ref |
ref |
ref |
ref |
LOH |
1.45 (0.91, 2.31) |
5.66 (0.59, 54.52) |
3.30 (0.93, 11.69) |
1.20 (0.58, 2.51) |
1.84 (0.70, 4.85) |
Nuclear expression |
|
|
|
|
|
High |
Ref |
ref |
ref |
ref |
ref |
Low |
1.70 (0.96, 3.00) |
0.28 (0.03, 2.34) |
2.09 (0.72, 6.10) |
2.67 (0.98, 7.32) |
0.44 (0.11, 1.79) |
Cytoplasmic expression |
|
|
|
|
|
Low |
Ref |
ref |
ref |
ref |
ref |
High |
1.39 (0.76, 2.56) |
1.34 (0.08, 21.48) |
4.71 (0.98, 22.65) |
1.22 (0.44, 3.39) |
0.65 (0.17, 2.44) |
Tumor grade |
|
|
|
|
|
Low |
Ref |
ref |
ref |
ref |
ref |
High |
1.80 (1.15, 2.83) |
0.48 (0.03, 6.68) |
1.56 (0.58, 4.20) |
1.28 (0.62, 2.65) |
5.80 (2.25, 14.94) |
Tumor location |
|
|
|
|
|
Rectum and distal colon |
Ref |
ref |
ref |
ref |
ref |
Proximal colon |
1.72 (1.11, 2.67) |
0.91 (0.11, 7.81) |
1.50 (0.59, 3.79) |
1.39 (0.68, 2.84) |
7.56 (2.18, 26.29) |
Tumor stage |
|
|
|
|
|
Stage I |
Ref |
|
|
|
|
Stage II |
1.61 (0.67, 3.84) |
|
|
|
|
Stage III |
3.20 (1.39, 7.36) |
|
|
|
|
Stage IV |
11.92 (4.99, 28.48) |
|
|
|
|
1Adjusted for the variables listed.
HR, hazard ratio;' CI, confidence interval.
DISCUSSION
First identified at the 18q21 locus in 1996 [3], Smad4 and has attracted interest as a candidate tumor suppressor gene for 18q21 allelic imbalance. Various studies have demonstrated associations between Smad4 mutations, SMAD4 protein expression, and 18q21 allelic imbalance at the Smad4 locus [10, 11, 32]. The results, however, have not been consistent. The prevalence of Smad4 mutations is about 50% [33, 34] in pancreatic carcinomas, but, in other tumor types, these mutations are less frequent [16, 35–38].
In CRCs, the frequency of loss of SMAD4 expression ranges from 9 to 67% [27, 39, 40]. Loss of SMAD4 expression in CRCs is associated with advanced-stage disease, presence of lymph node metastasis, and poor prognosis [15, 17, 24, 25, 27, 31]. Xie et al [25]. reported that loss of or reduced SMAD4 expression was associated with significantly shorter overall survival, but Kouvidou et al. [28] failed to find this relationship. The latter investigators demonstrated a significant correlation only between SMAD4 expression and tumor grade. Seshimo et al. [16] found that loss of SMAD4 expression was more frequent in poorly differentiated adenocarcinomas than in well- and moderately-differentiated carcinomas. In contrast, Bacman et al. [41] observed that lack of SMAD4 nuclear expression was not correlated with tumor grade or clinical outcome. SMAD4 expression is retained in high-grade CRCs, suggesting that loss of SMAD4 is a late event in carcinogenesis [28]. However, other investigators find no influence of reduced SMAD4 expression on prognosis [41]. There is a poor prognosis for CRCs with SMAD4, and high SMAD4 expression predicts a better prognosis for CRC patients with curative surgery [42]. Also, loss of SMAD4 expression is an independent prognostic factor, because it is associated with recurrence-free survival and overall survival in CRC patients [43]. In the current study, SMAD4 protein immunophenotic expression levels in the nuclear and cytoplasmic compartments of malignant cells were analyzed by IHC for 209 CRC patients with various stages of disease. Of these patients, 37% had low SMAD4 expression in the nuclei, whereas 26% had low SMAD4 expression in the cytoplasm. These proportions are in agreement with those of other studies [11, 28, 35, 39, 41, 44-49], indicating that Smad4 is a tumor suppressor gene and that its protein expression, especially in nuclei, is lost in most cases of CRC progression. Kaplan-Meier and multivariate analyses found poorer overall survival for stage III patients with low nuclear SMAD4 protein. In contrast, among stage II patients, high cytoplasmic expression of the protein was associated with worse survival. Further, a borderline significant shorter survival was found for those with low SMAD4 protein levels in the nuclei with Smad4 LOH positive in stage III CRC patients, indicating that Smad4 is a tumor suppressor gene for these 18q21 deletions at the Smad4 locus. SMAD4 levels decrease progressively from stage I through stage IV CRC, and the incidence of SMAD4 loss is higher in advanced stages of CRC [50, 51]. Our results demonstrate that low nuclear SMAD4 tumor protein in stage III and high cytoplasmic protein levels in stage II tumors are markers of poor prognosis for CRC patients. These findings can be considered in relation to reports that loss of expression of SMAD4 is more frequent for stage III than for stage II CRCs, that stage III CRCs that are microsatellite stable and exhibit loss of SMAD4 have a worse prognosis, and that time to recurrence after curative therapy is shorter for patients with stage III CRCs with SMAD4 loss [43, 50]. Together, these results can be useful in stratifying and predicting outcomes for patients with stage II or stage III CRC.
