
Background
Diabetes mellitus (DM) and cancer are among the most frequent causes of death worldwide. According to Global Burden of Disease data, from 1990 to 2013 mortality from DM increased by 90% [1]. Colorectal cancer (CRC) is among the top causes of cancer death. From 1990 to 2013 global deaths from CRC increased by 57%, [1]. In the United States, CRC is the second leading cause of cancer death in men and women combined (http://www.ccalliance.org/colorectal_cancer/statistics.html) [2]. A link between DM and cancer is now recognized in American Diabetes Association (ADA) guidelines, following a 2010 consensus report [3, 4]. If the association holds, the current worldwide diabetes epidemic, fueled by life-style changes, may trigger a wave of CRC diagnoses. However, this knowledge has had limited impact on clinical care in the form of specific diagnostic tests or therapeutic approaches supported by clinical guidelines. Furthermore, on a worldwide basis the prevalence of DM and the incidence of CRC are not correlated, suggesting that country-specific factors may play a role in the association between DM and CRC (Figure 1). Annual CRC incidence rates vary more than ten-fold worldwide, the highest rates being in developed countries such as Korea (age-standardized rate 45 per 100, 000), Australia and Ireland, and the lowest in Western Africa (e.g. Cameroon 3.3 per 100, 000) (http://globocan.iarc.fr). By contrast, DM prevalence is highest in Egypt and United Arab Emirates (20, 000 per 100, 000, and lowest in Australia (5, 100), Ireland (4, 400) and Western Africa (www.diabetesatlas.org/). A better understanding of the factors underlying regional differences may provide clues to the relationship between DM and CRC. We now review the epidemiological evidence, potential pathophysiological mechanisms and therapeutic implications of the association between DM and CRC and propose a research agenda that may impact clinical practice to prevent or treat CRC in DM patients. A Pubmed search with the key words “(diabetes OR insulin OR hyperglycemia) AND (colon OR colorectal) AND cancer” was performed with no time cut-off points and further references added from the reference list of the publications found or based on the authors own experience knowledge.
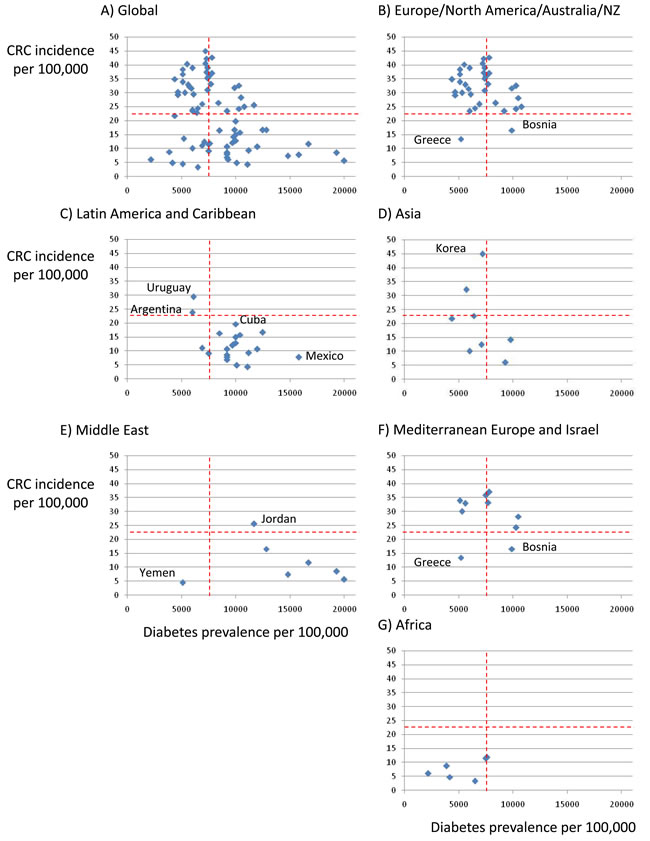
Figure 1: Relationship between incidence of colorectal cancer (CRC) and prevalence of DM in different parts of the world. A. Global, B. Europe, North America and Australia/New Zealand, C. Latin America and Caribbean, D. Asia, E. Middle East, F. European Mediterranean countries and Israel, G. Africa. IDF 2015 data for DM (www.diabetesatlas.org/) and Globocan 2012 data for colorectal cancer (http://globocan.iarc.fr/Pages/age-specific_table_sel.aspx). Discontinuous red lines represent median values for the global population. Regional differences can be identified by the location of countries within the four quadrants. Note regional differences as well as countries that differ from others in the region. Regions are more clearly separated by CRC incidence than by DM prevalence, Europe/North America/Australia/NZ is the only high DM/high CRC region. Latin America and Caribbean is a high DM/low CRC region with the exception of Argentina and Uruguay where meat intake is high, while in the opposite extreme Mexico a is very high DM/low CRC country. In the Middle East a high prevalence of DM is not associated with high CRC incidence, unlike in European Mediterranean countries which in general behave as the rest of Europe. Korea is an example of low DM/high CRC country in Asia.
Diabetes mellitus
DM is characterized by hyperglycemia resulting from defects in insulin secretion and/or insulin action. Chronic hyperglycemia is associated with injury to the kidneys, heart, nerves, eyes and blood vessels [5]. In type 1 DM (T1DM, 5-10% of DM cases), cell-mediated autoimmune destruction of pancreatic β-cells causes absolute insulin deficiency. Type 2 DM (T2DM) is characterized by insulin resistance and relative insulin deficiency. T2DM patients are frequently obese and older at DM onset than T1DM patients [6]. Obesity promotes insulin-resistance and is thought to be a major driver of the current DM epidemic. Mendelian-inherited genetic defects of β-cells or of the insulin signaling machinery also cause DM [5].
Mean age at diagnosis of DM is 54 years in the US (http://www.cdc.gov/diabetes/statistics/age/). Therapies for DM increase insulin availability (insulin or insulin analog administration or agents that promote insulin secretion), improve sens itivity to insulin, decrease glucose synthesis, delay the gut absorption of carbohydrate, or increase urinary glucose excretion (Supplementary Table 1). The preferred initial and most widely used pharmacological agent for T2DM is metformin, which decreases glucose production by inhibiting the mitochondrial glycerophosphate dehydrogenase (GPDH, GPD2) [7]. If adequate glucose control is not achieved within 3-6 months, a second oral agent, a Glucagon-like peptide-1 (GLP-1) receptor agonist, or insulin should be added [8, 9].
Colorectal cancer
CRC originates from colon epithelium [10]. Over 70% CRCs are sporadic, resulting from dietary and environmental factors. The incidence increases with age and they usually occur over the age of 50 years. True inheritable CRC (<10% of cases) may be associated or not to colonic polyps (Table 1) [11]. The familial type (25% of cases) is associated with a family history of CRC or large adenomas, in the absence of classic Mendelian inheritance [12]. Right- and left-sided CRCs exhibit different epidemiological patterns, sensitivities to chemotherapy and outcomes, probably related to different molecular characteristics and chromosomal instability with left-sided tumors [13].
Table 1: Genetics of colorectal cancer and potential impact of DM on colorectal cancer-related genes
Colorectal cancer |
Mutation |
Inheritance |
Impact of DM on gene expression * |
Reference |
Familial adenomatous polyposis |
Inactivating germline mutation in adenomatous polyposis coli (APC) |
Autosomal dominant |
Increased APC |
|
MUTYH-associated polyposis |
Inactivating germline mutation in MUTYH |
Autosomal recessive |
Unchanged MUTYH |
|
Peutz-Jeghers syndrome |
Inactivating germline mutation in serine threonine kinase 11 (STK11) |
Autosomal dominant |
Increased STK11 |
[285] |
Hereditary non-polyposis colorectal cancer (Lynch syndrome) |
Inactivating germline mutation in MLH1, MSH2, MSH6, or PMS2 |
Autosomal dominant |
Unchanged MLH1, PMS2 Increased MSH2, MSH6 |
[286] |
Chromosomal instability (frequent) |
Acquired accumulation of numerical (aneuploidy) or structural chromosomal abnormalities and mutations in specific oncogenes and tumor suppressor genes (e.g. APC, PIK3CA, SMAD4, KRAS, TP53, BRAF) |
Unchanged PIK3CA, SMAD4, BRAF Increased KRAS, TP53 |
* Kidney gene expression in human diabetic kidney disease transcriptomics (http://www.nephromine.org).
CRC is initiated by mutations in tumor suppressor genes (adenomatous polyposis coli or APC, CTNNB1, p53) and oncogenes (KRAS). Accumulation of multiple mutations leads to a selective growth advantage for transformed epithelial cells that is modulated by epigenetic changes [14, 15]. Diet, the microbiota and the inflammatory response to the microbiota are potential players [16-22]. Indeed, chronic gut inflammation (e.g. ulcerative colitis or Crohn´s disease) is associated with increased incidence of colon cancer. A major molecular pathway is Wnt signaling activation of the transcription factor β-catenin to promote expression of cell proliferation genes. Loss-of-function mutations or epigenetic silencing of APC leads to aberrant β-catenin accumulation and uncontrolled cell proliferation. The normal APC protein forms a complex with glycogen synthase kinase 3-beta (GSK-3β) that allows GSK-3β to phosphorylate β-catenin, targeting it for ubiquitination and proteasomal degradation, thus decreasing β-catenin-dependent transcriptional events [23].
Early-stage CRC is treated with surgery and locally advanced CRC (radically resected stage III and ‘high-risk’ stage II disease) with adjuvant chemotherapy on top of surgery. Rectal cancer with nodal disease standard treatment includes neoadjuvant chemo-radiation [24]. Adjuvant chemotherapy schemes contain 5-fluorouracil and oxaliplatin. Metastatic CRC is treated with irinotecan or oxaliplatin combined with a fluoropyrimidine and leucovorin (FOLFIRI or FOLFOX regimens) [25]. Addition of targeted therapies over the past 10 years has improved overall survival. Testing for KRAS, NRAS, BRAF, PIK3CA and PTEN mutations is used to assess the potential clinical benefit of anti-Epidermal Growth Factor Receptor (anti-EGFR) and panitumumab treatment. Meta-analyses suggest that mutation testing for KRAS exon 2 is the strongest biomarker of response. The addition of anti-Vascular Endothelial Growth Factor (anti-VEGF) agents (bevacizumab, regorafenib) to chemotherapy of metastatic CRC prolongs progression-free and overall survival in first- and second line therapy [26].
Table 2: Key risk factors for T2DM, colorectal cancer and DM complications (Diabetic kidney disease)
Risk factor |
T2DM |
Colorectal cancer |
Diabetic kidney disease |
Race |
African American, Native American |
African American |
African American, Native American |
Obesity |
Yes |
Yes |
Yes |
Inflammation |
Yes |
Yes |
Yes |
Microbiota |
Yes |
Yes |
Unknown |
Low vitamin D |
Yes |
Yes |
Yes |
High protein (meat protein) diet |
Yes |
Yes |
Yes |
Low fiber diet |
Yes |
Yes |
ND |
No Mediterranean diet |
Yes |
Yes |
ND |
Low magnesium intake/hypomagnesemia |
Yes |
Yes |
Yes |
Angiotensin II |
Yes |
Yes |
Yes |
Age |
Yes |
Yes |
Unclear |
Epidemiological association between diabetes and CRC
Epidemiological studies suggest that DM, especially T2DM, is associated with increased risk of cancer at several sites, including CRC [27] (Table 3). The first prospective association was reported in 1998 in US participants followed from 1960 to 1972 [28]. The adjusted incidence density ratio of CRC was 1.30 (95% confidence interval (CI) 1.03-1.65) for diabetic males, but not significant for females. The association was found only among non-smoker males. A more recent prospective US study followed an older cohort from 1995 to 2004 and observed an increased adjusted Hazard Ratio (HR) for CRC in both males and females [29]. Lifestyle changes from the 60s to the 90s may explain the change in female risk. A similar association has been reported in Japan [30], China [31], Australia [32] or certain European countries (e.g. Sweden) [33], among others. A recent umbrella review of meta-analyses of observational studies on T2DM and cancer updated to the end of 2013 concluded that CRC was one of only four cancer sites associated to T2DM with robust supporting evidence and without hints of bias [34]. Furthermore, in a meta-analysis of prospective cohort studies encompassing near a million participants, prediabetes (impaired fasting glucose and/or impaired glucose tolerance) was also associated with increased risk of CRC [35]. However, uncertainties remain. The presence of detection bias and/or reverse causation has been suggested by studies in Australia, Israel and the Netherlands that found a higher risk of cancer within 3 months of a DM diagnosis [32, 36, 37]. In this regard, in the US, respondents with diabetes were 22% more likely to be up-to-date on CRC screening than those without diabetes [38]. A higher risk of developing DM within 5 years of CRC diagnosis was also reported [39]. In addition, regional differences exist: in Norway and the Netherlands only diabetic females had a higher incidence of proximal colon cancer or CRC [40, 41], while no association was found in Tyrol. Unraveling the reasons underlying regional differences may provide clues to the association and to public health interventions. Potential differences in the use of specific antidiabetic drugs may play a role as discussed below. Furthermore, epidemiological data from developing countries are scarce. This is an important piece of missing information since almost 55% of CRC cases occur in more developed regions (http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx), while 80% of DM patients live in low- and middle-income countries (www.diabetesatlas.org/).
Table 3: Epidemiological association between DM and risk of CRC.
Country |
N (x1000) |
Mean age (years) |
Period (years) |
Location |
Males |
Females |
Overall |
Ref |
US* |
850 |
54 |
59-72 |
CRC |
1.30 (1.03-1.65) |
1.16 (0.87-1.53) |
Not available |
[28] |
US** |
484 |
62 |
95-06 |
CRC |
Colon 1.24 (1.12-1.38) Rectum 1.34 (1.14-1.57) |
Colon 1.37 (1.16-1.60) Rectum 1.43 (1.08-1.88) |
Colon 1.27 (1.17-1.39) Rectum 1.36 (1.18-1.56) |
[29] |
Japan*** |
335 |
N.A. |
N.A. |
CRC |
N.A. |
N.A. |
1.40 (1.19-1.64) |
[30] |
China**** |
327 |
60 |
07-13 |
CRC |
Colon 1.47 (1.29-1.67) Rectum 1.25 (1.09-1.43) |
Colon 1.33 (1.15-1.54) Rectum 1.29 (1.10-1.51) |
Colon 1.40 (1.27-1.55) Rectum1.26 (1.14-1.40) |
[31] |
Australia**** |
953 |
27 (T1DN) 60 (T2DN) |
97-08 |
CRC |
1.18 (1.15-1.21) |
1.16 (1.13-1.20) |
N.A. |
[32] |
Sweden**** |
2.9 1.4 |
N.A. |
64-10 |
CRC |
N.A. |
N.A. |
Colon 1.33 (1.28-1.38) Rectum 1.19 (1.13-1.25) |
[33] |
Norway*** |
751 pers/year |
71 |
84-96 |
CRC |
CRC 0.66 (0.35-1.34) |
CRC 1.55 (1.04-2.31) Colon 1.60 (1.02-2.51) Rectum 2.70 (1.29-5.61) |
N.A. |
[40] |
Tyrol**** |
5.7 |
58 |
88-10 |
CRC |
1.11 (0.81-1.49) |
0.94 (0.62-1.36) |
N.A. |
[290] |
Israel** |
2186 |
64 |
02-12 |
CRC |
1.45 (1.37-1.55) |
1.48 (1.39-1.57) |
N.A. |
[36] |
Netherlands** |
120 |
62 |
86-06 |
CRC |
CRC 0.95 (0.75–1.20) Proximal 1.13 (0.76-1.68) Distal 0.77 (0.49–1.21) Rectum 0.50 (0.21–1.22) |
CRC 1.08 (0.85–1.37) Proximal 1.44 (1.05–1.99) Distal 0.75 (0.44–1.27) Rectum 1.16 (0.54–2.48) |
N.A. |
[41] |
Meta-analysis*** |
8244 |
N.A. |
N.A. |
CRC |
N.A. |
N.A. |
1.27 (1.21-1.34) |
[34] |
95% confidence interval shown. * Adjusted incidence density ratio: ** Adjusted HR; ***RR. **** Standardized incidence ratios
N.A.: not available
Risk factors shared by CRC, DM, and DM target organ damage may be confounders in epidemiological studies (Table 2). Obesity is a major risk factor for T2DM, cancer and diabetic kidney disease (DKD) [42, 43]. However, key studies observing an association between DM and CRC were adjusted by BMI. In this regard, there may be a relationship between obesity, insulin resistance and CRC. In a prospective European study, lower CRC risk was observed for metabolically healthy/overweight individuals compared with metabolically unhealthy/overweight individuals, defined as individuals with higher C-peptide levels indicative of hyperinsulinaemia [44]. Diet may be another confounder. A high meat intake increases and a Mediterranean diet decreases both the risk of DM and of CRC [45].