SMAD4 acts as a central mediator in the TGF-ß superfamily signaling pathway. The receptor-regulated SMADS, i.e., phosphorylated SMAD2 and SMAD3, are mediated by SMAD4 to translocate into the nucleus, where they are involved in the regulation of expression of genes that are necessary for cell proliferation, differentiation, and apoptosis [52–54]. In the absence of TGF-ß signaling, SMAD4 shuttles between the cytoplasm and nucleus, suggesting that nuclear SMAD4 has a function in unstimulated cells [55]. In addition to the role of mediating and carrying SMAD2 and SMAD3 into the nucleus, SMAD4 may also cooperate with these factors in regulation of transcription within the nucleus [56]. SMAD4 is directly or indirectly involved in regulation of genes involved in cellular invasion, e.g., those for E-cadherin, P-cadherin [57], and UK Pan-1 [58]. In addition, SMAD4 may induce the expression of cyclin-dependent kinase inhibitors, including p21, p27, and p15 [59, 60]. This induction regulates the proliferation and apoptosis of cells [61]. Abnormally low SMAD4 IHC levels in the nucleus could indicate the interruption of shuttling and cause a deficiency in mediating nuclear translocation of other SMADS or in regulating gene expression in a more direct way. These functions of SMAD4 in nuclei may explain the association between the low levels of SMAD4 in the nuclei and poorer clinical outcomes for CRC patients, especially for those with stage III disease.
Deletions in the arm of chromosome 18q are among the most common genetic abnormalities found in CRCs [4, 62]. The relationship between mortality and loss of chromosome genetic material, however, is inconsistent. Some reports show that allelic loss of chromosome 18q21 has a negative impact on prognosis and survival; [5, 7, 29, 31, 63-66] other studies find no prognostic value [5, 8, 9, 43, 67, 68].
In the current study of 193 informative cases, 118 cases (61%) showed LOH of 18q21 at the Smad4 locus. This ratio is in agreement with a previous report [46]. In all 209 CRC cases (all stages included), there was no significant difference in survival between the cases with and without LOH of Smad4. However, an association between Smad4 LOH and mortality was demonstrated for stage II CRC patients in both bivariate and multivariate analyses. This finding is in agreement with previous reports [11, 51] suggesting that Smad4 LOH in stage II CRCs is a prognostic marker for cancer recurrence. For these high-risk stage II patients, the benefit of adjuvant chemotherapy following surgery should be assessed.
In cancer cells, allelic deletion of Smad4 is a mechanism attributed to the loss of TGF-β tumor-suppressive activities. As CRC progresses into later stages, TGF-ß switches from functioning as a tumor suppressor to a tumor promoter, and SMAD4 loss may be involved in this shift [69–71]. In CRCs and pancreatic cancers, allelic losses have a prevalence of approximately 40% to 55% [72, 73]. In normal epithelial cells and early-stage neoplasms, SMAD4 functions as a tumor suppressor [74]. When an allele is partially or completely lost, other intact allele generally compensate for the loss. In some cases, however, the mutant allele does not function normally and either directly inhibits the activity of the wild-type protein or inhibits the activity of another protein that is required for the normal function of the wild-type protein (a dominant-negative mutation) [75]. In other cases, after one functional allele is altered, the other normal allele cannot produce enough protein, a state (haploinsufficiency) that leads to disease [75].
As determined in the current study, only stage II patients with LOH of Smad4 demonstrated shorter survival. In addition, high cytoplasmic protein was associated with an increased risk of death for stage II patients, and, for stage III patients, low nuclear SMAD4 IHC expression was associated with an increased risk for cancer-specific mortality. Allelic imbalance of 18q21 at the Smad4 locus may be only a factor for early CRC progression and not a necessary cause for metastatic progression.
The present effort found only 4 somatic mutations in 53 tumors, a finding in agreement with the low number of mutations previously reported [10, 11, 16, 17, 45, 46]. Therefore, Smad4 mutational alternations appear to be involved only to a minor extent in CRC development.
Although adjuvant chemotherapy is considered efficacious for Stage III tumors, it is controversial for Stage II cancer patients [76]. Patients with stage III tumors with retained 18q who underwent adjuvant chemotherapy had a five-year, disease-free survival (DFS) advantage compared to those with LOH at 18q [77]. In a retrospective analysis of patients treated in two adjuvant trials (ECOG 2284 and INT 0035) for high-risk Stage II and Stage III colon cancer, patients with tumors containing 18q LOH were significantly associated with worse DFS [77]. However, other retrospective studies of adjuvant colon trials, CALGB 9581 and CALGB 89803, did not demonstrate this difference in survival [78]. Currently, a prospective study, ECOG 5202 (NCT00217737), is assessing the clinical utility of 18q LOH in recurrent stage II CRC. (https://clinicaltrials.gov/ct2/show/NCT00217737)
For CRCs, SMAD4 molecular alterations were predictive of failure to respond to fluoropyrimidine-based treatment [79] and of development of liver metastases [80]. Loss of SMAD4 in CRC tumors appears to induce resistance to 5FU-based therapy through regulation of a downstream apoptotic pathway and upregulation of PI3K/Akt and VEGF [14, 79, 81]. These findings were validated in other retrospective studies [81]. Thus, tumors with SMAD4 LOH may benefit from adjuvant treatment with non-5FU based regimens. Since assessing this molecular alteration to monitor the treatment in real-time may be challenging, the IHC findings of the present study will be useful in assessing Smad4 LOH as candidate predictive biomarker for therapeutic application.
In summary, LOH of Smad4 and high cytoplasmic protein in Stage II and low nuclear SMAD4 protein localization in Stage III CRCs are useful predictors of shortened patient survival.
MATERIALS AND METHODS
Patients and samples
The Institutional Review Board of the University of Alabama at Birmingham (UAB) approved this study. All samples were supplied by the Tissue Procurement Facility of the UAB Comprehensive Cancer Center. Formalin-fixed, paraffin-embedded (FFPE) archival tissue blocks were obtained for 209 CRC patients (Stage I, 34; Stage II, 70; Stage III, 64; Stage IV, 41) who had undergone surgical resection for a first primary sporadic CRC at the UAB Hospital from 1981 through 2002. None of these cases are Lynch syndrome patients and the microsatellite status (MSI) of these cases was not evaluated. Of the 209 cases, 53 had frozen tissues available (both CRCs and their corresponding normal control tissues).
The medical records of these patients were reviewed by two investigators (CKS & RH), and the surgical pathology reports were reviewed by two GI pathologists (CKS & NCJ). During the initial selection process, the following patients were excluded from the study population: those who died within a week of their surgery, those with inflammatory bowel disease, those with surgical-margin involvement or unspecified tumor location, those with multiple primary cancers within the colorectum or multiple malignancies, and those with a family or personal history of CRC. To control for treatment bias, those patients who underwent only surgery as a therapeutic intervention were included, and those who those who received any pre- or post-surgical chemotherapy were excluded.
Two GI pathologists (CKS & NCJ) independently reviewed slides (stained with hematoxylin and eosin) to determine the degree of histological differentiation of CRC tumors and, if there were discrepancies, reached a consensus. As in an earlier study [82], well and moderately differentiated CRCs were classified as “low” grade, and poorly differentiated tumors were classified as “high” grade pathological staging was performed according to the criteria of the American Joint Commission on Cancer (stages I, II, III, and IV) [83]. The International Classification of Diseases for Oncology (ICD-O) codes were used to specify anatomic locations of the tumors [84]. The anatomic sites were designated as proximal colon (cecum, ascending colon, and proximal 2/3 of the transverse colon), distal colon (distal 1/3 of the transverse colon, descending colon, and sigmoid colon), and rectum.
Patients were followed by the UAB Tumor Registry until their death or until the date of the last documented contact within the study time frame. The Tumor Registry ascertains outcome (mortality) information directly from patients (or living relatives) and from the physicians of the patients through telephone and mail contacts. This information is validated against State Death Lists. The Tumor Registry updates follow-up information every six months. Follow-up of the cohort ended in November 2008.
SMAD 4 immunohistochemical (IHC) staining
Tissue sections (5-μm) were cut from paraffin blocks 1-3 days prior to immunostaining to avoid potential problems in antigen recognition due to storage degradation of cut tissue sections on glass slides [85]. Immunostaining was performed as described in our earlier studies of antigen expression in various tissues [29, 30]. In brief, the sections were deparaffinized in xylene and rehydrated in graded alcohol. For antigen retrieval, the tissue sections in citrate buffer were boiled in a microwave oven, and the sections were treated with 3% H2O2 for 5 minutes to block endogenous peroxidase activity. After 1 hour of blocking with 3% goat serum, slides were incubated for 1 hour with 1:100 dilution of an anti-human monoclonal SMAD4 antibody (clone B-8; sc-7966; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA). The specificity of this antibody has been established [32–33]. Sections on which the primary antibody was not applied were utilized as negative controls. Secondary detection was accomplished with a multi-species detection system (Signet Lab Inc., Dedham, MA). The sections were exposed to biotinylated multispecies antibodies, including anti-mouse antibodies, for 20 minutes and then incubated with peroxidase-labeled streptavidin for 20 minutes. A diaminobenzidine tetrachloride super-sensitive substrate kit (BioGenex, San Ramon, CA) was used to visualize the antibody-antigen complexes. Each section was then counterstained with hematoxylin, dehydrated with graded alcohols, and soaked in xylene before application of coverslips.
The evaluation of staining of SMAD4 expression was as described in our earlier studies [29, 30]. To limit bias, evaluations were performed by two pathologists. In all cases, normal colon mucosa displayed positive staining, and both nuclear and cytoplasmic staining were evident. For tumor cells, the percentages of positive cells were evaluated separately for the nucleus and cytoplasm. In normal colonic epithelial tissues, strong IHC staining in both the nucleus and cytoplasm was evident. A semiquantitative immunostaining score (ISS) for SMAD4 was obtained by two pathologists who estimated the proportion of cells stained and the intensity of staining in the whole tissue section [86]. The intensity of immunostaining of individual cells was scored on a scale from 0 (no staining) to 4 (strongest intensity). Scores derived by the two investigators were combined to obtain average ISS values. The median staining value obtained for normal colon mucosa was 40%. An ISS of ≥ 0.5 and ≥ 40% percent of positive cells was chosen as a cut-off value for determining high vs. low expression of SMAD4. Based on the extent of SMAD4 staining in control tissues, the cut-off value was ≥ 0.5 for both the nucleus and cytoplasm. By use of this cut-off value and the percentage of positive cells (40%), the 209 CRC cases were separated into two groups: low protein expression of SMAD4 (< 0.5, < 40%) and high protein expression of SMAD4 (≥ 0.5, ≥ 40%). Representative pictures of SMAD4 immunostaining patterns are shown in Figure 3.