Potential molecular mechanisms of the association between DM and CRC
The association between DM and CRC may result from shared risk factors between T2DM and cancer but epidemiological data suggest a potential contribution of hyperinsulinemia, hyperglycemia or DM therapy [4, 46, 47] (Figure 2). Additionally, the DM microenvironment, such as advanced glycation end-products (AGEs), hyperlipidemia, local inflammation/oxidative stress, extracellular matrix alterations, and altered microbiota or ischemia due to vasculopathy may recruit secondary mediators of injury that may favor the development of both cancer and other complications of DM such as DKD.
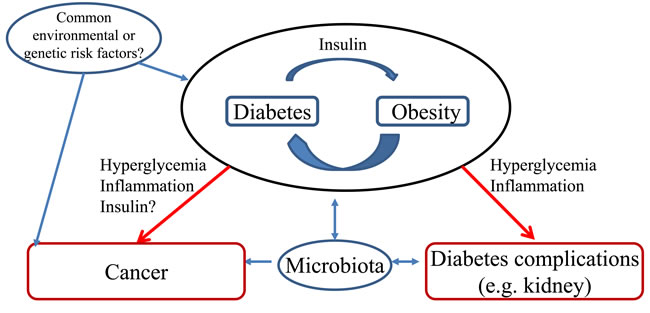
Figure 2: Hypotheses potentially explaining the association between diabetes and colorectal cancer. Two major potential relationships have been depicted. A. Common risk factors (e.g. diet, genetic) favor both diabetes and colorectal cancer; B. Diabetes favors cancer development. These potential relationships are put in context with the occurrence of other diabetes complications such a chronic kidney disease. Obesity is a known risk factor for both colorectal cancer and diabetic kidney disease.
Insulin
Insulin and insulin-like growth factor (IGF)-1 have growth factor and antiapoptotic properties in a variety of cultured tumor and non-tumor cell types, including normal colon epithelium and colon cancer cells [48, 49]. These actions have been interpreted as part of a putative tumor-enhancing effect of insulin [50, 51]. However, insulin signaling is also required for survival and function of healthy cells in vivo, such as podocytes, key cells in DKD, and selective podocyte insulin resistance reproduces features of DKD in the absence of hyperglycemia [52]. The mTOR and p21-activated protein kinase-1 (PAK-1)/Wnt/β-catenin intracellular pathways are involved in insulin-stimulated proto-oncogene expression in intestinal cells [53]. These molecular pathways also mediate diabetic complications, including DKD [54].
An increased incidence of azoxymethane-induced intestinal tract cancer was observed in preclinical models of obesity and T2DM, including obese Zucker rats and KK Ay, db/db and ob/ob mice [55-57]. The addition, the incidence and multiplicity of intestinal adenomas was higher in db/db mice with Apc mutations than in non-diabetic mice [58]. However, the relative role of hyperglycemia, hyperinsulinemia or obesity was not characterized.
The role of hyperinsulinemia was studied in a normoglycemic model of mammary cancer growth, but results do not necessarily extrapolate to CRC [59]. A tyrosine kinase inhibitor specific to the insulin and IGF-1 receptors aggravated hyperinsulinemia but prevented insulin signaling and cancer growth. However, tyrosine kinase inhibitors are promiscuous and are in clinical use as anti-tumor agents. Thus, the fact that members of an anti-tumor agent family decrease tumor growth is not definitive evidence for a role of insulin. CL-316243, a β3-adrenergic receptor agonist that sensitizes to insulin action, reduced hyperinsulinemia and phosphorylation of insulin and IGF-1 receptors and attenuated mammary tumor progression, supporting a role for hyperinsulinemia in T2DM associated tumor progression [60].
Hyperglycemia
Hyperglycemia has been implicated both in colon cancer growth and in DKD and some of the molecular mechanisms are shared by both diseases. High glucose levels and AGEs increase proliferation and migration of cultured colon cancer cells [61, 62]. High glucose levels also enhance resistance to 5-fluoruracil-induced apoptosis [63]. AGE-induced CRC cell proliferation requires carbohydrate response element-binding protein (ChREBP) [64], a key transcription factor also involved in DKD [65]. The polyol and hexosamine pathways, which increase glucose oxidation, are upregulated in diabetes target organ epithelial cells [54] and in colon cancer [66]. Hyperglycemia and AGEs induce oxidative stress and inflammation, which can damage cellular components and contribute to malignant cell transformation [67-69]. High glucose-induced oxidative stress plays a pivotal role in the development of diabetes complications by activating different pathways, such as the transcription factor nuclear factor-kappa B (NF-κB) [70, 71]. Indeed, bardoxolone methyl, a potent nuclear factor erythroid 2-related factor 2 (Nrf2) activator/NF-κB inhibitor, improved glomerular filtration in RCT in DKD [72]. Interestingly, the observation that bardoxolone increased glomerular filtration was first made in clinical trials exploring its anticancer potential.
The Warburg effect refers to the high glucose uptake and metabolism of glucose through glycolysis rather than aerobic phosphorylation in tumor cells despite the presence of oxygen [73, 74]. Glycolysis is less efficient but generates adenosine triphosphate (ATP) faster, conferring a growth advantage to tumor cells. Upregulation of insulin-independent glucose transporters such as glucotransporter-1 (Glut-1) favors glucose uptake by cancer cells [75, 76]. Glut overexpression is usually translated into higher proliferation rates. The diabetic milieu and transforming growth factor (TGF)-β1 upregulate renal cell Glut-1 and this is thought to contribute to the pathogenesis of DKD [77].
Few preclinical studies have addressed the impact of hyperglycemia per se (i.e. T1DM) on colon cancer. Streptozotocin-induced hyperglycemia, an insulin-deficiency DM model, increased liver metastasis of mouse colon cancer cells, while glycemic control with either insulin or gliclazide was protective [78]. These studies suggest that hyperglycemia per se may favor colorectal tumor growth and that hyperglycemia may be a more powerful stimulus for tumorigenesis than insulin in experimental animals.
Wnt/β-catenin is activated in CRC as a direct consequence of APC mutations and in kidney cells in DKD [79], protecting glomerular mesangial cells from high-glucose-mediated cell apoptosis [80] but causing podocyte dysfunction and proteinuria [79]. β-catenin expression and altered phosphorylation, and cell proliferation were higher in normal colon epithelium surrounding tumor tissue in diabetic than in non-diabetic patients [81]. VDR activation antagonizes Wnt/β-catenin signaling [82] (Figure 3). The nephroprotective action of VDR activators has been related to Wnt/β-catenin inhibition [83]. Vitamin D deficiency is common in DM [84] and has also been associated with increased risk of CRC [85, 86]. High-glucose-induced inflammatory and fibrogenic responses in kidney cells contribute to DKD and are prevented by vitamin D receptor (VDR) activation [87-90]
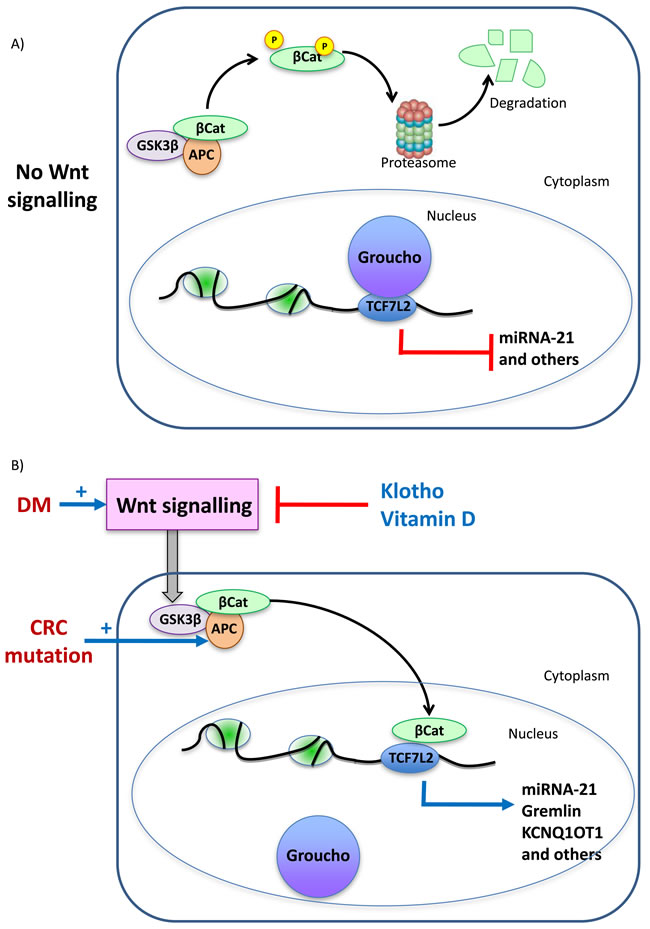
Figure 3: Key molecular pathways potentially linking diabetes and colorectal cancer. The example of β-catenin activation. A. In the absence of Wnt signaling, APC-bound glycogen synthase kinase 3-beta (GSK-3β) phosphorylates β-catenin (βCat), targeting it for ubiquitination and proteasomal degradation. In the absence of nuclear β-catenin, Groucho binds to transcription factors of the TCF family, repressing transcription. The TCF family includes TCF7L2 which has been associated to DM, DM complications and colon cancer by GWAS studies. B. Colon cancer is characterized by loss of function mutations of APC and in DM Wnt signaling is activated. Klotho and vitamin D prevent Wnt signaling and are protective against tumors and against DM complications. Wnt signaling prevents β-catenin phosphorylation and degradation allowing its nuclear migration, where it displaces Groucho and promotes transcription of genes involved in cell proliferation as well as other genes such as miR-21. miR21 contributes to tumorigenesis and to diabetes complications such as kidney injury. GWAS identified a GREM1 SNP associated with CRC susceptibility that facilitates TCF7L2 binding to DNA, leading to stronger GREM1 gene expression. A GREM1 SNP also associate with diabetic kidney disease. The gene product, Gremlin, promotes kidney injury in DM as well as colon cancer cell migration. KCNQ1 was associated with T2DM by GWAS. This locus encodes KCNQ1OT1, a β-catenin target upregulated in CRC.
EGFR signaling contributes to tumorigenesis and tumor progression of CRC and EGFR-targeted cetuximab is used to treat CRC. Genetic or pharmacological EGFR blockade slows experimental renal disease progression [91]. High-glucose, AGE, angiotensin II, and pro-inflammatory cytokines, such as TWEAK and parathyroid hormone-related protein (PTHrP) AGE promote EGFR transactivation in kidney cells [92-96]In this regard, TWEAK targeting antibodies are undergoing clinical trials in kidney disease, while targeting the TWEAK receptor Fn14 reduced colon cancer metastasis in experimental animals [95, 97]. Inhibition of EGFR with erlotinib attenuates DKD in experimental T1DM, through inhibition of mTOR [98]. Indeed, mTOR is activated in diabetic podocytes and mTOR targeting protects from DKD [99]. CCN2 is a novel EGFR ligand that promotes kidney inflammation and DKD progression [100, 101] and in CRC cells, regulates cell migration and prevents apoptosis [102].
Klotho is an anti-aging hormone of kidney origin with anti-inflammatory and anti-fibrotic properties [103, 104]. Experimental and human diabetes, inflammation and hyperlipidemia are associated with decreased Klotho expression [105-108]. Loss of Klotho contributes to kidney injury by de-repression of Wnt/β-catenin signaling [109] and similar mechanisms may be active in colon cancer cells. In this regard, Klotho suppresses growth and invasion of colon cancer cells through inhibition of the IGF1R-mediated PI3K/Akt pathway [110] and is frequently inactivated through promoter hypermethylation in CRC [111].
Inflammation and microbiota
Inflammation is a critical component of diabetes-induced target organ injury and of CRC initiation and progression [112, 113]. In preclinical models of T2DM, inflammation contributed to carcinogenesis and tumor growth, which were prevented by TNF-neutralizing monoclonal antibodies [57].
Multiple signaling pathways are involved in the inflammatory response, including MAPK, NF-κB, janus kinase/signal transducer and activator of transcription (JAK/STAT) and hypoxia-inducible factor-1α [68, 114-117]. Persistent NF-κB/IL-6/STAT3 activation promotes colitis associated CRC [118]. The non-canonical NF-κB pathway has also been implicated in diabetes complications and cancer [119-121]. The upstream kinase of this pathway, NIK, contributes to β cell failure in diet-induced obesity [122], promotes kidney injury [123] and underlies the sensitivity of Nlrp12-/- mice to gut inflammation and tumorigenesis [124]. These intracellular pathways amplify inflammatory responses and promote angiogenesis, cancer growth and invasiveness of malignant cells [125, 126], as well as progression of diabetes target organ injury such as DKD [117].
The interaction between colon epithelial cells and the microbiota may confer susceptibility to both colon cancer and obesity. The inflammasome regulates the microbiota and the inflammatory response of epithelial cells to the microbiota. Deficiency in inflammasome components (e.g. Nlrp6) is associated with an abnormal microbiota, exacerbated gut inflammatory responses [127] and colon tumorigenesis [128] dependent on microbiota-induced activation of epithelial IL-6 signaling [17]. Microbiota-dependent inflammatory responses may contribute to non-Mendelian familial aggregation of colon cancer since in preclinical models the risk of cancer was transmissible between co-housed individuals with the microbiota. The gut microbiota also impacts host metabolism, facilitating obesity, insulin resistance and T2DM [129]. Thus, inflammasome deficiency-related changes in gut microbiota are associated with insulin resistance and obesity [130]. In this regard, T2DM is one of three models of microbiome-associated human conditions to be studied by the Integrative Human Microbiome Project (iHMP, http://hmp2.org) [131]. Human T2DM and CRC share some microbiota features, such a decrease in the abundance of butyrate-producing bacteria [18, 132]. Butyrate is a breakdown product of dietary fiber that has anti-tumorigenic properties and is associated with decreased incidence of CRC [18]. In mice, the microbiota potential for butyrate production negatively correlated with tumor count [133]. Butyrate also has nephroprotective properties in DKD [134].
Iron metabolism. Altered iron metabolism facilitates rapid proliferation in cancer cells [135]. Indeed, constitutive Wnt/β-catenin signaling in colon cancer cells is iron-dependent [136] and iron chelation limits cell proliferation and has anti-inflammatory effects through NF-κB blockade [137]. Iron overload causes DM and is present in target organs of diabetes, such as the kidneys, while iron depletion upregulates glucose uptake and insulin signaling in liver and decreases kidney inflammation in experimental diabetes [138-140]. Indeed, the Trial to Assess Chelation Therapy (TACT) disclosed a benefit of ethylenediaminetetraacetic acid (EDTA), a chelator that also binds iron, on cardiovascular outcomes, especially in DM patients [141]. Thus, excess cellular iron may facilitate CRC growth, DM and DM complications. Heme iron may be the common denominator in the association of red meat intake with both DM and CRC [142, 143].
Epigenetic changes
CRC and DM also share some epigenetic changes. Thus, both CRC and DM were associated with a positive septin 9 (SEPT9) DNA-methylation assay (Epi-proColon) result [144]. In this regard, SEPT9 is differentially methylated in human T2DM islet cells and was shown to perturb insulin and glucagon secretion [145].
miRNAs are small non-coding RNA molecules that regulate gene expression. Pathogenic miRNAs may be shared by CRC and DKD [146-148]. In murine DKD, renal miR-21 expression was increased and miR-21 knockdown ameliorated renal damage [149]. The pathogenic potential of miR-21 is supported by some, but not all additional reports [150, 151]. miR-21 is also part of a six-miRNA-based classifier that reliably predicts CRC recurrence [148, 152]. Functional studies support a role for miR-21 in colon cancer proliferation and invasion [153, 154] and targeting miR-21 enhanced the sensitivity of human colon cancer cells to chemoradiotherapy and reduced angiogenesis [154, 155]. Metformin synergy with 5-fluorouracil and oxaliplatin to induce death of chemoresistant colon cancer cells was also associated with a reduction in miR-21 [156].
Additional information from systems biology approaches
Genome-wide association studies (GWAS) have identified susceptibility genes for DM or CRC that provide insights into potentially shared pathogenic pathways, such as TCF7L2, KCNQ1, HMGA2, RHPN2 and GREM1.
TCF7L2 harbors common genetic variants with the strongest effect on T2DM risk [157-159] and on DM complications such as DKD [160] and is also susceptibility locus for CRC loci in East Asians [161]. TCF7L2 is a transcription factor and β-catenin transcriptional partner in the Wnt-signaling pathway. DNA-bound TCFs repress gene transcription in the absence of β-catenin, but are required for β-catenin transcriptional activity [162]. TCF7L2 also promotes miR-21 expression [163]. Another CRC-associated Single Nucleotide Polymorphism (SNP), rs6983267, is located at a TCF7L2 binding site and the risk allele results in stronger TCF7L2 binding, facilitating Wnt signaling [164]. A common GREM1 SNP, rs16969681, associated with CRC susceptibility facilitates TCF7L2 binding to DNA leading to stronger gene expression [165]. A germline duplication upstream of GREM1 causes hereditary mixed polyposis syndrome and Mendelian-dominant predisposition to CRC through ectopic GREM1 overexpression in the intestinal epithelium [166, 167]. GREM1 was initially identified as one of the most upregulated genes in cultured mesangial cells exposed to high glucose [168] and GREM1 gene variants also associate with DKD [169]. Gremlin, the protein codified by GREM1, has been proposed as a key mediator of DKD [170-173]Gremlin promotes the motility of CRC cells [174] and the epithelial to mesenchymal transition in kidney tubular cells, also associated with increased motility [175, 176]. The precise role of TCF7L2 in CRC should be further defined. Thus, TCF7L2 mutations identified in cancer samples abolish its ability to function as a transcriptional regulator and result in increased CRC cell growth [177]. Given the multitude of target genes, this is not surprising.