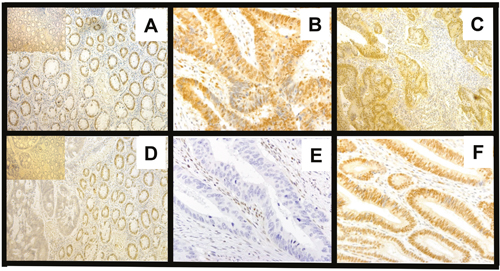
Figure 3: SMAD4 immunostaining patterns in CRCs based on Smad4 LOH status. A. Normal strong positive (200 μm); inset, control (without primary antibody) B & C. CRCs with cytoplasm and nuclei both high (600, 400 μm) D. Normal positive, tumor cytoplasm low and nuclei negative. Inset, control E. Tumor, cytoplasm and nuclei negative F. Tumor, with nuclei positive
LOH analysis at the Smad4 locus
Three polymorphic microsatellite markers (D18S363, D18S474, and D18S46), which are within 0.3 Mb of the Smad4 gene in the 18q21.1 region, were used to analyze the LOH status. A description of these markers is in Table 4. Samples of genomic DNA (~100 ng) extracted from CRCs and matching normal tissues were used as templates in the polymerase chain reaction (PCR). The PCR reaction volume (25 μl) contained 10X PCR buffer, 1 mM of each dNTP, 15 mM of MgCl2, 10 pmoles of each marker, and 0.3 μl (1.5 units) of Platinum Tag Polymerase (Invitrogen Life Technologies, Carlsbad, CA). Amplification proceeded under the following conditions: 94°C for 5 minutes, 35 cycles of 94°C for 30 seconds, 55°C for 30 seconds, and 70°C for 1 minute. The final extension was 70°C for 7 minutes. The PCR product (2 μl) was mixed with 1 μl of GeneScan-500 ROX standard (Applied Biosystems, Foster City, CA) in 12 μl of Hi-Di formamide (Applied Biosystems), denatured for 5 minutes at 88°C, and cooled on ice. The samples were subjected to capillary electrophoresis with an ABI PRISM™ 3100 Genetic Analyzer (Applied Biosystems). The results were analyzed with Genotyper 2.1 software (Applied Biosystems). Incomplete allelic losses were commonly observed because of contamination by normal cells or tumor heterogeneity. As previously described [87], one of the tumor alleles decreased by > 40% when the calculated ratio was >1.49 or < 0.5; thus, we counted this ratio value as an indication of LOH positivity. Homozygous cases were considered as non-informative for LOH.
Table 4: Three markers of LOH at the Smad4 locus
D18S46 |
F: HEX GAA TAG CAG GAC CTA TCA AAG AGC |
R: CAG ATT AAG TGA AAA CAG CAT ATG TG |
|
D18S363 |
F: 6FAM TTG GGA ACT GCT CTA CAT TC |
R: GCT TCA TTC TCT CAC TGG AT |
|
D18S474 |
F: NED TGG GGT GTT TAC CAG CAT C |
R: TGG CTT TCA ATG TCA GAA GG |
Sequences of primers for direct sequencing of Smad4 |
|
|---|---|
Smad4P1F |
ATGGACAATATGTCTATTACGAAT |
Smad4P2F |
GACAGCCATCGTTGTCCA |
Smad4P3F |
AGGTAGGAGAGACATTTA |
Smad4P4F |
TGCACCTGGAGATGCTGT |
Smad4P1R |
ACCTCAGTCTAAAGGTTGTGGGTC |
Smad4P2R |
CACCTTTGCCTATGTGCA |
Smad4P3R |
CAACTCGTTCGTAGTGAT |
Sequence of RT-PCR for expression of Smad4 mRNA |
|
|---|---|
Smad4 |
F: GTGTGAATCCATATCACTACGAACG |
R: CATACTTGATGGAGCATTACTCTGC |
|
β-Actin |
F: TAAGTAGGCGCACAGTAGGTCTGA |
R: AAGTGCAAAGAACACGGCTAAG |
|
Mutation analysis of Smad4
RNA extraction, RT-PCR, and PCR amplification
Easy Mini RNA isolation kits (Qiagen, Valencia, CA) were used to isolate RNA from frozen tissues. The cDNAs were prepared from purified RNA (~100 ng/μl) with 200 units/μl of superscript III (Invitrogen Life Technologies), 4 μl 5x RT buffer, 1 μl of 10 mM dNTP mixture, 1 μl of 50 μm oligo dT, 2 μl of 0.1M DTT, 4 μl of 25 mM MgCl2, and 40 units/μl of RNase OUT. The reverse transcription reaction was performed by incubating samples at 50°C for 50 minutes and then heating at 70°C for 15 minutes to inactivate superscript III. The RNA template from the cDNA-RNA hybrid molecule was removed by digesting with RNase H at 37°C for 20 minutes. The final volume of the cDNA reaction mixture was 25 μl. cDNA (6 μl) was used as a template to amplify the Smad4 gene by PCR. The forward primer sequence was 5’-ATGGACAATATGTCTATTACGAAT-3’, and the reverse primer sequence was 5’-ACCTCAGTCTAAAGGTTGTGGGTC-3’.
Mutational analysis by direct DNA sequencing
The resulting PCR products were sequenced by use of an ABI 3100 sequence detector with a set of primers covering the entire Smad4 coding region (exon 1 through 11; codons 1 through 552). The set included four forward primers and three reverse primers (Table 4). To detect gene mutations in Smad4, sequencing data were analyzed with DNA Star software.
Genomic DNA extraction from paraffin blocks and PCR with specific exon primers
To detect mutations, paraffin sections of tissue blocks of CRCs and their matched normal samples were examined under a microscope, and genomic DNA samples were extracted. This was followed by PCR amplification using exon-specific primers and direct DNA sequencing. The PCR forward primer sequence was 5’ –AGGCATTGGTTTTTAATGTATG-3’, and the reverse primer sequence was 5’-CTGCTCAAAGAAACTTAATCAAC-3’ (exon 10).
Statistical analyses
Characteristics of the study population were compared according to their Smad4 LOH status and Smad4 protein expression in the nucleus and cytoplasm. Chi-square statistics were used to determine the statistical significance of the observed versus expected distributions of demographic and tumor variables. Kaplan-Meier overall survival curves were plotted for LOH status and for protein expression by use of PRISM software (GraphPad Software, Inc., La Jolla, CA). Patients were stratified into phase II and phase III groups, and log-rank tests were used to determine if survival differed by levels of nuclear protein expression, cytoplasmic expression, and LOH status.