KCNQ1 was associated with T2DM [178]. This locus encodes both KCNQ1 and the long noncoding RNAs (lncRNAs) KCNQ1OT1, which is a β-catenin target dysregulated in CRC [179]. In human CRC, low KCNQ1 expression was associated with poor survival and mutation of the murine homologue Kcnq1 increased the risk for intestinal tumors [180].
HMGA2 is a further gene associated to risk of T2DM and DKD in GWAS [158, 181]. HMGA2 expression is increased in and promotes the malignant behavior of CRC [182, 183]. Conversely, CRC GWAS identified RHPN2 as a susceptibility loci and RHPN2 expression is upregulated in experimental DKD [184, 185].
Pathway-based enrichment analysis of 23 independent gene expression profiling studies on prognosis of CRC observed overrepresentation of the oxidative phosphorylation chain, the extracellular matrix receptor interaction category, and a general category related to cell proliferation and apoptosis [186]. These categories are functionally related with cancer progression. Eight of the genes were also present in a previous meta-analysis of ten expression profiling studies of differentially expressed genes in CRC with good versus bad prognosis, including IQGAP1, YWHAH and TP53. [186]. IQGAP1 is part of the podocyte filter for proteins and regulates the occurrence of proteinuria, the hallmark of DKD [187] and YWHAH expression was increased in human DKD transcriptomics studies [188, 189]. Furthermore, human DKD transcriptomics revealed that 25% of apoptosis-related genes were differentially regulated in kidney tissue [190-192]. Some of the specific factors identified by human DKD transcriptomics and functionally characterized to contribute to kidney injury, also promote CRC growth, such as the MIF/CD74 system which is under study as a therapeutic target in colon cancer [193, 194]. Furthermore, elements of the JAK/STAT, VEGFR signaling and inflammation-related pathways were also overrepresented in human DKD [184, 189]. JAK/STAT, VEGF and inflammation are therapeutic targets in cancer.
As part of the Human Proteome Project, the Biology/Disease-driven Human Proteome Project (B/D-HPP) consortium leads specific projects on diabetes (HDPP) and cancer that may shed some additional light on the relationship between both diseases [195]. Protein candidate markers responding to CRC existence (diagnosis), stratification (different response related to stage) or prognosis (survival/metastasis) have been identified [196-199]. Most studies compared normal (healthy) tissue with tumor. The top four regulated proteins in a systematic review of CRC were 60-kDa heat shock protein (HSP60) and Nucleoside diphosphate kinase A (nm23-H1), up-regulated, and Selenium-binding protein 1 (SELENBP1) and Carbonic anhydrase I (CAI), down-regulated [200]. Interestingly, expression of the HSPD1 gene encoding HSP60 was upregulated and SELENBP1 downregulated in human DKD, according to the Nephromine database, further suggesting potential common pathogenic pathways between DKD and colon cancer (http://www.nephromine.org/).
Bioinformatics approaches may be used to integrate the growing systems biology databases. One such approach, the Drug-specific Signaling Pathway Network (DSPathNet) was used to tentatively identify seven genes (CDKN1A, ESR1, MAX, MYC, PPARGC1A, SP1, and STK11) and one novel MYC-centered pathway that might play a role in metformin antidiabetic and anticancer effects [201]. Interestingly, PPARGC1A protects from kidney injury and the expression is downregulated by inflammation [202].
Implications for therapy
Given the high and increasing incidence and prevalence of DM and CRC, it is likely that, independently from any common pathogenic pathways or associations, many DM patients will develop CRC. This brings the question whether physicians need to modify the approach to therapy of DM or CRC in diabetic patients with both conditions. In this regard, a diagnosis of cancer is frequently associated to a subsequent decrease in adherence to antidiabetic medication [203].
Choice of antidiabetic agent in the patient with CRC
The ADA Standards of Medical Care in Diabetes indicates that patients with DM should be encouraged to undergo recommended age- and sex-appropriate cancer screenings and to reduce their modifiable cancer risk factors (obesity, smoking, physical inactivity) [9]. In the presence of cancer, higher HbA1c goals should be considered: <8% in the absence of metastases and <8.5% for patients with metastatic cancer. If indeed hyperglycemia underlies the higher incidence of colon cancer in DM, these higher thresholds may theoretically impair cancer-related outcomes.
ADA 2016 does not provide recommendations on the choice of antidiabetic treatment in patients with cancer or CRC [204]. However, the initial antidiabetic agent recommended for standard T2DM patients, metformin, has been associated with decreased incidence or better outcomes in cancer patients [205-207]. Thus, even if prospective clinical studies confirmed the superiority of metformin on cancer incidence or outcome, this would not change the current standard therapeutic approach for T2DM in the cancer patient. The debate about the association of specific antidiabetic drugs and cancer risk has been marred by the lack of properly designed studies.
Although observational studies suggest that the choice of treatment for DM may modify cancer risk [208], no prospective studies have been specifically designed to address this issue. Thus, no firm conclusions can be reached at this point. The crux of the debate has been whether insulin or analogs are associated to an increased risk of CRC (and cancer in general) [209] and whether metformin is associated with a decreased risk of CRC [210]. This may represent two sides of the same coin: if one drug does modify the risk of CRC, by comparison the other may appear to modify the risk in the opposite direction. Confounders may exist. Thus, insulin is generally prescribed and metformin remains formally contraindicated in advanced chronic kidney disease (CKD), a late event in the course of T2DM, despite recent clinical recommendations [211]. Renal insufficiency is associated with higher risk for all-cause cancer [212], although this association has not been demonstrated for CRC [213].
Recent meta-analyses have attempted to unravel the potential relationship between antidiabetic therapy and cancer or colorectal cancer. However, meta-analysis results heavily depend on the quality of the included studies. A recent meta-analysis involving approximately 7.6 million and 137, 540 patients with diabetes from observational studies and randomized controlled trials (RCTs), respectively, suggested that metformin or thiazolidinediones were associated with a lower risk of all cancer incidence, while insulin, sulfonylureas and alpha glucosidase inhibitors were associated with an increased risk of cancer incidence [214]. Another large (491, 384 individuals) meta-analysis addressing specifically the impact of insulin, found it to be associated with a significant 69% increased risk of CRC in T2DM only in case-control but not in cohort studies [215]. The Barcelona nested case-control study of 275, 164 T2DM patients did not find an increased risk of cancer for any insulin or oral antidiabetic agent [216]. Finally, a metaanalysis of 19 publications representing data for 1, 332, 120 individuals, insulin had no effect and insulin glargine was associated with a decreased risk of CRC [217].
Metformin use has been associated with a decreased risk of colon cancer and increased survival [210, 218, 219]. A systematic review of 12 randomized controlled trials (21, 595 patients) and 41 observational studies (1, 029, 389 patients) found that in observational studies the risk of CRC was 17% lower among DM patients treated with metformin than in those not on metformin [210]. In a meta-analysis of 21 observational studies metformin was associated with a reduction in cancer-specific mortality, including a reduction in mortality for colon cancer (4 studies, HR 0.65, 0.56-0.76) [205, 220]. Several mechanisms may account for the antitumor effect of metformin. It reduces circulating insulin, promotes weight loss and activates 5’ adenosine monophosphate-activated protein kinase (AMPK), thus inhibiting growth of colon cancer cells [221, 222]. In mice with Apc mutations, metformin suppressed polyp growth [223] and in diabetic mice metformin, alone or in combination with oxaliplatin, reduced the severity of colorectal tumors [224]. Older literature described increased expression of mitochondrial GPDH, the target of metformin, in rapidly growing, undifferentiated tumors [225, 226]. However, there are no data on CRC expression of mitochondrial GPDH. In non-diabetic subjects, oral short-term low-dose metformin suppressed the development of colorectal aberrant crypt foci in a clinical trial [227]. In phase 3 RCT, low-dose (250 mg/day) metformin was safe and reduced the prevalence and number of metachronous adenomas or polyps after polypectomy in non-diabetic patients [228].
Conflicting results are available on thiazolidinediones and cancer. A systematic review and meta-analysis of 840, 787 diabetic patients did not support an association between thiazolidinediones and CRC [229, 230]. In a 6-year population-based cohort study, thiazolidinediones were associated with decreased cancer risk including CRC and the association was dose-dependent [231]. Thiazolidinediones have cytostatic effects and inhibit growth and metastasis of colon cancer cells as they induce differentiation and modulate the E-cadherin/β-catenin system [232-234]. However, some studies point to a mitogenic potential of troglitazone which induced colon tumors in normal C57BL/6J mice and increased colon carcinogenesis in Apc1638 N/+Mlh1+/− double mutant mice [235].
In systematic meta-analyses, sulphonylureas were associated with increased risk of pancreatic and hepatocellular cancer but not of CRC [229, 236, 237]. A cohort of 275, 164 T2DM patients found no evidence for altered cancer risk for repaglinide or α-glucosidase inhibitors compared to insulin-based therapies or other oral glucose-lowering drugs [216]. In other reports, acarbose was associated with reduced the risk of incident CRC in patients with diabetes in a dose-dependent manner [238, 239]. Acarbose may alter the microbiota [240] and decreased the size of gastrointestinal adenomas in Apc knockout mice [241].
Empaglifozin dramatically decreased mortality and slowed DKD progression and sodium-linked glucose transporter-2 (SGLT2) inhibitors may soon become the new standard of therapy [242]. A safety warning was issued by the FDA regarding bladder and breast cancer risk from early clinical trials of dapagliflozin but not for CRC (http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/UCM262994.pdf). Adenocarcinomas express SGLT2 and SGLT2 inhibitors blocked glucose uptake and reduced growth of tumor xenografts [243]. Whether this applies to CRC is unknown.
No relationship between GLP-1-based therapies and CRC have been reported [244, 245]. However, exenatide inhibited proliferation and induced apoptosis in cultured murine CT26 colon cancer cells [246, 247].
Choice of chemotherapy for colorectal rectal cancer in patients with diabetes
Studies addressing chemotherapy efficacy or safety in DM are very limited and there is no evidence supporting specific chemotherapy approaches for CRC patients with DM. No differences in the survival benefit or severe adverse effects associated to chemotherapy were observed in 5, 330 elderly CRC patients with (n=950) and without (n=4, 380) DM [248]. By contrast, a cohort study within the INT-0089 randomized adjuvant chemotherapy trial of 3, 759 patients with high-risk stage II/III colon cancer concluded that in DM patients overall mortality and cancer recurrence were higher than in non-diabetic patients [249]. Treatment-related toxicities were similar between DM and non-DM patients, except for a higher risk of treatment-related diarrhea among DM patients [249]. However, disease-free survival was lower and neurotoxicity more frequent in DM patients treated with capecitabine and oxaliplatin (CAPOX) chemotherapy than in non-diabetics [250]. It is likely that whether DM modifies the risk of severe adverse effects that limit chemotherapy depends on the specific chemotherapeutic regimen.
Agents in the pipeline targeting both CRC and diabetic complications
Some therapeutic targets are undergoing or have undergone RCTs in both diabetes complications (e.g. DKD) and cancer or are in clinical use in one condition and have been successfully used for the other condition in preclinical settings. These include statins, renin angiotensin aldosterone system (RAAS) blockers, endothelin receptor antagonists, VDR activators, mTOR inhibitors, anti-inflammatory molecules and inhibitors of EGF ligands/receptors (Table 4) [251, 252].
Table 4: Examples of agents in the pipeline targeting both cancer and diabetic target organ complications exemplified by diabetic kidney disease
Activity |
Agent |
Successful in animal models of cancer |
Successful in experimental DKD |
RCT in human cancer |
RCT in human DKD |
Refs |
HMGCoA reductase inhibitors |
statins |
Yes |
Yes |
Yes |
Yes |
|
RAAS targeting drugs |
ACE inhibitors, ARBs |
Yes |
Yes |
No |
Yes |
|
VDR activator |
Paricalcitol |
Yes |
Yes |
Yes |
Yes |
|
Endothelin receptor antagonists |
Atrasentan and others |
Yes |
Yes |
Yes |
Yes |
|
Anti-fibrotic agents |
Anti-CTGF mAb FG3019 |
Yes |
Yes |
Yes |
Yes |
|
Anti- TGF-β1 mAb. |
Yes |
Yes |
Yes |
Yes |
[304] |
|
Anti-inflammatory agents |
Chemokine targeting agents |
Yes |
Yes |
Yes (anti-CXCR4) |
Yes (anti-CCL2 and others ) |
[270] |
JAK/STAT inhibitors |
Yes |
Yes |
Yes |
Yes |
||
Inhibitors of epidermal growth factor Receptor/ligands |
Several agents |
Yes |
Yes |
Anti-EGFR antibodies (cetuximab) |
Anti-TGF-α/epiregulin antibody (LY3016859) |
|
mTOR inhibitors |
Several agents |
Yes |
Yes |
Yes |
No (Yes in non-DKD CKD) |
DKD: diabetic kidney disease, CKD: chronic kidney disease, HMGCoA: 3-hydroxy-3-methylglutaryl coenzyme A, RAAS: renin angiotensin aldosterone system, ACE: angiotensin converting enzyme, ARB: angiotensin receptor blocker; CTGF: Connective tissue growth factor, TGF-beta: Transforming growth factor beta, EGFR: Epidermal growth factor receptor, CXCR4: Chemokine Receptor type 4, CCL2: Chemokine Ligand 2
Statins are commonly used to treat hyperlipidemia and have been linked with a small reduction in the risk for colon cancer in diabetic patients [253] and improved prognosis of curatively resected CRC [254]. In an obesity-related colon cancer model associated with hyperlipidemia and hyperinsulinemia, pitavastatin prevented carcinogenesis and inhibited colon proliferation and inflammation [255], while simvastatin inhibited the release of inflammatory cytokines by colorectal cell lines [256]. Clinical trials are exploring statins in the treatment of human CRC.
RAAS blockers are the mainstay of therapy for human DKD [252]. Angiotensin-converting enzyme inhibitors and angiotensin-II type 1 receptor blockers suppress chemically-induced colonic preneoplasic lesions in diabetic animals [257-259]. However, their clinical use to prevent colon cancer is not being pursued.
The endothelin receptor antagonist atrasentan is undergoing RCTs for DKD [260, 261], and as add-on to docetaxel and prednisone for stage IV hormone therapy-resistant prostate cancer bone metastases (NCT00134056) [262-264]. However, no trial is exploring CRC.
Paricalcitol is a VDR activator that may have antiproteinuric effects on DKD as suggested by RCTs [265] and may also slow cancer cell growth [266]. Phase I trials have tested combinations of paricalcitol and chemotherapeutic agents (NCT00217477). Additionally, vitamin D has been explored for colon cancer prevention. However, a combination of calcitriol, aspirin, and calcium carbonate or vitamin D/calcium did not prevent recurrence of colorectal adenomas over a 3- to 5-year period [267, 268].
mTOR inhibitors are used as anticancer agents and also improve experimental DKD [99, 269]. The mTOR inhibitors RAD001 (NCT01058655) and everolimus and the dual PI3K/mTOR inhibitor PF-05212384 (NCT01937715, NCT01154335) are undergoing clinical trials for metastatic CRC.
Several agents targeting cytokines and chemokines have been tested both in T2DM and cancer [251, 252, 270-273]. Plerixafor is a CXCR4 antagonist undergoing trials for advanced CRC (NCT02179970). Although not specifically tested in DM, a selective CXCR4 antagonist AMD3465 decreased mineralocorticoid-dependent renal fibrosis in mice [274] and targeting CXCR4 prevented glomerular injury associated to high podocyte CXCR4 expression in mice [275].
JAK2 targeting prevented high-glucose-induced fibrogenic responses in renal cells and prevented kidney and vascular injury in experimental diabetes [276-279]. An ongoing phase II RCT is testing the JAK1 and JAK2 inhibitor baricitinib as add-on to RAS blockade in patients with DKD (NCT01683409) while another will explore the JAK2/FLT3 inhibitor pacritinib in patients with refractory CRC and KRAS mutations (NCT02277093).
Unanswered questions and the way forward
Table 5 summarizes the key points of the review. The association between DM and CRC is recognized by scientific consensus [4]. However, a number of issues require more detailed studies (Table 6).
An overview of T2DM and CRC country-based prevalence/incidence suggests that environmental, development-associated or other factors may interact with the T2DM milieu to increase the risk of CRC. Identification of these putative factors and whether DM associates with increased CRC risk in different cultures and countries may provide further insights into mechanisms underlying the relationship between DM and cancer.