The Cox proportional hazards model was used to obtain hazard ratios (HRs) with 95% confidence intervals (CIs) for the bivariate association of Smad4 LOH, phenotypic protein expression of SMAD4, and other covariates with death due to CRC. Cox multivariate models were constructed to obtain adjusted HRs for the association of Smad4 (LOH and phenotypic expression) with cancer-specific mortality. For selection of variables, in addition to tumor stage, those variables associated with mortality at p < 0.20 in the bivariate analysis were considered potential confounders for the association of SMAD4 with mortality and were included in the initial multivariate model. To obtain the final model, the least significant variable was removed in a step-wise manner. Once the final model was obtained, the statistical significance of all two-way interactions between LOH status, nuclear expression, and tumor stage was assessed. The proportional hazards assumption was evaluated and met for Smad4 LOH and protein expression of SMAD4. Patients were again stratified by stage into four subgroups, and similar multivariable analyses were accomplished for each group.
ACKNOWLEDGMENTS
We thank Donald L. Hill, Ph.D., Department of Preventive Medicine, University of Alabama at Birmingham, for his critical review of this manuscript.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest with any of the material submitted in this manuscript.
GRANT SUPPORT
This work is supported in part by grants from the National Institute of Health/National Cancer Institute (RO1-CA98932-01 and U54-CA118948) to Dr. U. Manne.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66:7-30.
2. Fearon ER, Cho KR, Nigro JM, Kern SE, Simons JW, Ruppert JM, Hamilton SR, Preisinger AC, Thomas G, Kinzler KW and et al. Identification of a chromosome 18q gene that is altered in colorectal cancers. Science. 1990; 247:49-56.
3. Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996; 271:350-353.
4. Mitelman F, Mertens F, Johansson B. A breakpoint map of recurrent chromosomal rearrangements in human neoplasia. Nat Genet. 1997; 15 Spec No:417-474.
5. Jen J, Kim H, Piantadosi S, Liu ZF, Levitt RC, Sistonen P, Kinzler KW, Vogelstein B, Hamilton SR. Allelic loss of chromosome 18q and prognosis in colorectal cancer. N Engl J Med. 1994; 331:213-221.
6. Arai T, Akiyama Y, Yamamura A, Hosoi T, Shibata T, Saitoh K, Okabe S, Yuasa Y. Allelotype analysis of early colorectal cancers with lymph node metastasis. Int J Cancer. 1998; 79:418-423.
7. Lanza G, Matteuzzi M, Gafa R, Orvieto E, Maestri I, Santini A and del Senno L. Chromosome 18q allelic loss and prognosis in stage II, III colon cancer. Int J Cancer. 1998; 79:390-395.
8. Watanabe T, Wu TT, Catalano PJ, Ueki T, Satriano R, Haller DG, Benson AB, 3rd and Hamilton SR. Molecular predictors of survival after adjuvant chemotherapy for colon cancer. N Engl J Med. 2001; 344:1196-1206.
9. Cohn KH, Ornstein DL, Wang F, LaPaix FD, Phipps K, Edelsberg C, Zuna R, Mott LA, Dunn JL. The significance of allelic deletions and aneuploidy in colorectal carcinoma. Results of a 5-year follow-up study. Cancer. 1997; 79:233-244.
10. Koyama M, Ito M, Nagai H, Emi M, Moriyama Y. Inactivation of both alleles of the DPC4/SMAD4 gene in advanced colorectal cancers: identification of seven novel somatic mutations in tumors from Japanese patients. Mutat Res. 1999; 406:71-77.
11. Miyaki M, Iijima T, Konishi M, Sakai K, Ishii A, Yasuno M, Hishima T, Koike M, Shitara N, Iwama T, Utsunomiya J, Kuroki T, Mori T. Higher frequency of Smad4 gene mutation in human colorectal cancer with distant metastasis. Oncogene. 1999; 18:3098-3103.
12. Takaku K, Oshima M, Miyoshi H, Matsui M, Seldin MF, Taketo MM. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell. 1998; 92:645-656.
13. Mikami T, Ookawa K, Shimoyama T, Fukuda S, Saito H, Munakata A. KAI1, CAR, and Smad4 expression in the progression of colorectal tumor. J Gastroenterol. 2001; 36:465-469.
14. Alhopuro P, Alazzouzi H, Sammalkorpi H, Davalos V, Salovaara R, Hemminki A, Jarvinen H, Mecklin JP, Schwartz S Jr, Aaltonen LA, Arango D. SMAD4 levels and response to 5-fluorouracil in colorectal cancer. Clin Cancer Res. 2005; 11:6311-6316.
15. Voorneveld PW, Jacobs RJ, Kodach LL, Hardwick JC. A Meta-Analysis of SMAD4 Immunohistochemistry as a Prognostic Marker in Colorectal Cancer. Transl Oncol. 2015; 8:18-24.
16. Seshimo I, Yamamoto H, Mishima H, Kurata A, Suzuki R, Ezumi K, Takemasa I, Ikeda M, Fukushima T, Tsujinaka T, Sekimoto M, Kikkawa N, Takenoshita S, et al. Expression and mutation of SMAD4 in poorly differentiated carcinoma and signet-ring cell carcinoma of the colorectum. J Exp Clin Cancer Res. 2006; 25:433-442.
17. Alazzouzi H, Alhopuro P, Salovaara R, Sammalkorpi H, Jarvinen H, Mecklin JP, Hemminki A, Schwartz S Jr, Aaltonen LA, Arango D. SMAD4 as a prognostic marker in colorectal cancer. Clin Cancer Res. 2005; 11:2606-2611.
18. Fleming NI, Jorissen RN, Mouradov D, Christie M, Sakthianandeswaren A, Palmieri M, Day F, Li S, Tsui C, Lipton L, Desai J, Jones IT, McLaughlin S, et al. SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer research. 2013; 73:725-735.
19. De Bosscher K, Hill CS, Nicolas FJ. Molecular and functional consequences of Smad4 C-terminal missense mutations in colorectal tumour cells. Biochem J. 2004; 379:209-216.
20. Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001; 29:117-129.
21. Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000; 19:1745-1754.
22. Whitman M. Smads and early developmental signaling by the TGFbeta superfamily. Genes Dev. 1998; 12:2445-2462.
23. Riggins GJ, Thiagalingam S, Rozenblum E, Weinstein CL, Kern SE, Hamilton SR, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B. Mad-related genes in the human. Nat Genet. 1996; 13:347-349.
24. Maitra A, Molberg K, Albores-Saavedra J, Lindberg G. Loss of Dpc4 expression in colonic adenocarcinomas correlates with the presence of metastatic disease. Am J Pathol. 2000; 157:1105-1111.
25. Xie W, Rimm DL, Lin Y, Shih WJ, Reiss M. Loss of Smad signaling in human colorectal cancer is associated with advanced disease and poor prognosis. Cancer J. 2003; 9:302-312.
26. Reinacher-Schick A, Baldus SE, Romdhana B, Landsberg S, Zapatka M, Monig SP, Holscher AH, Dienes HP, Schmiegel W, Schwarte-Waldhoff I. Loss of Smad4 correlates with loss of the invasion suppressor E-cadherin in advanced colorectal carcinomas. J Pathol. 2004; 202:412-420.
27. Isaksson-Mettavainio M, Palmqvist R, Forssell J, Stenling R, Oberg A. SMAD4/DPC4 expression and prognosis in human colorectal cancer. Anticancer Res. 2006; 26:507-510.
28. Kouvidou C, Latoufis C, Lianou E, Kouvatseas G, Kakouri E, Anagnostakis D, Vrettou-Aravani V, Betsi E, Karatapanis S. Expression of Smad4 and TGF-beta2 in colorectal carcinoma. Anticancer Res. 2006; 26:2901-2907.
29. Ogunbiyi OA, Goodfellow PJ, Herfarth K, Gagliardi G, Swanson PE, Birnbaum EH, Read TE, Fleshman JW, Kodner IJ, Moley JF. Confirmation that chromosome 18q allelic loss in colon cancer is a prognostic indicator. J Clin Oncol. 1998; 16:427-433.
30. Tanaka T, Watanabe T, Kazama Y, Tanaka J, Kanazawa T, Kazama S, Nagawa H. Chromosome 18q deletion and Smad4 protein inactivation correlate with liver metastasis: A study matched for T- and N- classification. Br J Cancer. 2006; 95:1562-1567.
31. Tanaka T, Watanabe T, Kazama Y, Tanaka J, Kanazawa T, Kazama S, Nagawa H. Loss of Smad4 protein expression and 18qLOH as molecular markers indicating lymph node metastasis in colorectal cancer--a study matched for tumor depth and pathology. J Surg Oncol. 2008; 97:69-73.
32. Howe JR, Roth S, Ringold JC, Summers RW, Jarvinen HJ, Sistonen P, Tomlinson IP, Houlston RS, Bevan S, Mitros FA, Stone EM, Aaltonen LA. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science. 1998; 280:1086-1088.
33. Hahn SA, Hoque AT, Moskaluk CA, da Costa LT, Schutte M, Rozenblum E, Seymour AB, Weinstein CL, Yeo CJ, Hruban RH, Kern SE. Homozygous deletion map at 18q21.1 in pancreatic cancer. Cancer Res. 1996; 56:490-494.
34. Hruban RH, Offerhaus GJ, Kern SE, Goggins M, Wilentz RE, Yeo CJ. Tumor-suppressor genes in pancreatic cancer. J Hepatobiliary Pancreat Surg. 1998; 5:383-391.
35. Riggins GJ, Kinzler KW, Vogelstein B, Thiagalingam S. Frequency of Smad gene mutations in human cancers. Cancer Res. 1997; 57:2578-2580.
36. Powell SM, Harper JC, Hamilton SR, Robinson CR, Cummings OW. Inactivation of Smad4 in gastric carcinomas. Cancer Res. 1997; 57:4221-4224.
37. Zhou Y, Kato H, Shan D, Minami R, Kitazawa S, Matsuda T, Arima T, Barrett JC, Wake N. Involvement of mutations in the DPC4 promoter in endometrial carcinoma development. Mol Carcinog. 1999; 25:64-72.
38. Yakicier MC, Irmak MB, Romano A, Kew M, Ozturk M. Smad2 and Smad4 gene mutations in hepatocellular carcinoma. Oncogene. 1999; 18:4879-4883.
39. Salovaara R, Roth S, Loukola A, Launonen V, Sistonen P, Avizienyte E, Kristo P, Jarvinen H, Souchelnytskyi S, Sarlomo-Rikala M, Aaltonen LA. Frequent loss of SMAD4/DPC4 protein in colorectal cancers. Gut. 2002; 51:56-59.
40. Royce SG, Alsop K, Haydon A, Mead L, Smith LD, Tesoriero AA, Giles GG, Jenkins MA, Hopper JL, Southey MC. The role of SMAD4 in early-onset colorectal cancer. Colorectal Dis. 2010; 12:213-219.
41. Bacman D, Merkel S, Croner R, Papadopoulos T, Brueckl W, Dimmler A. TGF-beta receptor 2 downregulation in tumour-associated stroma worsens prognosis and high-grade tumours show more tumour-associated macrophages and lower TGF-beta1 expression in colon carcinoma: a retrospective study. BMC Cancer. 2007; 7:156.
42. Isaksson-Mettavainio M, Palmqvist R, Dahlin AM, Van Guelpen B, Rutegard J, Oberg A, Henriksson ML. High SMAD4 levels appear in microsatellite instability and hypermethylated colon cancers, and indicate a better prognosis. Int J Cancer. 2012; 131:779-788.