Table 5: Key points

The case for a causal association should be strengthened by the characterization of the DM-initiated molecular pathways involved. This information may also lead to the development of specific preventive or therapeutic approaches. Studies should address the relationship between DM-associated CRC and the development of other DM-associated complications, i.e., whether there is a patient profile prone to develop any DM-related complications. If this were the case, tools should be developed for the early identification of such patients. Urine proteomics holds promise in this regard, as it allows identification of DKD at earlier stages than currently available methods and predicts progression [280, 281] and may also be useful for the diagnosis of cancer outside the urogenital system [282]. Early identification of the subpopulation of DM patients at highest risk for developing cancer or classical complications may allow enrollment in trials assessing the efficacy of drugs targeting shared molecular mechanisms for prevention and/or therapy. Additional systems biology approaches may also contribute to define molecular pathways leading to DM-associated cancer or target organ damage. The most promising approaches should undergo clinical trial testing, ideally in high-risk populations or in early disease stages identified by the study of specific molecular signatures.
Research is needed to define the optimal therapeutic approach for the patient with T2DM and CRC. Studies of the impact of different antidiabetic agents on cancer incidence are marred by the fact that both sides of the comparison may theoretically modulate cancer incidence. Additionally, there are potential biases related to the indication of the specific agent. These research efforts have the potential to decrease the incidence of DM-associated complications and to improve outcomes. The DiabetesCancerConnect Consortium funded by the Spanish Government is attempting to answer some of these questions.
Table 6: Standing questions on the relationship between DM and colorectal cancer
Standing question |
Relevance |
What is required to address it |
Is there an association between T2DM and colorectal cancer across all countries and cultures? |
Provides insights into etiologic and pathophysiologic factors, may prevent a colorectal cancer epidemic in the developing world |
Head-to-head comparison between developed and developing country cohorts |
Is there an association between development of cancer and development of other complications of DM? |
Provides the epidemiological basis to search for common mediators of disease |
Epidemiological studies, ideally prospective |
What molecular mediators explain the association between DM and cancer? Are they shared by other complications of DM? |
Identification of potential diagnostic signatures and therapeutic targets |
Interventional preclinical models that address function of key molecules. These may have been identified by non-biased systems biology approaches and hypothesis-driven studies designed from the analysis of available literature |
Has DM-associated colorectal cancer a specific molecular signature? |
This may identify diagnostic signatures and therapeutic targets specific for DM-associated colorectal cancer |
Systems biology comparison between DM and non-DM associated colorectal cancer with DM and non-DM healthy colon as control |
Can DM patients at high risk for cancer development be identified by diagnostic tests? |
Early diagnosis of risk or cancer |
Prospective systems biology approach to relevant biological samples (feces, urine, blood or others) |
Can DM patients at high risk or early colorectal cancer be treated by specific, DM-tailored approaches? Do these approaches also prevent/treat other diabetic complications? |
New preventive/therapeutic approaches that address both cancer and non-cancer DM complications |
Early identification of patients at high risk or with early disease Unraveling of common pathogenic pathways |
Are there common microbiota signatures for colorectal cancer and other DM complications? |
New preventive/therapeutic approaches that address both cancer and non-cancer DM complications |
Metagenomic studies |
What is the optimal therapeutic approach for colorectal cancer in diabetic individuals and the optimal therapeutic approaches for DM in colorectal cancer patients? |
Therapy individualization and improved outcomes |
Hypothesis-generating observational studies followed by randomized clinical trials |
Abbreviations
DM-Diabetes mellitus
CRC-Colorectal cancer
ADA- American Diabetes Association
T1DM- Type 1 DM
T2DM- Type 2 DM
MODY─ Maturity-onset diabetes of the young
GLP-1- Glucagon like peptide-1
GPDH- Glycerophosphate dehydrogenase
GPD2- Glycerol-3-phosphate dehydrogenase 2
GSK-3β- Glycogen synthase kinase 3-beta
EGFR- Epidermal growth factor receptor
HR- Hazard Ratio
SIR- Standardized incidence ratios
RR- Relative risk
CI- Confidence interval
BMI- Body mass index
OR- Odds Ratio
HbA1c- Glycated hemoglobin
AGEs- Advanced Glycation End-products
RELMβ- Resistin-like molecule β
IGF-1- Insulin-like growth factor
MAPK- Mitogen activated protein kinase
PI3K- Phosphatidyl-inositol-3-kinase
PAK-1- Activated protein kinase-1
mRNA- Messenger RNA
ChREBP- Carbohydrate response element-binding protein
NF-κB- Factor-kappa B
Nrf2- Nuclear factor erythroid 2–related factor 2
ATP- Adenosine triphosphate
Glut-1- Glucotransporter-1
TGF- Transforming growth factor
VDR- Vitamin D receptor
PTHrP- Parathyroid hormone-related protein-
Grem1- Gremlin
S1P- Sphingosine-1-phosphate
iHMP- Integrative Human Microbiome Project
TACT- Trial to Assess Chelation Therapy
EDTA- Ethylenediaminetetraacetic acid
miRNAs- MicroRNAs
GWAS- Genome-wide association studies
lncRNA- Long non-coding RNA
SNP- Single Nucleotide Polymorphism
eQTL- Expression quantitative trait loci
KEGG- Kyoto Encyclopedia of Genes and Genomes
VEGFR- Vascular endothelial growth factor receptor
B/D-HPP- Biology/Disease-driven Human Proteome Project
HDPP- Human Diabetes Proteome Project
SELENBP1- Selenium-binding protein 1
CAI- Carbonic anhydrase I
DSPathNet- Drug-specific Signaling Pathway Network
RCTs- Randomized controlled trials
CKD- Chronic kidney disease
ROS- Reactive Oxygen species
PPARγ- peroxisome proliferator–activated receptor γ
DNA- Deoxyribonucleic acid
RNA- Ribonucleic acid
FDA- Food and Drug Administration
CAPOX- Capecitabine and oxaliplatin
RAAS- Renin angiotensin aldosterone system
mAb- Monoclonal antibody
HG- Hyperglycemia
DKD- Diabetic kidney disease
ARB- Angiotensin receptor blocker
HMGCoA- 3-hydroxy-3-methylglutaryl coenzyme A
ACE- Angiotensin converting enzyme-
iNOS- Inducible nitric oxide synthase
NF-κB- Nuclear factor kappa-light-chain-enhancer of activated B cells
ERK- Extracellular Signal-regulated Kinase
CTGF- Connective tissue growth factor
TGF-beta- Transforming growth factor beta
CXCR4- Chemokine Receptor type 4
CCL2- Chemokine Ligand 2.
Acknowledgments
Members of the DiabetesCancerConnect Consortium: Zaida Moreno Villegas, Maria Estrella Martin-Crespo Aznar, Alberto Ortiz, Marta Ruiz Ortega, Maria Jose Trujillo Tiebas, Alvaro Conrado Ucero Herreria, Maria Concepcion Izquierdo Carnero, Irene Gutierrez Rojas, Rebeca Manso Alonso, Cristina Chamizo Garcia, Alfonso Rubio Navarro, Marta Corton Perez, Carmen Gomez Guerrero, Manuel Jesus Hernandez Perez, Matilde Alique Aguilar, Socorro Maria Rodriguez Pinilla, Gloria Alvarez Llamas, Oscar Aguilera Martinez, Maria Posada Ayala, Sergio Portal Nuñez, Jesus Egido De Los Rios, Jesus Miguel Garcia-Foncillas Lopez, Federico Gustavo Rojo Todo, Juan Madoz Gurpide, Carlos Antonio Tarin Cerezo, Iolanda Lazaro Lopez, Juan Antonio Moreno Gutierrez, Maria Rodriguez Remirez, Aurea Borrero Palacios, Patricia Fernandez San Jose, Jonay Poveda Nuñez, Rocio Sanchez Alcudia, Clara Isabel Gomez Sanchez, Ana Belen Sanz Bartolome, Fernando Vivanco Martinez, Maria Esther Martin Aparicio, Oscar Lorenzo Gonzalez, Pedro Esbrit Arguelles, Beatriz Fernandez Fernandez, Sandra Zazo Hernandez, Ruth Fernandez Sanchez, Fiona Blanco Kelly, Raquel Perez Carro, Juan Antonio Ardura Rodriguez, Carmen Ayuso Garcia, Laura Del Puerto Nevado, Almudena Avila Fernandez, Ana Maria Ramos Verde, Carlos Pastor Vargas, Nieves Gonzalez Gomez, Iker Sanchez Navarro, Javier Martinez Useros, Rosa Riveiro Alvarez, Laura Gonzalez Calero, Catalina Martin Cleary, Olga Ruiz Andres, Luis Carlos Tabara Rodriguez, Paula Gonzalez Alonso, Marta Martin Lorenzo, Ion Cristobal Yoldi, Elena Burillo Ipiens, Ainhoa Oguiza Bilbao, Carlota Recio Cruz, Sorina Daniela Tatu, Adrian Ramos Cortassa, Jorge Enrique Rojas Rivera, Liliana Gonzalez Espinoza, Carolina Lavoz Barria, Maria Vanessa Perez Gomez, Pablo Minguez Paniagua, Sebastian Mas Fontao, Ana María Díez Rodríguez.
Funding
Research was supported by the grants FIS/FEDER PI14/01650, PI13/00047, PI14/00386, PIE13/00051, PI13/01873, PI13/00802, PI14/00883, PI15/00298, PI15/01460, PI16/02057, PI16/01900, CP09/00229, CP14/00133, CPII15/00027, SAF2012-38830, CP12/03262 ISCIII-RETIC REDinREN RD12/0021 RD16/0009 and RETICEF RD12/0043/0008, CIBER in Diabetes and Associated Metabolic Disorders (CIBERDEM, ISCIII), Biobanco IIS-FJD PT13/0010/0012, FP7-HEALTH-2013-INNOVATION-1- 602422 e-PREDICE, Comunidad de Madrid S2010/BMD-2378, CYTED IBERERC, Programa Intensificación Actividad Investigadora (ISCIII) to AO and CA, Sociedad Española de Nefrología y Fundación Renal Iñigo Alvarez de Toledo to JAM, Programa Miguel Servet to NG, MC, JAM, MDSN, ABS and GALL, and Programa Joan Rodes to BFF.
Conflict of Interests
The authors have no competing financial interests.
References
1. GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet (London, England). 2015; 385: 117–71. doi: 10.1016/S0140-6736(14)61682-2.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66: 7–30. doi: 10.3322/caac.21332.
3. American Diabetes Association. 3. Foundations of Care and Comprehensive Medical Evaluation. Diabetes Care. 2016; 39 Suppl 1: S23-35. doi: 10.2337/dc16-S006.
4. Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, Pollak M, Regensteiner JG, Yee D. Diabetes and cancer: a consensus report. Diabetes Care. 2010; 33: 1674–85. doi: 10.2337/dc10-0666.
5. American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2004; 27 Suppl 1: S5–10. Available from http://www.ncbi.nlm.nih.gov/pubmed/14693921.
6. Rimm AA, Werner LH, Yserloo B V, Bernstein RA. Relationship of ovesity and disease in 73,532 weight-conscious women. Public Health Rep. 1975; 90: 44–51. Available from http://www.ncbi.nlm.nih.gov/pubmed/803696.
7. Madiraju AK, Erion DM, Rahimi Y, Zhang X-M, Braddock DT, Albright RA, Prigaro BJ, Wood JL, Bhanot S, MacDonald MJ, Jurczak MJ, Camporez J-P, Lee H-Y, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014; 510: 542–6. doi: 10.1038/nature13270.
8. Bennett WL, Maruthur NM, Singh S, Segal JB, Wilson LM, Chatterjee R, Marinopoulos SS, Puhan MA, Ranasinghe P, Block L, Nicholson WK, Hutfless S, Bass EB, et al. Comparative effectiveness and safety of medications for type 2 diabetes: an update including new drugs and 2-drug combinations. Ann Intern Med. 2011; 154: 602–13. doi: 10.7326/0003-4819-154-9-201105030-00336.
9. American Diabetes Association. Standards of medical care in diabetes--2014. Diabetes Care. 2014; 37 Suppl 1: S14-80. doi: 10.2337/dc14-S014.
10. Chan AT, Giovannucci EL. Primary prevention of colorectal cancer. Gastroenterology. 2010; 138: 2029–2043.e10. doi: 10.1053/j.gastro.2010.01.057.
11. Jo W-S, Chung DC. Genetics of hereditary colorectal cancer. Semin Oncol. 2005; 32: 11–23. doi: 10.1053/j.seminoncol.2004.09.034.
12. Imperiale TF, Ransohoff DF. Risk for colorectal cancer in persons with a family history of adenomatous polyps: a systematic review. Ann Intern Med. 2012; 156: 703–9. doi: 10.7326/0003-4819-156-10-201205150-00006.
13. Iacopetta B. Are there two sides to colorectal cancer? Int J cancer. 2002; 101: 403–8. doi: 10.1002/ijc.10635.
14. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990; 61: 759–67. Available from http://www.ncbi.nlm.nih.gov/pubmed/2188735.
15. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487: 330–7. doi: 10.1038/nature11252.
16. Vetrano S, Danese S. Colitis, microbiota, and colon cancer: an infernal triangle. Gastroenterology. 2013; 144: 461–3. doi: 10.1053/j.gastro.2012.12.016.
17. Hu B, Elinav E, Huber S, Strowig T, Hao L, Hafemann A, Jin C, Wunderlich C, Wunderlich T, Eisenbarth SC, Flavell RA. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc Natl Acad Sci U S A. 2013; 110: 9862–7. doi: 10.1073/pnas.1307575110.
18. Abreu MT, Peek RM. Gastrointestinal malignancy and the microbiome. Gastroenterology. 2014; 146: 1534–1546.e3. doi: 10.1053/j.gastro.2014.01.001.
19. Sears CL, Garrett WS. Microbes, microbiota, and colon cancer. Cell Host Microbe. 2014; 15: 317–28. doi: 10.1016/j.chom.2014.02.007.
20. Song X, Gao H, Lin Y, Yao Y, Zhu S, Wang J, Liu Y, Yao X, Meng G, Shen N, Shi Y, Iwakura Y, Qian Y. Alterations in the microbiota drive interleukin-17C production from intestinal epithelial cells to promote tumorigenesis. Immunity. 2014; 40: 140–52. doi: 10.1016/j.immuni.2013.11.018.
21. Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, Thangaraju M, Prasad PD, Manicassamy S, Munn DH, Lee JR, Offermanns S, Ganapathy V. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014; 40: 128–39. doi: 10.1016/j.immuni.2013.12.007.
22. Belcheva A, Irrazabal T, Robertson SJ, Streutker C, Maughan H, Rubino S, Moriyama EH, Copeland JK, Kumar S, Green B, Geddes K, Pezo RC, Navarre WW, et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell. 2014; 158: 288–99. doi: 10.1016/j.cell.2014.04.051.
23. Tezcan G, Tunca B, Ak S, Cecener G, Egeli U. Molecular approach to genetic and epigenetic pathogenesis of early-onset colorectal cancer. World J Gastrointest Oncol. 2016; 8: 83–98. doi: 10.4251/wjgo.v8.i1.83.
24. McCarthy K, Pearson K, Fulton R, Hewitt J. Pre-operative chemoradiation for non-metastatic locally advanced rectal cancer. Cochrane database Syst Rev. 2012; 12: CD008368. doi: 10.1002/14651858.CD008368.pub2.
25. André T, Boni C, Mounedji-Boudiaf L, Navarro M, Tabernero J, Hickish T, Topham C, Zaninelli M, Clingan P, Bridgewater J, Tabah-Fisch I, de Gramont A, Multicenter International Study of Oxaliplatin/5-Fluorouracil/Leucovorin in the Adjuvant Treatment of Colon Cancer (MOSAIC) Investigators. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med. 2004; 350: 2343–51. doi: 10.1056/NEJMoa032709.
26. Therkildsen C, Bergmann TK, Henrichsen-Schnack T, Ladelund S, Nilbert M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol. 2014; 53: 852–64. doi: 10.3109/0284186X.2014.895036.
27. Suh S, Kim K-W. Diabetes and cancer: is diabetes causally related to cancer? Diabetes Metab J. 2011; 35: 193–8. doi: 10.4093/dmj.2011.35.3.193.
28. Will JC, Galuska DA, Vinicor F, Calle EE. Colorectal cancer: another complication of diabetes mellitus? Am J Epidemiol. 1998; 147: 816–25. Available from http://www.ncbi.nlm.nih.gov/pubmed/9583711.
29. Jarvandi S, Davidson NO, Schootman M. Increased risk of colorectal cancer in type 2 diabetes is independent of diet quality. PLoS One. 2013; 8: e74616. doi: 10.1371/journal.pone.0074616.
30. Kasuga M, Ueki K, Tajima N, Noda M, Ohashi K, Noto H, Goto A, Ogawa W, Sakai R, Tsugane S, Hamajima N, Nakagama H, Tajima K, et al. Report of the Japan Diabetes Society/Japanese Cancer Association Joint Committee on Diabetes and Cancer. Cancer Sci. 2013; 104: 965–76. doi: 10.1111/cas.12203.
31. Wang M, Hu R-Y, Wu H-B, Pan J, Gong W-W, Guo L-H, Zhong J-M, Fei F-R, Yu M. Cancer risk among patients with type 2 diabetes mellitus: a population-based prospective study in China. Sci Rep. 2015; 5: 11503. doi: 10.1038/srep11503.
32. Harding JL, Shaw JE, Peeters A, Cartensen B, Magliano DJ. Cancer risk among people with type 1 and type 2 diabetes: disentangling true associations, detection bias, and reverse causation. Diabetes Care. 2015; 38: 264–70. doi: 10.2337/dc14-1996.
33. Liu X, Hemminki K, Försti A, Sundquist K, Sundquist J, Ji J. Cancer risk in patients with type 2 diabetes mellitus and their relatives. Int J cancer. 2015; 137: 903–10. doi: 10.1002/ijc.29440.
34. Tsilidis KK, Kasimis JC, Lopez DS, Ntzani EE, Ioannidis JPA. Type 2 diabetes and cancer: umbrella review of meta-analyses of observational studies. BMJ. 2015; 350: g7607. Available from http://www.ncbi.nlm.nih.gov/pubmed/25555821.
35. Huang Y, Cai X, Qiu M, Chen P, Tang H, Hu Y, Huang Y. Prediabetes and the risk of cancer: a meta-analysis. Diabetologia. 2014; 57: 2261–9. doi: 10.1007/s00125-014-3361-2.
36. Dankner R, Boffetta P, Balicer RD, Boker LK, Sadeh M, Berlin A, Olmer L, Goldfracht M, Freedman LS. Time-Dependent Risk of Cancer After a Diabetes Diagnosis in a Cohort of 2.3 Million Adults. Am J Epidemiol. 2016; 183: 1098–106. doi: 10.1093/aje/kwv290.
37. De Bruijn KMJ, Ruiter R, de Keyser CE, Hofman A, Stricker BH, van Eijck CHJ. Detection bias may be the main cause of increased cancer incidence among diabetics: results from the Rotterdam Study. Eur J Cancer. 2014; 50: 2449–55. doi: 10.1016/j.ejca.2014.06.019.
38. Porter NR, Eberth JM, Samson ME, Garcia-Dominic O, Lengerich EJ, Schootman M. Diabetes Status and Being Up-to-Date on Colorectal Cancer Screening, 2012 Behavioral Risk Factor Surveillance System. Prev Chronic Dis. 2016; 13: E19. doi: 10.5888/pcd13.150391.
39. Singh S, Earle CC, Bae SJ, Fischer HD, Yun L, Austin PC, Rochon PA, Anderson GM, Lipscombe L. Incidence of Diabetes in Colorectal Cancer Survivors. J Natl Cancer Inst . 2016; 108: djv402. doi: 10.1093/jnci/djv402.
40. Nilsen TI, Vatten LJ. Prospective study of colorectal cancer risk and physical activity, diabetes, blood glucose and BMI: exploring the hyperinsulinaemia hypothesis. Br J Cancer. 2001; 84: 417–22. doi: 10.1054/bjoc.2000.1582.
41. de Kort S, Simons CCJM, van den Brandt PA, Goldbohm RAS, Arts ICW, de Bruine AP, Janssen-Heijnen MLG, Sanduleanu S, Masclee AAM, Weijenberg MP. Diabetes mellitus type 2 and subsite-specific colorectal cancer risk in men and women: results from the Netherlands Cohort Study on diet and cancer. Eur J Gastroenterol Hepatol. 2016; 28: 896–903. doi: 10.1097/MEG.0000000000000626.
42. Bardou M, Barkun AN, Martel M. Obesity and colorectal cancer. Gut. 2013; 62: 933–47. doi: 10.1136/gutjnl-2013-304701.
43. Bhaskaran K, Douglas I, Forbes H, dos-Santos-Silva I, Leon DA, Smeeth L. Body-mass index and risk of 22 specific cancers: a population-based cohort study of 5·24 million UK adults. Lancet (London, England). 2014; 384: 755–65. doi: 10.1016/S0140-6736(14)60892-8.
44. Murphy N, Cross AJ, Abubakar M, Jenab M, Aleksandrova K, Boutron-Ruault M-C, Dossus L, Racine A, Kühn T, Katzke VA, Tjønneland A, Petersen KEN, Overvad K, et al. A Nested Case-Control Study of Metabolically Defined Body Size Phenotypes and Risk of Colorectal Cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC). PLoS Med. 2016; 13: e1001988. doi: 10.1371/journal.pmed.1001988.
45. Bloomfield HE, Koeller E, Greer N, MacDonald R, Kane R, Wilt TJ. Effects on Health Outcomes of a Mediterranean Diet With No Restriction on Fat Intake: A Systematic Review and Meta-analysis. Ann Intern Med. 2016; 165: 491–500. doi: 10.7326/M16-0361.
46. Gallagher EJ, LeRoith D. Obesity and Diabetes: The Increased Risk of Cancer and Cancer-Related Mortality. Physiol Rev. 2015; 95: 727–48. doi: 10.1152/physrev.00030.2014.
47. Hua F, Yu J-J, Hu Z-W. Diabetes and cancer, common threads and missing links. Cancer Lett. 2016; 374: 54–61. doi: 10.1016/j.canlet.2016.02.006.
48. Björk J, Nilsson J, Hultcrantz R, Johansson C. Growth-regulatory effects of sensory neuropeptides, epidermal growth factor, insulin, and somatostatin on the non-transformed intestinal epithelial cell line IEC-6 and the colon cancer cell line HT 29. Scand J Gastroenterol. 1993; 28: 879–84. Available from http://www.ncbi.nlm.nih.gov/pubmed/7505479.
49. Nagel JM, Staffa J, Renner-Müller I, Horst D, Vogeser M, Langkamp M, Hoeflich A, Göke B, Kolligs FT, Mantzoros CS. Insulin glargine and NPH insulin increase to a similar degree epithelial cell proliferation and aberrant crypt foci formation in colons of diabetic mice. Horm Cancer. 2010; 1: 320–30. doi: 10.1007/s12672-010-0020-z.
50. Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev. 2009; 30: 586–623. doi: 10.1210/er.2008-0047.
51. Wolf I, Sadetzki S, Catane R, Karasik A, Kaufman B. Diabetes mellitus and breast cancer. Lancet Oncol. 2005; 6: 103–11. doi: 10.1016/S1470-2045(05)01736-5.
52. Welsh GI, Hale LJ, Eremina V, Jeansson M, Maezawa Y, Lennon R, Pons DA, Owen RJ, Satchell SC, Miles MJ, Caunt CJ, McArdle CA, Pavenstädt H, et al. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab. 2010; 12: 329–40. doi: 10.1016/j.cmet.2010.08.015.
53. Sun J, Khalid S, Rozakis-Adcock M, Fantus IG, Jin T. P-21-activated protein kinase-1 functions as a linker between insulin and Wnt signaling pathways in the intestine. Oncogene. 2009; 28: 3132–44. doi: 10.1038/onc.2009.167
54. Reidy K, Kang HM, Hostetter T, Susztak K. Molecular mechanisms of diabetic kidney disease. J Clin Invest. 2014; 124: 2333–40. doi: 10.1172/JCI72271.
55. Jain SS, Bird RP. Elevated expression of tumor necrosis factor-alpha signaling molecules in colonic tumors of Zucker obese (fa/fa) rats. Int J cancer. 2010; 127: 2042–50. doi: 10.1002/ijc.25232.
56. Teraoka N, Mutoh M, Takasu S, Ueno T, Nakano K, Takahashi M, Imai T, Masuda S, Sugimura T, Wakabayashi K. High susceptibility to azoxymethane-induced colorectal carcinogenesis in obese KK-Ay mice. Int J cancer. 2011; 129: 528–35. doi: 10.1002/ijc.25711.
57. Flores MBS, Rocha GZ, Damas-Souza DM, Osório-Costa F, Dias MM, Ropelle ER, Camargo JA, de Carvalho RB, Carvalho HF, Saad MJA, Carvalheira JBC. Obesity-induced increase in tumor necrosis factor-α leads to development of colon cancer in mice. Gastroenterology. 2012; 143: 741-53-4. doi: 10.1053/j.gastro.2012.05.045.
58. Hata K, Kubota M, Shimizu M, Moriwaki H, Kuno T, Tanaka T, Hara A, Hirose Y. C57BL/KsJ-db/db-Apc mice exhibit an increased incidence of intestinal neoplasms. Int J Mol Sci. 2011; 12: 8133–45. doi: 10.3390/ijms12118133.
59. LeRoith D. Can endogenous hyperinsulinaemia explain the increased risk of cancer development and mortality in type 2 diabetes: evidence from mouse models. Diabetes Metab Res Rev. 2010; 26: 599–601. doi: 10.1002/dmrr.1139.
60. Fierz Y, Novosyadlyy R, Vijayakumar A, Yakar S, LeRoith D. Insulin-sensitizing therapy attenuates type 2 diabetes-mediated mammary tumor progression. Diabetes. 2010; 59: 686–93. doi: 10.2337/db09-1291.
61. Masur K, Vetter C, Hinz A, Tomas N, Henrich H, Niggemann B, Zänker KS. Diabetogenic glucose and insulin concentrations modulate transcriptome and protein levels involved in tumour cell migration, adhesion and proliferation. Br J Cancer. 2011; 104: 345–52. doi: 10.1038/sj.bjc.6606050.
62. Tomas NM, Masur K, Piecha JC, Niggemann B, Zänker KS. Akt and phospholipase Cγ are involved in the regulation of growth and migration of MDA-MB-468 breast cancer and SW480 colon cancer cells when cultured with diabetogenic levels of glucose and insulin. BMC Res Notes. 2012; 5: 214. doi: 10.1186/1756-0500-5-214.
63. Ma Y-S, Yang I-P, Tsai H-L, Huang C-W, Juo S-HH, Wang J-Y. High glucose modulates antiproliferative effect and cytotoxicity of 5-fluorouracil in human colon cancer cells. DNA Cell Biol. 2014; 33: 64–72. doi: 10.1089/dna.2013.2161.
64. Chen H, Wu L, Li Y, Meng J, Lin N, Yang D, Zhu Y, Li X, Li M, Xu Y, Wu Y, Tong X, Su Q. Advanced glycation end products increase carbohydrate responsive element binding protein expression and promote cancer cell proliferation. Mol Cell Endocrinol. 2014; 395: 69–78. doi: 10.1016/j.mce.2014.07.021.
65. Park M-J, Kim D-I, Lim S-K, Choi J-H, Han H-J, Yoon K-C, Park S-H. High glucose-induced O-GlcNAcylated carbohydrate response element-binding protein (ChREBP) mediates mesangial cell lipogenesis and fibrosis: the possible role in the development of diabetic nephropathy. J Biol Chem. 2014; 289: 13519–30. doi: 10.1074/jbc.M113.530139.
66. Uzozie A, Nanni P, Staiano T, Grossmann J, Barkow-Oesterreicher S, Shay JW, Tiwari A, Buffoli F, Laczko E, Marra G. Sorbitol dehydrogenase overexpression and other aspects of dysregulated protein expression in human precancerous colorectal neoplasms: a quantitative proteomics study. Mol Cell Proteomics. 2014; 13: 1198–218. doi: 10.1074/mcp.M113.035105.
67. Wu Y, Zhou BP. Inflammation: a driving force speeds cancer metastasis. Cell Cycle. 2009; 8: 3267–73. doi: 10.4161/cc.8.20.9699.
68. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008; 454: 436–44. doi: 10.1038/nature07205.
69. Rojas A, Figueroa H, Morales E. Fueling inflammation at tumor microenvironment: the role of multiligand/RAGE axis. Carcinogenesis. 2010; 31: 334–41. doi: 10.1093/carcin/bgp322.
70. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010; 107: 1058–70. doi: 10.1161/CIRCRESAHA.110.223545.
71. Yao D, Brownlee M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes. 2010; 59: 249–55. doi: 10.2337/db09-0801.
72. de Zeeuw D, Akizawa T, Audhya P, Bakris GL, Chin M, Christ-Schmidt H, Goldsberry A, Houser M, Krauth M, Lambers Heerspink HJ, McMurray JJ, Meyer CJ, Parving H-H, et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med. 2013; 369: 2492–503. doi: 10.1056/NEJMoa1306033.
73. WARBURG O. On the origin of cancer cells. Science. 1956; 123: 309–14. Available from http://www.ncbi.nlm.nih.gov/pubmed/13298683.
74. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324: 1029–33. doi: 10.1126/science.1160809.
75. Macheda ML, Rogers S, Best JD. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol. 2005; 202: 654–62. doi: 10.1002/jcp.20166.
76. Szablewski L. Expression of glucose transporters in cancers. Biochim Biophys Acta. 2013; 1835: 164–9. doi: 10.1016/j.bbcan.2012.12.004.
77. Gnudi L, Thomas SM, Viberti G. Mechanical forces in diabetic kidney disease: a trigger for impaired glucose metabolism. J Am Soc Nephrol. 2007; 18: 2226–32. doi: 10.1681/ASN.2006121362.
78. Luo Y, Ohmori H, Shimomoto T, Fujii K, Sasahira T, Chihara Y, Kuniyasu H. Anti-angiotensin and hypoglycemic treatments suppress liver metastasis of colon cancer cells. Pathobiology. 2011; 78: 285–90. doi: 10.1159/000330169.
79. Dai C, Stolz DB, Kiss LP, Monga SP, Holzman LB, Liu Y. Wnt/beta-catenin signaling promotes podocyte dysfunction and albuminuria. J Am Soc Nephrol. 2009; 20: 1997–2008. doi: 10.1681/ASN.2009010019.
80. Lin C-L, Wang J-Y, Huang Y-T, Kuo Y-H, Surendran K, Wang F-S. Wnt/beta-catenin signaling modulates survival of high glucose-stressed mesangial cells. J Am Soc Nephrol . 2006; 17: 2812–20. doi: 10.1681/ASN.2005121355.
81. Li J-Y, Yu T, Xia Z-S, Chen G-C, Yuan Y-H, Zhong W, Zhao L-N, Chen Q-K. Enhanced proliferation in colorectal epithelium of patients with type 2 diabetes correlates with β-catenin accumulation. J Diabetes Complications. 2016; 28: 689–97. doi: 10.1016/j.jdiacomp.2014.05.001.
82. Larriba MJ, González-Sancho JM, Barbáchano A, Niell N, Ferrer-Mayorga G, Muñoz A. Vitamin D Is a Multilevel Repressor of Wnt/b-Catenin Signaling in Cancer Cells. Cancers (Basel). 2013; 5: 1242–60. doi: 10.3390/cancers5041242.
83. He W, Kang YS, Dai C, Liu Y. Blockade of Wnt/β-catenin signaling by paricalcitol ameliorates proteinuria and kidney injury. J Am Soc Nephrol. 2011; 22: 90–103. doi: 10.1681/ASN.2009121236.
84. Holick MF. Vitamin D deficiency. N Engl J Med. 2007; 357: 266–81. doi: 10.1056/NEJMra070553.
85. Feldman D, Krishnan A V, Swami S, Giovannucci E, Feldman BJ. The role of vitamin D in reducing cancer risk and progression. Nat Rev Cancer. 2014; 14: 342–57. doi: 10.1038/nrc3691.
86. Pereira F, Larriba MJ, Muñoz A. Vitamin D and colon cancer. Endocr Relat Cancer. 2012; 19: R51-71. doi: 10.1530/ERC-11-0388.
87. Ortiz A, Ziyadeh FN, Neilson EG. Expression of apoptosis-regulatory genes in renal proximal tubular epithelial cells exposed to high ambient glucose and in diabetic kidneys. J Investig Med. 1997; 45: 50–6. Available from http://www.ncbi.nlm.nih.gov/pubmed/9084575.
88. Sanchez-Niño M-D, Bozic M, Córdoba-Lanús E, Valcheva P, Gracia O, Ibarz M, Fernandez E, Navarro-Gonzalez JF, Ortiz A, Valdivielso JM. Beyond proteinuria: VDR activation reduces renal inflammation in experimental diabetic nephropathy. Am J Physiol Renal Physiol. 2012; 302: F647-57. doi: 10.1152/ajprenal.00090.2011.
89. Sanchez-Niño M-D, Benito-Martin A, Ortiz A. New paradigms in cell death in human diabetic nephropathy. Kidney Int. 2010; 78: 737–44. doi: 10.1038/ki.2010.270
90. Pérez-Gómez MV, Ortiz-Arduán A, Lorenzo-Sellares V. Vitamin D and proteinuria: a critical review of molecular bases and clinical experience. Nefrol publicación Of la Soc Española Nefrol. 2013; 33: 716–26. doi: 10.3265/Nefrologia.pre2013.Apr.12025.