43. Roth AD, Delorenzi M, Tejpar S, Yan P, Klingbiel D, Fiocca R, d'Ario G, Cisar L, Labianca R, Cunningham D, Nordlinger B, Bosman F, Van Cutsem E. Integrated analysis of molecular and clinical prognostic factors in stage II/III colon cancer. J Natl Cancer Inst. 2012; 104:1635-1646.
44. Korchynskyi O, Landstrom M, Stoika R, Funa K, Heldin CH, ten Dijke P, Souchelnytskyi S. Expression of Smad proteins in human colorectal cancer. International journal of cancer. 1999; 82:197-202.
45. Takagi Y, Kohmura H, Futamura M, Kida H, Tanemura H, Shimokawa K, Saji S. Somatic alterations of the DPC4 gene in human colorectal cancers in vivo. Gastroenterology. 1996; 111:1369-1372.
46. Thiagalingam S, Lengauer C, Leach FS, Schutte M, Hahn SA, Overhauser J, Willson JK, Markowitz S, Hamilton SR, Kern SE, Kinzler KW, Vogelstein B. Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat Genet. 1996; 13:343-346.
47. MacGrogan D, Pegram M, Slamon D, Bookstein R. Comparative mutational analysis of DPC4 (Smad4) in prostatic and colorectal carcinomas. Oncogene. 1997; 15:1111-1114.
48. Schutte M, Hruban RH, Hedrick L, Cho KR, Nadasdy GM, Weinstein CL, Bova GS, Isaacs WB, Cairns P, Nawroz H, Sidransky D, Casero RA Jr, Meltzer PS, et al. DPC4 gene in various tumor types. Cancer Res. 1996; 56:2527-2530.
49. Tarafa G, Villanueva A, Farre L, Rodriguez J, Musulen E, Reyes G, Seminago R, Olmedo E, Paules AB, Peinado MA, Bachs O, Capella G. DCC and SMAD4 alterations in human colorectal and pancreatic tumor dissemination. Oncogene. 2000; 19:546-555.
50. Ahn BK, Jang SH, Paik SS, Lee KH. Smad4 may help to identify a subset of colorectal cancer patients with early recurrence after curative therapy. Hepatogastroenterology. 2011; 58:1933-1936.
51. Voorneveld PW, Jacobs RJ, De Miranda NF, Morreau H, van Noesel CJ, Offerhaus GJ, Kodach LL, Hardwick JC. Evaluation of the prognostic value of pSMAD immunohistochemistry in colorectal cancer. Eur J Cancer Prev. 2013.
52. Wang CY, Eshleman JR, Willson JK, Markowitz S. Both transforming growth factor-beta and substrate release are inducers of apoptosis in a human colon adenoma cell line. Cancer Res. 1995; 55:5101-5105.
53. Markowitz SD, Myeroff L, Cooper MJ, Traicoff J, Kochera M, Lutterbaugh J, Swiriduk M, Willson JK. A benign cultured colon adenoma bears three genetically altered colon cancer oncogenes, but progresses to tumorigenicity and transforming growth factor-beta independence without inactivating the p53 tumor suppressor gene. J Clin Invest. 1994; 93:1005-1013.
54. Markowitz S. TGF-beta receptors and DNA repair genes, coupled targets in a pathway of human colon carcinogenesis. Biochimica et biophysica acta. 2000; 1470:M13-20.
55. Pierreux CE, Nicolas FJ, Hill CS. Transforming growth factor beta-independent shuttling of Smad4 between the cytoplasm and nucleus. Mol Cell Biol. 2000; 20:9041-9054.
56. Fink SP, Mikkola D, Willson JK, Markowitz S. TGF-beta-induced nuclear localization of Smad2 and Smad3 in Smad4 null cancer cell lines. Oncogene. 2003; 22:1317-1323.
57. Muller N, Reinacher-Schick A, Baldus S, van Hengel J, Berx G, Baar A, van Roy F, Schmiegel W, Schwarte-Waldhoff I. Smad4 induces the tumor suppressor E-cadherin and P-cadherin in colon carcinoma cells. Oncogene. 2002; 21:6049-6058.
58. Zhao S, Ammanamanchi S, Brattain M, Cao L, Thangasamy A, Wang J, Freeman JW. Smad4-dependent TGF-beta signaling suppresses RON receptor tyrosine kinase-dependent motility and invasion of pancreatic cancer cells. The Journal of biological chemistry. 2008; 283:11293-11301.
59. Feng XH, Lin X, Derynck R. Smad2, Smad3 and Smad4 cooperate with Sp1 to induce p15(Ink4B) transcription in response to TGF-beta. Embo J. 2000; 19:5178-5193.
60. Pardali K, Kurisaki A, Moren A, ten Dijke P, Kardassis D, Moustakas A. Role of Smad proteins and transcription factor Sp1 in p21(Waf1/Cip1) regulation by transforming growth factor-beta. The Journal of biological chemistry. 2000; 275:29244-29256.
61. Subramanian G, Schwarz RE, Higgins L, McEnroe G, Chakravarty S, Dugar S, Reiss M. Targeting endogenous transforming growth factor beta receptor signaling in SMAD4-deficient human pancreatic carcinoma cells inhibits their invasive phenotype1. Cancer Res. 2004; 64:5200-5211.
62. Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988; 319:525-532.
63. Carethers JM, Hawn MT, Greenson JK, Hitchcock CL, Boland CR. Prognostic significance of allelic lost at chromosome 18q21 for stage II colorectal cancer. Gastroenterology. 1998; 114:1188-1195.