91. Lautrette A, Li S, Alili R, Sunnarborg SW, Burtin M, Lee DC, Friedlander G, Terzi F. Angiotensin II and EGF receptor cross-talk in chronic kidney diseases: a new therapeutic approach. Nat Med. 2005; 11: 867–74. doi: 10.1038/nm1275.
92. Cai W, He JC, Zhu L, Lu C, Vlassara H. Advanced glycation end product (AGE) receptor 1 suppresses cell oxidant stress and activation signaling via EGF receptor. Proc Natl Acad Sci U S A. 2006; 103: 13801–6. doi: 10.1073/pnas.0600362103.
93. Taniguchi K, Xia L, Goldberg HJ, Lee KWK, Shah A, Stavar L, Masson EAY, Momen A, Shikatani EA, John R, Husain M, Fantus IG. Inhibition of Src kinase blocks high glucose-induced EGFR transactivation and collagen synthesis in mesangial cells and prevents diabetic nephropathy in mice. Diabetes. 2013; 62: 3874–86. doi: 10.2337/db12-1010.
94. Rayego-Mateos S, Morgado-Pascual JL, Sanz AB, Ramos AM, Eguchi S, Batlle D, Pato J, Keri G, Egido J, Ortiz A, Ruiz-Ortega M. TWEAK transactivation of the epidermal growth factor receptor mediates renal inflammation. J Pathol. 2013; 231: 480–94. doi: 10.1002/path.4250.
95. Sanz AB, Izquierdo MC, Sanchez-Niño MD, Ucero AC, Egido J, Ruiz-Ortega M, Ramos AM, Putterman C, Ortiz A. TWEAK and the progression of renal disease: clinical translation. Nephrol Dial Transplant. 2014; 29 Suppl 1: i54–62. doi: 10.1093/ndt/gft342.
96. Ardura JA, Rayego-Mateos S, Rámila D, Ruiz-Ortega M, Esbrit P. Parathyroid hormone-related protein promotes epithelial-mesenchymal transition. J Am Soc Nephrol. 2010; 21: 237–48. doi: 10.1681/ASN.2009050462.
97. Trebing J, Lang I, Chopra M, Salzmann S, Moshir M, Silence K, Riedel SS, Siegmund D, Beilhack A, Otto C, Wajant H. A novel llama antibody targeting Fn14 exhibits anti-metastatic activity in vivo. MAbs. 2016; 6: 297–308. doi: 10.4161/mabs.26709.
98. Zhang M-Z, Wang Y, Paueksakon P, Harris RC. Epidermal growth factor receptor inhibition slows progression of diabetic nephropathy in association with a decrease in endoplasmic reticulum stress and an increase in autophagy. Diabetes. 2014; 63: 2063–72. doi: 10.2337/db13-1279.
99. Gödel M, Hartleben B, Herbach N, Liu S, Zschiedrich S, Lu S, Debreczeni-Mór A, Lindenmeyer MT, Rastaldi M-P, Hartleben G, Wiech T, Fornoni A, Nelson RG, et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest . 2011; 121: 2197–209. doi: 10.1172/JCI44774.
100. Rayego-Mateos S, Rodrigues-Díez R, Morgado-Pascual JL, Rodrigues Díez RR, Mas S, Lavoz C, Alique M, Pato J, Keri G, Ortiz A, Egido J, Ruiz-Ortega M. Connective tissue growth factor is a new ligand of epidermal growth factor receptor. J Mol Cell Biol. 2013; 5: 323–35. doi: 10.1093/jmcb/mjt030.
101. Guha M, Xu Z-G, Tung D, Lanting L, Natarajan R. Specific down-regulation of connective tissue growth factor attenuates progression of nephropathy in mouse models of type 1 and type 2 diabetes. FASEB J. 2007; 21: 3355–68. doi: 10.1096/fj.06-6713com.
102. Chang C-C, Lin B-R, Wu T-S, Jeng Y-M, Kuo M-L. Input of microenvironmental regulation on colorectal cancer: role of the CCN family. World J Gastroenterol. 2014; 20: 6826–31. doi: 10.3748/wjg.v20.i22.6826.
103. Izquierdo MC, Perez-Gomez M V, Sanchez-Niño MD, Sanz AB, Ruiz-Andres O, Poveda J, Moreno JA, Egido J, Ortiz A. Klotho, phosphate and inflammation/ageing in chronic kidney disease. Nephrol Dial Transplant. 2012; 27 Suppl 4: iv6-10. doi: 10.1093/ndt/gfs426.
104. Sanchez-Niño MD, Sanz AB, Ortiz A. Klotho to treat kidney fibrosis. J Am Soc Nephrol. 2013; 24: 687–9. doi: 10.1681/ASN.2013030294.
105. Wu C, Wang Q, Lv C, Qin N, Lei S, Yuan Q, Wang G. The changes of serum sKlotho and NGAL levels and their correlation in type 2 diabetes mellitus patients with different stages of urinary albumin. Diabetes Res Clin Pract. 2014; 106: 343–50. doi: 10.1016/j.diabres.2014.08.026.
106. Cheng M-F, Chen L-J, Cheng J-T. Decrease of Klotho in the kidney of streptozotocin-induced diabetic rats. J Biomed Biotechnol. 2010; 2010: 513853. doi: 10.1155/2010/513853.
107. Moreno JA, Izquierdo MC, Sanchez-Niño MD, Suárez-Alvarez B, Lopez-Larrea C, Jakubowski A, Blanco J, Ramirez R, Selgas R, Ruiz-Ortega M, Egido J, Ortiz A, Sanz AB. The inflammatory cytokines TWEAK and TNFα reduce renal klotho expression through NFκB. J Am Soc Nephrol. 2011; 22: 1315–25. doi: 10.1681/ASN.2010101073.
108. Sastre C, Rubio-Navarro A, Buendía I, Gómez-Guerrero C, Blanco J, Mas S, Egido J, Blanco-Colio LM, Ortiz A, Moreno JA. Hyperlipidemia-associated renal damage decreases Klotho expression in kidneys from ApoE knockout mice. PLoS One. 2013; 8: e83713. doi: 10.1371/journal.pone.0083713.
109. Zhou L, Li Y, Zhou D, Tan RJ, Liu Y. Loss of Klotho contributes to kidney injury by derepression of Wnt/β-catenin signaling. J Am Soc Nephrol. 2013; 24: 771–85. doi: 10.1681/ASN.2012080865.
110. Li X-X, Huang L-Y, Peng J-J, Liang L, Shi D-B, Zheng H-T, Cai S-J. Klotho suppresses growth and invasion of colon cancer cells through inhibition of IGF1R-mediated PI3K/AKT pathway. Int J Oncol. 2014; 45: 611–8. doi: 10.3892/ijo.2014.2430.
111. Pan J, Zhong J, Gan LH, Chen SJ, Jin HC, Wang X, Wang LJ. Klotho, an anti-senescence related gene, is frequently inactivated through promoter hypermethylation in colorectal cancer. Tumour Biol. 2011; 32: 729–35. doi: 10.1007/s13277-011-0174-5.
112. Odegaard JI, Chawla A. Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science. 2013; 339: 172–7. doi: 10.1126/science.1230721.
113. Nguyen A V, Wu Y-Y, Lin EY. STAT3 and sphingosine-1-phosphate in inflammation-associated colorectal cancer. World J Gastroenterol. 2014; 20: 10279–87. doi: 10.3748/wjg.v20.i30.10279.
114. Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004; 431: 461–6. doi: 10.1038/nature02924.
115. Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE. Stat3 as an oncogene. Cell. 1999; 98: 295–303. Available from http://www.ncbi.nlm.nih.gov/pubmed/10458605.
116. O’Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med. 2013; 368: 161–70. doi: 10.1056/NEJMra1202117.
117. Sanz AB, Sanchez-Niño MD, Ramos AM, Moreno JA, Santamaria B, Ruiz-Ortega M, Egido J, Ortiz A. NF-kappaB in renal inflammation. J Am Soc Nephrol. 2010; 21: 1254–62. doi: 10.1681/ASN.2010020218.
118. Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang W-C, Hait NC, Allegood JC, Price MM, Avni D, Takabe K, Kordula T, Milstien S, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell. 2013; 23: 107–20. doi: 10.1016/j.ccr.2012.11.013.
119. Sheng L, Zhou Y, Chen Z, Ren D, Cho KW, Jiang L, Shen H, Sasaki Y, Rui L. NF-κB–inducing kinase (NIK) promotes hyperglycemia and glucose intolerance in obesity by augmenting glucagon action. Nat Med. 2012; 18: 943–9. doi: 10.1038/nm.2756.
120. Rosebeck S, Madden L, Jin X, Gu S, Apel IJ, Appert A, Hamoudi RA, Noels H, Sagaert X, Van Loo P, Baens M, Du M-Q, Lucas PC, et al. Cleavage of NIK by the API2-MALT1 fusion oncoprotein leads to noncanonical NF-kappaB activation. Science. 2011; 331: 468–72. doi: 10.1126/science.1198946.
121. Gochman E, Mahajna J, Reznick AZ. NF-κB activation by peroxynitrite through IκBα-dependent phosphorylation versus nitration in colon cancer cells. Anticancer Res. 2011; 31: 1607–17. Available from http://www.ncbi.nlm.nih.gov/pubmed/21617217.
122. Malle EK, Zammit NW, Walters SN, Koay YC, Wu J, Tan BM, Villanueva JE, Brink R, Loudovaris T, Cantley J, McAlpine SR, Hesselson D, Grey ST. Nuclear factor κB-inducing kinase activation as a mechanism of pancreatic β cell failure in obesity. J Exp Med. 2015; 212: 1239–54. doi: 10.1084/jem.20150218.
123. Ortiz A, Husi H, Gonzalez-Lafuente L, Valiño-Rivas L, Fresno M, Sanz AB, Mullen W, Albalat A, Mezzano S, Vlahou T, Mischak H, Sanchez-Niño MD. Mitogen-Activated Protein Kinase 14 Promotes AKI. J Am Soc Nephrol. 2016.
124. Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, Woodford R-MT, Davis BK, Uronis JM, Herfarth HH, Jobin C, Rogers AB, Ting JP-Y. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity. 2012; 36: 742–54. doi: 10.1016/j.immuni.2012.03.012.
125. Sciacca L, Vigneri R, Tumminia A, Frasca F, Squatrito S, Frittitta L, Vigneri P. Clinical and molecular mechanisms favoring cancer initiation and progression in diabetic patients. Nutr Metab Cardiovasc Dis. 2013; 23: 808–15. doi: 10.1016/j.numecd.2013.05.006.
126. He G, Karin M. NF-κB and STAT3 - key players in liver inflammation and cancer. Cell Res. 2011; 21: 159–68. doi: 10.1038/cr.2010.183.
127. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, Flavell RA. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011; 145: 745–57. doi: 10.1016/j.cell.2011.04.022.
128. Chen GY, Liu M, Wang F, Bertin J, Núñez G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol. 2011; 186: 7187–94. doi: 10.4049/jimmunol.1100412.
129. Khan MT, Nieuwdorp M, Bäckhed F. Microbial modulation of insulin sensitivity. Cell Metab. 2014; 20: 753–60. doi: 10.1016/j.cmet.2014.07.006.
130. Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, Camporez J-P, Shulman GI, Gordon JI, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012; 482: 179–85. doi: 10.1038/nature10809.
131. Integrative HMP (iHMP) Research Network Consortium. The Integrative Human Microbiome Project: dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe. 2014; 16: 276–89. doi: 10.1016/j.chom.2014.08.014.
132. Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012; 490: 55–60. doi: 10.1038/nature11450.
133. Baxter NT, Zackular JP, Chen GY, Schloss PD. Structure of the gut microbiome following colonization with human feces determines colonic tumor burden. Microbiome. 2014; 2: 20. doi: 10.1186/2049-2618-2-20.
134. Khan S, Jena G. Sodium butyrate, a HDAC inhibitor ameliorates eNOS, iNOS and TGF-β1-induced fibrogenesis, apoptosis and DNA damage in the kidney of juvenile diabetic rats. Food Chem Toxicol. 2014; 73: 127–39. doi: 10.1016/j.fct.2014.08.010.
135. Lui GYL, Kovacevic Z, Richardson V, Merlot AM, Kalinowski DS, Richardson DR. Targeting cancer by binding iron: Dissecting cellular signaling pathways. Oncotarget. 2015; 6: 18748–79. doi: 10.18632/oncotarget.4349.
136. Song S, Christova T, Perusini S, Alizadeh S, Bao R-Y, Miller BW, Hurren R, Jitkova Y, Gronda M, Isaac M, Joseph B, Subramaniam R, Aman A, et al. Wnt inhibitor screen reveals iron dependence of β-catenin signaling in cancers. Cancer Res. 2011; 71: 7628–39. doi: 10.1158/0008-5472.CAN-11-2745.
137. Banerjee A, Mifsud NA, Bird R, Forsyth C, Szer J, Tam C, Kellner S, Grigg A, Motum P, Bentley M, Opat S, Grigoriadis G. The oral iron chelator deferasirox inhibits NF-κB mediated gene expression without impacting on proximal activation: implications for myelodysplasia and aplastic anaemia. Br J Haematol. 2015; 168: 576–82. doi: 10.1111/bjh.13151.
138. Dongiovanni P, Valenti L, Ludovica Fracanzani A, Gatti S, Cairo G, Fargion S. Iron depletion by deferoxamine up-regulates glucose uptake and insulin signaling in hepatoma cells and in rat liver. Am J Pathol. 2008; 172: 738–47. doi: 10.2353/ajpath.2008.070097.
139. Fernández-Real JM, McClain D, Manco M. Mechanisms Linking Glucose Homeostasis and Iron Metabolism Toward the Onset and Progression of Type 2 Diabetes. Diabetes Care. 2015; 38: 2169–76. doi: 10.2337/dc14-3082.
140. Morita T, Nakano D, Kitada K, Morimoto S, Ichihara A, Hitomi H, Kobori H, Shiojima I, Nishiyama A. Chelation of dietary iron prevents iron accumulation and macrophage infiltration in the type I diabetic kidney. Eur J Pharmacol. 2015; 756: 85–91. doi: 10.1016/j.ejphar.2015.03.053.
141. Lamas GA, Goertz C, Boineau R, Mark DB, Rozema T, Nahin RL, Lindblad L, Lewis EF, Drisko J, Lee KL, TACT Investigators. Effect of disodium EDTA chelation regimen on cardiovascular events in patients with previous myocardial infarction: the TACT randomized trial. JAMA. 2013; 309: 1241–50. doi: 10.1001/jama.2013.2107.
142. Bastide NM, Chenni F, Audebert M, Santarelli RL, Taché S, Naud N, Baradat M, Jouanin I, Surya R, Hobbs DA, Kuhnle GG, Raymond-Letron I, Gueraud F, et al. A central role for heme iron in colon carcinogenesis associated with red meat intake. Cancer Res. 2015; 75: 870–9. doi: 10.1158/0008-5472.CAN-14-2554.
143. Kim Y, Keogh J, Clifton P. A review of potential metabolic etiologies of the observed association between red meat consumption and development of type 2 diabetes mellitus. Metabolism. 2015; 64: 768–79. doi: 10.1016/j.metabol.2015.03.008.
144. Ørntoft M-BW, Nielsen HJ, Ørntoft TF, Andersen CL, Danish Study Group on Early Detection of Colorectal Cancer. Performance of the colorectal cancer screening marker Sept9 is influenced by age, diabetes and arthritis: a nested case-control study. BMC Cancer. 2015; 15: 819. doi: 10.1186/s12885-015-1832-6.
145. Dayeh T, Volkov P, Salö S, Hall E, Nilsson E, Olsson AH, Kirkpatrick CL, Wollheim CB, Eliasson L, Rönn T, Bacos K, Ling C. Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 2014; 10: e1004160. doi: 10.1371/journal.pgen.1004160.
146. Trionfini P, Benigni A, Remuzzi G. MicroRNAs in kidney physiology and disease. Nat Rev Nephrol. 2015; 11: 23–33. doi: 10.1038/nrneph.2014.202.
147. Goossens-Beumer IJ, Derr RS, Buermans HPJ, Goeman JJ, Böhringer S, Morreau H, Nitsche U, Janssen K-P, van de Velde CJH, Kuppen PJK. MicroRNA classifier and nomogram for metastasis prediction in colon cancer. Cancer Epidemiol Biomarkers Prev. 2015; 24: 187–97. doi: 10.1158/1055-9965.EPI-14-0544-T.
148. Zhang J-X, Song W, Chen Z-H, Wei J-H, Liao Y-J, Lei J, Hu M, Chen G-Z, Liao B, Lu J, Zhao H-W, Chen W, He Y-L, et al. Prognostic and predictive value of a microRNA signature in stage II colon cancer: a microRNA expression analysis. Lancet Oncol. 2013; 14: 1295–306. doi: 10.1016/S1470-2045(13)70491-1.
149. Zhong X, Chung ACK, Chen HY, Dong Y, Meng XM, Li R, Yang W, Hou FF, Lan HY. miR-21 is a key therapeutic target for renal injury in a mouse model of type 2 diabetes. Diabetologia. 2013; 56: 663–74. doi: 10.1007/s00125-012-2804-x.
150. Dey N, Das F, Mariappan MM, Mandal CC, Ghosh-Choudhury N, Kasinath BS, Choudhury GG. MicroRNA-21 orchestrates high glucose-induced signals to TOR complex 1, resulting in renal cell pathology in diabetes. J Biol Chem. 2011; 286: 25586–603. doi: 10.1074/jbc.M110.208066.
151. Zhang Z, Peng H, Chen J, Chen X, Han F, Xu X, He X, Yan N. MicroRNA-21 protects from mesangial cell proliferation induced by diabetic nephropathy in db/db mice. FEBS Lett. 2009; 583: 2009–14. doi: 10.1016/j.febslet.2009.05.021.
152. Hansen TF, Kjær-Frifeldt S, Christensen RD, Morgenthaler S, Blondal T, Lindebjerg J, Sørensen FB, Jakobsen A. Redefining high-risk patients with stage II colon cancer by risk index and microRNA-21: results from a population-based cohort. Br J Cancer. 2014; 111: 1285–92. doi: 10.1038/bjc.2014.409.
153. Zhang J, Xiao Z, Lai D, Sun J, He C, Chu Z, Ye H, Chen S, Wang J. miR-21, miR-17 and miR-19a induced by phosphatase of regenerating liver-3 promote the proliferation and metastasis of colon cancer. Br J Cancer. 2012; 107: 352–9. doi: 10.1038/bjc.2012.251.
154. Deng J, Lei W, Fu J-C, Zhang L, Li J-H, Xiong J-P. Targeting miR-21 enhances the sensitivity of human colon cancer HT-29 cells to chemoradiotherapy in vitro. Biochem Biophys Res Commun. 2014; 443: 789–95. doi: 10.1016/j.bbrc.2013.11.064.
155. Song M-S, Rossi JJ. The anti-miR21 antagomir, a therapeutic tool for colorectal cancer, has a potential synergistic effect by perturbing an angiogenesis-associated miR30. Front Genet. 2014; 4: 301. doi: 10.3389/fgene.2013.00301.
156. Nangia-Makker P, Yu Y, Vasudevan A, Farhana L, Rajendra SG, Levi E, Majumdar APN. Metformin: a potential therapeutic agent for recurrent colon cancer. PLoS One. 2014; 9: e84369. doi: 10.1371/journal.pone.0084369.
157. Grant SFA, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, Helgason A, Stefansson H, Emilsson V, Helgadottir A, Styrkarsdottir U, Magnusson KP, Walters GB, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006; 38: 320–3. doi: 10.1038/ng1732.
158. Saxena R, Elbers CC, Guo Y, Peter I, Gaunt TR, Mega JL, Lanktree MB, Tare A, Castillo BA, Li YR, Johnson T, Bruinenberg M, Gilbert-Diamond D, et al. Large-scale gene-centric meta-analysis across 39 studies identifies type 2 diabetes loci. Am J Hum Genet. 2012; 90: 410–25. doi: 10.1016/j.ajhg.2011.12.022.
159. Ng MCY, Shriner D, Chen BH, Li J, Chen W-M, Guo X, Liu J, Bielinski SJ, Yanek LR, Nalls MA, Comeau ME, Rasmussen-Torvik LJ, Jensen RA, et al. Meta-analysis of genome-wide association studies in African Americans provides insights into the genetic architecture of type 2 diabetes. PLoS Genet. 2014; 10: e1004517. doi: 10.1371/journal.pgen.1004517.
160. Franceschini N, Shara NM, Wang H, Voruganti VS, Laston S, Haack K, Lee ET, Best LG, Maccluer JW, Cochran BJ, Dyer TD, Howard B V, Cole SA, et al. The association of genetic variants of type 2 diabetes with kidney function. Kidney Int. 2012; 82: 220–5. doi: 10.1038/ki.2012.107.
161. Zhang B, Jia W-H, Matsuda K, Kweon S-S, Matsuo K, Xiang Y-B, Shin A, Jee SH, Kim D-H, Cai Q, Long J, Shi J, Wen W, et al. Large-scale genetic study in East Asians identifies six new loci associated with colorectal cancer risk. Nat Genet. 2014; 46: 533–42. doi: 10.1038/ng.2985.
162. Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012; 149: 1192–205. doi: 10.1016/j.cell.2012.05.012
163. Lan F, Yue X, Han L, Shi Z, Yang Y, Pu P, Yao Z, Kang C. Genome-wide identification of TCF7L2/TCF4 target miRNAs reveals a role for miR-21 in Wnt-driven epithelial cancer. Int J Oncol. 2012; 40: 519–26. doi: 10.3892/ijo.2011.1215.
164. Tuupanen S, Turunen M, Lehtonen R, Hallikas O, Vanharanta S, Kivioja T, Björklund M, Wei G, Yan J, Niittymäki I, Mecklin J-P, Järvinen H, Ristimäki A, et al. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat Genet. 2009; 41: 885–90. doi: 10.1038/ng.406.
165. Lewis A, Freeman-Mills L, de la Calle-Mustienes E, Giráldez-Pérez RM, Davis H, Jaeger E, Becker M, Hubner NC, Nguyen LN, Zeron-Medina J, Bond G, Stunnenberg HG, Carvajal JJ, et al. A polymorphic enhancer near GREM1 influences bowel cancer risk through differential CDX2 and TCF7L2 binding. Cell Rep. 2014; 8: 983–90. doi: 10.1016/j.celrep.2014.07.020.
166. Jaeger E, Leedham S, Lewis A, Segditsas S, Becker M, Cuadrado PR, Davis H, Kaur K, Heinimann K, Howarth K, East J, Taylor J, Thomas H, et al. Hereditary mixed polyposis syndrome is caused by a 40-kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nat Genet. 2012; 44: 699–703. doi: 10.1038/ng.2263.
167. Davis H, Irshad S, Bansal M, Rafferty H, Boitsova T, Bardella C, Jaeger E, Lewis A, Freeman-Mills L, Giner FC, Rodenas-Cuadrado P, Mallappa S, Clark S, et al. Aberrant epithelial GREM1 expression initiates colonic tumorigenesis from cells outside the stem cell niche. Nat Med. 2015; 21: 62–70. doi: 10.1038/nm.3750.
168. McMahon R, Murphy M, Clarkson M, Taal M, Mackenzie HS, Godson C, Martin F, Brady HR. IHG-2, a mesangial cell gene induced by high glucose, is human gremlin. Regulation by extracellular glucose concentration, cyclic mechanical strain, and transforming growth factor-beta1. J Biol Chem. 2000; 275: 9901–4. Available from http://www.ncbi.nlm.nih.gov/pubmed/10744662.
169. McKnight AJ, Patterson CC, Pettigrew KA, Savage DA, Kilner J, Murphy M, Sadlier D, Maxwell AP, Warren 3/U.K. Genetics of Kidneys in Diabetes (GoKinD) Study Group. A GREM1 gene variant associates with diabetic nephropathy. J Am Soc Nephrol. 2010; 21: 773–81. doi: 10.1681/ASN.2009070773.
170. Wada J, Sun L, Kanwar YS. Discovery of genes related to diabetic nephropathy in various animal models by current techniques. Contrib Nephrol. 2011; 169: 161–74. doi: 10.1159/000313951.
171. Marchant V, Droguett A, Valderrama G, Burgos ME, Carpio D, Kerr B, Ruiz-Ortega M, Egido J, Mezzano S. Tubular overexpression of Gremlin in transgenic mice aggravates renal damage in diabetic nephropathy. Am J Physiol Renal Physiol. 2015; 309: F559-68. doi: 10.1152/ajprenal.00023.2015.
172. Lavoz C, Alique M, Rodrigues-Diez R, Pato J, Keri G, Mezzano S, Egido J, Ruiz-Ortega M. Gremlin regulates renal inflammation via the vascular endothelial growth factor receptor 2 pathway. J Pathol. 2015; 236: 407–20. doi: 10.1002/path.4537.
173. Roxburgh SA, Kattla JJ, Curran SP, O’Meara YM, Pollock CA, Goldschmeding R, Godson C, Martin F, Brazil DP. Allelic depletion of grem1 attenuates diabetic kidney disease. Diabetes. 2009; 58: 1641–50. doi: 10.2337/db08-1365.
174. Karagiannis GS, Berk A, Dimitromanolakis A, Diamandis EP. Enrichment map profiling of the cancer invasion front suggests regulation of colorectal cancer progression by the bone morphogenetic protein antagonist, gremlin-1. Mol Oncol. 2013; 7: 826–39. doi: 10.1016/j.molonc.2013.04.002.
175. Rodrigues-Diez R, Lavoz C, Carvajal G, Rayego-Mateos S, Rodrigues Diez RR, Ortiz A, Egido J, Mezzano S, Ruiz-Ortega M. Gremlin is a downstream profibrotic mediator of transforming growth factor-beta in cultured renal cells. Nephron Exp Nephrol. 2012; 122: 62–74. doi: 10.1159/000346575.
176. Malnutrition and tissue injury in the chronic alcoholic. A symposium. London, U.K., 16 October 1984. Alcohol Alcohol. 1985; 20: 87–249. Available from http://www.ncbi.nlm.nih.gov/pubmed/4052161.
177. Tang W, Dodge M, Gundapaneni D, Michnoff C, Roth M, Lum L. A genome-wide RNAi screen for Wnt/beta-catenin pathway components identifies unexpected roles for TCF transcription factors in cancer. Proc Natl Acad Sci U S A. 2008; 105: 9697–702. doi: 10.1073/pnas.0804709105.
178. Yasuda K, Miyake K, Horikawa Y, Hara K, Osawa H, Furuta H, Hirota Y, Mori H, Jonsson A, Sato Y, Yamagata K, Hinokio Y, Wang H-Y, et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat Genet. 2008; 40: 1092–7. doi: 10.1038/ng.207.
179. Sunamura N, Ohira T, Kataoka M, Inaoka D, Tanabe H, Nakayama Y, Oshimura M, Kugoh H. Regulation of functional KCNQ1OT1 lncRNA by β-catenin. Sci Rep. 2016; 6: 20690. doi: 10.1038/srep20690.
180. Than BLN, Goos JACM, Sarver AL, O’Sullivan MG, Rod A, Starr TK, Fijneman RJA, Meijer GA, Zhao L, Zhang Y, Largaespada DA, Scott PM, Cormier RT. The role of KCNQ1 in mouse and human gastrointestinal cancers. Oncogene. 2014; 33: 3861–8. doi: 10.1038/onc.2013.350.
181. Alkayyali S, Lajer M, Deshmukh H, Ahlqvist E, Colhoun H, Isomaa B, Rossing P, Groop L, Lyssenko V. Common variant in the HMGA2 gene increases susceptibility to nephropathy in patients with type 2 diabetes. Diabetologia. 2013; 56: 323–9. doi: 10.1007/s00125-012-2760-5.
182. Fedele M, Bandiera A, Chiappetta G, Battista S, Viglietto G, Manfioletti G, Casamassimi A, Santoro M, Giancotti V, Fusco A. Human colorectal carcinomas express high levels of high mobility group HMGI(Y) proteins. Cancer Res. 1996; 56: 1896–901. Available from http://www.ncbi.nlm.nih.gov/pubmed/8620511.
183. Wu J, Wang Y, Xu X, Cao H, Sahengbieke S, Sheng H, Huang Q, Lai M. Transcriptional activation of FN1 and IL11 by HMGA2 promotes the malignant behavior of colorectal cancer. Carcinogenesis. 2016; 37: 511–21. doi: 10.1093/carcin/bgw029.
184. Hodgin JB, Nair V, Zhang H, Randolph A, Harris RC, Nelson RG, Weil EJ, Cavalcoli JD, Patel JM, Brosius FC, Kretzler M. Identification of cross-species shared transcriptional networks of diabetic nephropathy in human and mouse glomeruli. Diabetes. 2013; 62: 299–308. doi: 10.2337/db11-1667.
185. Kang BW, Jeon H-S, Chae YS, Lee SJ, Park JY, Choi JE, Park JS, Choi GS, Kim JG. Association between GWAS-identified genetic variations and disease prognosis for patients with colorectal cancer. PLoS One. 2015; 10: e0119649. doi: 10.1371/journal.pone.0119649.
186. Lascorz J, Chen B, Hemminki K, Försti A. Consensus pathways implicated in prognosis of colorectal cancer identified through systematic enrichment analysis of gene expression profiling studies. PLoS One. 2011; 6: e18867. doi: 10.1371/journal.pone.0018867.
187. Rigothier C, Auguste P, Welsh GI, Lepreux S, Deminière C, Mathieson PW, Saleem MA, Ripoche J, Combe C. IQGAP1 interacts with components of the slit diaphragm complex in podocytes and is involved in podocyte migration and permeability in vitro. PLoS One. 2012; 7: e37695. doi: 10.1371/journal.pone.0037695.
188. Schmid H, Boucherot A, Yasuda Y, Henger A, Brunner B, Eichinger F, Nitsche A, Kiss E, Bleich M, Gröne H-J, Nelson PJ, Schlöndorff D, Cohen CD, et al. Modular activation of nuclear factor-kappaB transcriptional programs in human diabetic nephropathy. Diabetes. 2006; 55: 2993–3003. doi: 10.2337/db06-0477.
189. Woroniecka KI, Park ASD, Mohtat D, Thomas DB, Pullman JM, Susztak K. Transcriptome analysis of human diabetic kidney disease. Diabetes. 2011; 60: 2354–69. doi: 10.2337/db10-1181.
190. Lorz C, Benito-Martín A, Boucherot A, Ucero AC, Rastaldi MP, Henger A, Armelloni S, Santamaría B, Berthier CC, Kretzler M, Egido J, Ortiz A. The death ligand TRAIL in diabetic nephropathy. J Am Soc Nephrol. 2008; 19: 904–14. doi: 10.1681/ASN.2007050581.
191. Sanchez-Niño MD, Sanz AB, Lorz C, Gnirke A, Rastaldi MP, Nair V, Egido J, Ruiz-Ortega M, Kretzler M, Ortiz A. BASP1 promotes apoptosis in diabetic nephropathy. J Am Soc Nephrol. 2010; 21: 610–21. doi: 10.1681/ASN.2009020227.
192. Sanchez-Niño MD, Sanz AB, Ihalmo P, Lassila M, Holthofer H, Mezzano S, Aros C, Groop P-H, Saleem M a, Mathieson PW, Langham R, Kretzler M, Nair V, et al. The MIF receptor CD74 in diabetic podocyte injury. J Am Soc Nephrol. 2009; 20: 353–62. doi: 10.1681/ASN.2008020194.
193. Ioannou K, Cheng KF, Crichlow G V, Birmpilis AI, Lolis EJ, Tsitsilonis OE, Al-Abed Y. ISO-66, a novel inhibitor of macrophage migration, shows efficacy in melanoma and colon cancer models. Int J Oncol. 2014; 45: 1457–68. doi: 10.3892/ijo.2014.2551.
194. Sanchez-Niño MD, Sanz AB, Ruiz-Andres O, Poveda J, Izquierdo MC, Selgas R, Egido J, Ortiz A. MIF, CD74 and other partners in kidney disease: tales of a promiscuous couple. Cytokine Growth Factor Rev. 2013; 24: 23–40. doi: 10.1016/j.cytogfr.2012.08.001.
195. Aebersold R, Bader GD, Edwards AM, van Eyk JE, Kussmann M, Qin J, Omenn GS. The biology/disease-driven human proteome project (B/D-HPP): enabling protein research for the life sciences community. J Proteome Res. 2013; 12: 23–7. doi: 10.1021/pr301151m.
196. Alvarez-Chaver P, Otero-Estévez O, Páez de la Cadena M, Rodríguez-Berrocal FJ, Martínez-Zorzano VS. Proteomics for discovery of candidate colorectal cancer biomarkers. World J Gastroenterol. 2014; 20: 3804–24. doi: 10.3748/wjg.v20.i14.3804.
197. Jimenez CR, Knol JC, Meijer GA, Fijneman RJA. Proteomics of colorectal cancer: overview of discovery studies and identification of commonly identified cancer-associated proteins and candidate CRC serum markers. J Proteomics. 2010; 73: 1873–95. doi: 10.1016/j.jprot.2010.06.004.
198. Wang K, Huang C, Nice EC. Proteomics, genomics and transcriptomics: their emerging roles in the discovery and validation of colorectal cancer biomarkers. Expert Rev Proteomics. 2014; 11: 179–205. doi: 10.1586/14789450.2014.894466.
199. de Wit M, Fijneman RJA, Verheul HMW, Meijer GA, Jimenez CR. Proteomics in colorectal cancer translational research: biomarker discovery for clinical applications. Clin Biochem. 2013; 46: 466–79. doi: 10.1016/j.clinbiochem.2012.10.039.
200. Ma Y, Zhang P, Wang F, Qin H. Searching for consistently reported up- and down-regulated biomarkers in colorectal cancer: a systematic review of proteomic studies. Mol Biol Rep. 2012; 39: 8483–90. doi: 10.1007/s11033-012-1702-0.