64. Martinez-Lopez E, Abad A, Font A, Monzo M, Ojanguren I, Pifarre A, Sanchez JJ, Martin C, Rosell R. Allelic loss on chromosome 18q as a prognostic marker in stage II colorectal cancer. Gastroenterology. 1998; 114:1180-1187.
65. Jernvall P, Makinen MJ, Karttunen TJ, Makela J, Vihko P. Loss of heterozygosity at 18q21 is indicative of recurrence and therefore poor prognosis in a subset of colorectal cancers. Br J Cancer. 1999; 79:903-908.
66. Chan TL, Curtis LC, Leung SY, Farrington SM, Ho JW, Chan AS, Lam PW, Tse CW, Dunlop MG, Wyllie AH, Yuen ST. Early-onset colorectal cancer with stable microsatellite DNA and near-diploid chromosomes. Oncogene. 2001; 20:4871-4876.
67. Laurent-Puig P, Olschwang S, Delattre O, Remvikos Y, Asselain B, Melot T, Validire P, Muleris M, Girodet J, Salmon RJ and et al. Survival and acquired genetic alterations in colorectal cancer. Gastroenterology. 1992; 102:1136-1141.
68. Diep CB, Thorstensen L, Meling GI, Skovlund E, Rognum TO, Lothe RA. Genetic tumor markers with prognostic impact in Dukes' stages B and C colorectal cancer patients. J Clin Oncol. 2003; 21:820-829.
69. Bierie B, Moses HL. TGF-beta and cancer. Cytokine & growth factor reviews. 2006; 17:29-40.
70. Elliott RL, Blobe GC. Role of transforming growth factor Beta in human cancer. J Clin Oncol. 2005; 23:2078-2093.
71. Zhang B, Halder SK, Kashikar ND, Cho YJ, Datta A, Gorden DL, Datta PK. Antimetastatic role of Smad4 signaling in colorectal cancer. Gastroenterology. 2010; 138:969-980 e961-963.
72. Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008; 3:157-188.
73. Popat S, Zhao D, Chen Z, Pan H, Shao Y, Chandler I, Houlston RS. Relationship between chromosome 18q status and colorectal cancer prognosis: a prospective, blinded analysis of 280 patients. Anticancer Res. 2007; 27:627-633.
74. Tang Y, Katuri V, Srinivasan R, Fogt F, Redman R, Anand G, Said A, Fishbein T, Zasloff M, Reddy EP, Mishra B, Mishra L. Transforming growth factor-beta suppresses nonmetastatic colon cancer through Smad4 and adaptor protein ELF at an early stage of tumorigenesis. Cancer research. 2005; 65:4228-4237.
75. Yang G, Yang X. Smad4-mediated TGF-beta signaling in tumorigenesis. Int J Biol Sci. 2010; 6:1-8.
76. Dotan E, Cohen SJ. Challenges in the management of stage II colon cancer. Semin Oncol. 2011; 38:511-520.
77. Watanabe T, Wu T-T, Catalano PJ, Ueki T, Satriano R, Haller DG, Benson AB, Hamilton SR. Molecular Predictors of Survival after Adjuvant Chemotherapy for Colon Cancer. New England Journal of Medicine. 2001; 344:1196-1206.
78. Bertagnolli MM, Redston M, Compton CC, Niedzwiecki D, Mayer RJ, Goldberg RM, Colacchio TA, Saltz LB, Warren RS. Microsatellite instability and loss of heterozygosity at chromosomal location 18q: prospective evaluation of biomarkers for stages II and III colon cancer--a study of CALGB 9581 and 89803. J Clin Oncol. 2011; 29:3153-3162.
79. Zhang B, Zhang B, Chen X, Bae S, Singh K, Washington MK, Datta PK. Loss of Smad4 in colorectal cancer induces resistance to 5-fluorouracil through activating Akt pathway. Br J Cancer. 2014; 110:946-957.
80. Tanaka T, Watanabe T, Kitayama J, Kanazawa T, Kazama Y, Tanaka J, Kazama S, Nagawa H. Chromosome 18q deletion as a novel molecular predictor for colorectal cancer with simultaneous hepatic metastasis. Diagnostic molecular pathology. 2009; 18:219-225.
81. Kozak MM, von Eyben R, Pai J, Vossler SR, Limaye M, Jayachandran P, Anderson EM, Shaffer JL, Longacre T, Pai RK, Koong AC, Chang DT. Smad4 inactivation predicts for worse prognosis and response to fluorouracil-based treatment in colorectal cancer. J Clin Pathol. 2015; 68:341-345.
82. Compton C, Fenoglio-Preiser CM, Pettigrew N, Fielding LP. American Joint Committee on Cancer Prognostic Factors Consensus Conference: Colorectal Working Group. Cancer. 2000; 88:1739-1757.
83. Greene F, Balch C, Page D, Haller D, Fleming I, Morrow M, Fritz A. (2002). AJCC Cancer Staging Manual. (New York: Springer).
84. National Cancer Institute (U.S.). (2001). ICD-O International Classification of Diseases for Oncology, Third Edition, 2000.
85. Jacobs TW, Prioleau JE, Stillman IE, Schnitt SJ. Loss of tumor marker-immunostaining intensity on stored paraffin slides of breast cancer. J Natl Cancer Inst. 1996; 88:1054-1059.
86. Manne U, Myers RB, Moron C, Poczatek RB, Dillard S, Weiss H, Brown D, Srivastava S, Grizzle WE. Prognostic significance of Bcl-2 expression and p53 nuclear accumulation in colorectal adenocarcinoma. Int J Cancer. 1997; 74:346-358.
87. Demopoulos K, Arvanitis DA, Vassilakis DA, Siafakas NM, Spandidos DA. MYCL1, FHIT, SPARC, p16(INK4) and TP53 genes associated to lung cancer in idiopathic pulmonary fibrosis. J Cell Mol Med. 2002; 6:215-222.