201. Sun J, Zhao M, Jia P, Wang L, Wu Y, Iverson C, Zhou Y, Bowton E, Roden DM, Denny JC, Aldrich MC, Xu H, Zhao Z. Deciphering Signaling Pathway Networks to Understand the Molecular Mechanisms of Metformin Action. PLoS Comput Biol. 2015; 11: e1004202. doi: 10.1371/journal.pcbi.1004202.
202. Ruiz-Andres O, Suarez-Alvarez B, Sánchez-Ramos C, Monsalve M, Sanchez-Niño MD, Ruiz-Ortega M, Egido J, Ortiz A, Sanz AB. The inflammatory cytokine TWEAK decreases PGC-1α expression and mitochondrial function in acute kidney injury. Kidney Int. 2016; 89: 399–410. doi: 10.1038/ki.2015.332.
203. Zanders MMJ, Haak HR, van Herk-Sukel MPP, van de Poll-Franse L V, Johnson JA. Impact of cancer on adherence to glucose-lowering drug treatment in individuals with diabetes. Diabetologia. 2015; 58: 951–60. doi: 10.1007/s00125-015-3497-8.
204. Chamberlain JJ, Rhinehart AS, Shaefer CF, Neuman A. Diagnosis and Management of Diabetes: Synopsis of the 2016 American Diabetes Association Standards of Medical Care in Diabetes. Ann Intern Med. 2016; 164: 542–52. doi: 10.7326/M15-3016.
205. Mei Z-B, Zhang Z-J, Liu C-Y, Liu Y, Cui A, Liang Z-L, Wang G-H, Cui L. Survival benefits of metformin for colorectal cancer patients with diabetes: a systematic review and meta-analysis. PLoS One. 2014; 9: e91818. doi: 10.1371/journal.pone.0091818.
206. Rizos C V, Elisaf MS. Metformin and cancer. Eur J Pharmacol. 2013; 705: 96–108. doi: 10.1016/j.ejphar.2013.02.038.
207. Morales DR, Morris AD. Metformin in cancer treatment and prevention. Annu Rev Med. 2015; 66: 17–29. doi: 10.1146/annurev-med-062613-093128.
208. Wild SH. Diabetes, treatments for diabetes and their effect on cancer incidence and mortality: attempts to disentangle the web of associations. Diabetologia. 2011; 54: 1589–92. doi: 10.1007/s00125-011-2169-6.
209. Yang Y-X, Hennessy S, Lewis JD. Insulin therapy and colorectal cancer risk among type 2 diabetes mellitus patients. Gastroenterology. 2004; 127: 1044–50. Available from http://www.ncbi.nlm.nih.gov/pubmed/15480982.
210. Franciosi M, Lucisano G, Lapice E, Strippoli GFM, Pellegrini F, Nicolucci A. Metformin therapy and risk of cancer in patients with type 2 diabetes: systematic review. PLoS One. 2013; 8: e71583. doi: 10.1371/journal.pone.0071583.
211. Guideline development group. Clinical Practice Guideline on management of patients with diabetes and chronic kidney disease stage 3b or higher (eGFR. Nephrol Dial Transplant. 2015; 30 Suppl 2: ii1-142. doi: 10.1093/ndt/gfv100.
212. Ortiz A, Covic A, Fliser D, Fouque D, Goldsmith D, Kanbay M, Mallamaci F, Massy ZA, Rossignol P, Vanholder R, Wiecek A, Zoccali C, London GM, et al. Epidemiology, contributors to, and clinical trials of mortality risk in chronic kidney failure. Lancet (London, England). 2014; 383: 1831–43. doi: 10.1016/S0140-6736(14)60384-6.
213. Maisonneuve P, Agodoa L, Gellert R, Stewart JH, Buccianti G, Lowenfels AB, Wolfe RA, Jones E, Disney AP, Briggs D, McCredie M, Boyle P. Cancer in patients on dialysis for end-stage renal disease: an international collaborative study. Lancet (London, England). 1999; 354: 93–9. Available from http://www.ncbi.nlm.nih.gov/pubmed/10408483.
214. Wu L, Zhu J, Prokop LJ, Murad MH. Pharmacologic Therapy of Diabetes and Overall Cancer Risk and Mortality: A Meta-Analysis of 265 Studies. Sci Rep. 2015; 5: 10147. doi: 10.1038/srep10147.
215. Yin S, Bai H, Jing D. Insulin therapy and colorectal cancer risk among type 2 diabetes mellitus patients: a systemic review and meta-analysis. Diagn Pathol. 2014; 9: 91. doi: 10.1186/1746-1596-9-91.
216. Simó R, Plana-Ripoll O, Puente D, Morros R, Mundet X, Vilca LM, Hernández C, Fuentes I, Procupet A, Tabernero JM, Violán C. Impact of glucose-lowering agents on the risk of cancer in type 2 diabetic patients. The Barcelona case-control study. PLoS One. 2013; 8: e79968. doi: 10.1371/journal.pone.0079968.
217. Colmers IN, Bowker SL, Tjosvold LA, Johnson JA. Insulin use and cancer risk in patients with type 2 diabetes: a systematic review and meta-analysis of observational studies. Diabetes Metab. 2012; 38: 485–506. doi: 10.1016/j.diabet.2012.08.011.
218. Sehdev A, Shih Y-CT, Vekhter B, Bissonnette MB, Olopade OI, Polite BN. Metformin for primary colorectal cancer prevention in patients with diabetes: a case-control study in a US population. Cancer. 2015; 121: 1071–8. doi: 10.1002/cncr.29165.
219. Rokkas T, Portincasa P. Colon neoplasia in patients with type 2 diabetes on metformin: A meta-analysis. Eur J Intern Med. 2016; 33: 60–6. doi: 10.1016/j.ejim.2016.05.027.
220. Lega IC, Shah PS, Margel D, Beyene J, Rochon PA, Lipscombe LL. The effect of metformin on mortality following cancer among patients with diabetes. Cancer Epidemiol Biomarkers Prev. 2014; 23: 1974–84. doi: 10.1158/1055-9965.EPI-14-0327.
221. Lee A, Morley JE. Metformin decreases food consumption and induces weight loss in subjects with obesity with type II non-insulin-dependent diabetes. Obes Res. 1998; 6: 47–53. Available from http://www.ncbi.nlm.nih.gov/pubmed/9526970.
222. Zakikhani M, Dowling RJO, Sonenberg N, Pollak MN. The effects of adiponectin and metformin on prostate and colon neoplasia involve activation of AMP-activated protein kinase. Cancer Prev Res (Phila). 2008; 1: 369–75. doi: 10.1158/1940-6207.CAPR-08-0081.
223. Tomimoto A, Endo H, Sugiyama M, Fujisawa T, Hosono K, Takahashi H, Nakajima N, Nagashima Y, Wada K, Nakagama H, Nakajima A. Metformin suppresses intestinal polyp growth in ApcMin/+ mice. Cancer Sci. 2008; 99: 2136–41. doi: 10.1111/j.1349-7006.2008.00933.x.
224. Zaafar DK, Zaitone SA, Moustafa YM. Role of metformin in suppressing 1,2-dimethylhydrazine-induced colon cancer in diabetic and non-diabetic mice: effect on tumor angiogenesis and cell proliferation. PLoS One. 2014; 9: e100562. doi: 10.1371/journal.pone.0100562.
225. Hunt SM, Osnos M, Rivlin RS. Thyroid hormone regulation of mitochondrial alpha-glycerophosphate dehydrogenase in liver and hepatoma. Cancer Res. 1970; 30: 1764–8. Available from http://www.ncbi.nlm.nih.gov/pubmed/4318703.
226. Karsten U, Sydow G, Wollenberger A, Graffi A. Rat liver glycerolphosphate dehydrogenases: activity changes and induction by thyroid hormone of the mitochondrial enzyme in hepatomas and in precancerous and growing liver. Acta Biol Med Ger. 1971; 26: 1131–40. Available from http://www.ncbi.nlm.nih.gov/pubmed/4340892.
227. Hosono K, Endo H, Takahashi H, Sugiyama M, Sakai E, Uchiyama T, Suzuki K, Iida H, Sakamoto Y, Yoneda K, Koide T, Tokoro C, Abe Y, et al. Metformin suppresses colorectal aberrant crypt foci in a short-term clinical trial. Cancer Prev Res (Phila). 2010; 3: 1077–83. doi: 10.1158/1940-6207.CAPR-10-0186.
228. Higurashi T, Hosono K, Takahashi H, Komiya Y, Umezawa S, Sakai E, Uchiyama T, Taniguchi L, Hata Y, Uchiyama S, Hattori A, Nagase H, Kessoku T, et al. Metformin for chemoprevention of metachronous colorectal adenoma or polyps in post-polypectomy patients without diabetes: a multicentre double-blind, placebo-controlled, randomised phase 3 trial. Lancet Oncol. 2016; 17: 475–83. doi: 10.1016/S1470-2045(15)00565-3.
229. Singh S, Singh H, Singh PP, Murad MH, Limburg PJ. Antidiabetic medications and the risk of colorectal cancer in patients with diabetes mellitus: a systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2013; 22: 2258–68. doi: 10.1158/1055-9965.EPI-13-0429.
230. Govindarajan R, Ratnasinghe L, Simmons DL, Siegel ER, Midathada M V, Kim L, Kim PJ, Owens RJ, Lang NP. Thiazolidinediones and the risk of lung, prostate, and colon cancer in patients with diabetes. J Clin Oncol. 2007; 25: 1476–81. doi: 10.1200/JCO.2006.07.2777.
231. Lin HC, Hsu YT, Kachingwe BH, Hsu CY, Uang YS, Wang LH. Dose effect of thiazolidinedione on cancer risk in type 2 diabetes mellitus patients: a six-year population-based cohort study. J Clin Pharm Ther. 2014; 39: 354–60. doi: 10.1111/jcpt.12151.
232. Yoshizumi T, Ohta T, Ninomiya I, Terada I, Fushida S, Fujimura T, Nishimura G-I, Shimizu K, Yi S, Miwa K. Thiazolidinedione, a peroxisome proliferator-activated receptor-gamma ligand, inhibits growth and metastasis of HT-29 human colon cancer cells through differentiation-promoting effects. Int J Oncol. 2004; 25: 631–9. Available from http://www.ncbi.nlm.nih.gov/pubmed/15289864.
233. Kitamura S, Miyazaki Y, Shinomura Y, Kondo S, Kanayama S, Matsuzawa Y. Peroxisome proliferator-activated receptor gamma induces growth arrest and differentiation markers of human colon cancer cells. Jpn J Cancer Res. 1999; 90: 75–80. Available from http://www.ncbi.nlm.nih.gov/pubmed/10076568.
234. Su W, Necela BM, Fujiwara K, Kurakata S, Murray NR, Fields AP, Thompson EA. The high affinity peroxisome proliferator-activated receptor-gamma agonist RS5444 inhibits both initiation and progression of colon tumors in azoxymethane-treated mice. Int J cancer. 2008; 123: 991–7. doi: 10.1002/ijc.23640.
235. Yang K, Fan K-H, Lamprecht SA, Edelmann W, Kopelovich L, Kucherlapati R, Lipkin M. Peroxisome proliferator-activated receptor gamma agonist troglitazone induces colon tumors in normal C57BL/6J mice and enhances colonic carcinogenesis in Apc1638 N/+ Mlh1+/- double mutant mice. Int J cancer. 2005; 116: 495–9. doi: 10.1002/ijc.21018.
236. Singh S, Singh PP, Singh AG, Murad MH, Sanchez W. Anti-diabetic medications and the risk of hepatocellular cancer: a systematic review and meta-analysis. Am J Gastroenterol. 2013; 108: 881–91; quiz 892. doi: 10.1038/ajg.2013.5.
237. Singh S, Singh PP, Singh AG, Murad MH, McWilliams RR, Chari ST. Anti-diabetic medications and risk of pancreatic cancer in patients with diabetes mellitus: a systematic review and meta-analysis. Am J Gastroenterol. 2013; 108: 510–9; quiz 520. doi: 10.1038/ajg.2013.7.
238. Tseng Y-H, Tsan Y-T, Chan W-C, Sheu WH-H, Chen P-C. Use of an α-Glucosidase Inhibitor and the Risk of Colorectal Cancer in Patients With Diabetes: A Nationwide, Population-Based Cohort Study. Diabetes Care. 2015; 38: 2068–74. doi: 10.2337/dc15-0563.
239. Lin C-M, Huang H-L, Chu F-Y, Fan H-C, Chen H-A, Chu D-M, Wu L-W, Wang C-C, Chen W-L, Lin S-H, Ho S-Y. Association between Gastroenterological Malignancy and Diabetes Mellitus and Anti-Diabetic Therapy: A Nationwide, Population-Based Cohort Study. PLoS One. 2015; 10: e0125421. doi: 10.1371/journal.pone.0125421.
240. Su B, Liu H, Li J, Sunli Y, Liu B, Liu D, Zhang P, Meng X. Acarbose treatment affects the serum levels of inflammatory cytokines and the gut content of bifidobacteria in Chinese patients with type 2 diabetes mellitus. J Diabetes. 2015; 7: 729–39. doi: 10.1111/1753-0407.12232.
241 Quesada CF, Kimata H, Mori M, Nishimura M, Tsuneyoshi T, Baba S. Piroxicam and acarbose as chemopreventive agents for spontaneous intestinal adenomas in APC gene 1309 knockout mice. Jpn J Cancer Res. 1998; 89: 392–6. Available from http://www.ncbi.nlm.nih.gov/pubmed/9617344.
242. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE, EMPA-REG OUTCOME Investigators. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N Engl J Med. 2015; 373: 2117–28. doi: 10.1056/NEJMoa1504720.
243. Scafoglio C, Hirayama BA, Kepe V, Liu J, Ghezzi C, Satyamurthy N, Moatamed NA, Huang J, Koepsell H, Barrio JR, Wright EM. Functional expression of sodium-glucose transporters in cancer. Proc Natl Acad Sci U S A. 2015; 112: E4111-9. doi: 10.1073/pnas.1511698112.
244. Elashoff M, Matveyenko A V, Gier B, Elashoff R, Butler PC. Pancreatitis, pancreatic, and thyroid cancer with glucagon-like peptide-1-based therapies. Gastroenterology. 2011; 141: 150–6. doi: 10.1053/j.gastro.2011.02.018.
245. Lee PH, Stockton MD, Franks AS. Acute pancreatitis associated with liraglutide. Ann Pharmacother. 2011; 45: e22. doi: 10.1345/aph.1P714.
246. Koehler JA, Kain T, Drucker DJ. Glucagon-like peptide-1 receptor activation inhibits growth and augments apoptosis in murine CT26 colon cancer cells. Endocrinology. 2011; 152: 3362–72. doi: 10.1210/en.2011-1201.
247. Ligumsky H, Wolf I, Israeli S, Haimsohn M, Ferber S, Karasik A, Kaufman B, Rubinek T. The peptide-hormone glucagon-like peptide-1 activates cAMP and inhibits growth of breast cancer cells. Breast Cancer Res Treat. 2012; 132: 449–61. doi: 10.1007/s10549-011-1585-0.
248. Gross CP, McAvay GJ, Guo Z, Tinetti ME. The impact of chronic illnesses on the use and effectiveness of adjuvant chemotherapy for colon cancer. Cancer. 2007; 109: 2410–9. doi: 10.1002/cncr.22726.
249. Meyerhardt JA, Catalano PJ, Haller DG, Mayer RJ, Macdonald JS, Benson AB, Fuchs CS. Impact of diabetes mellitus on outcomes in patients with colon cancer. J Clin Oncol. 2003; 21: 433–40. Available from http://www.ncbi.nlm.nih.gov/pubmed/12560431.
250. Ottaiano A, Nappi A, Tafuto S, Nasti G, De Divitiis C, Romano C, Cassata A, Casaretti R, Silvestro L, Avallone A, Capuozzo M, Capozzi M, Maiolino P, et al. Diabetes and Body Mass Index Are Associated with Neuropathy and Prognosis in Colon Cancer Patients Treated with Capecitabine and Oxaliplatin Adjuvant Chemotherapy. Oncology. 2016; 90: 36–42. doi: 10.1159/000442527.
251. Perez-Gomez MV, Sanchez-Niño MD, Sanz AB, Martín-Cleary C, Ruiz-Ortega M, Egido J, Navarro-González JF, Ortiz A, Fernandez-Fernandez B. Horizon 2020 in Diabetic Kidney Disease: The Clinical Trial Pipeline for Add-On Therapies on Top of Renin Angiotensin System Blockade. J Clin Med. 2015; 4: 1325–47. doi: 10.3390/jcm4061325.
252. Fernandez-Fernandez B, Ortiz A, Gomez-Guerrero C, Egido J. Therapeutic approaches to diabetic nephropathy--beyond the RAS. Nat Rev Nephrol. 2014; 10: 325–46. doi: 10.1038/nrneph.2014.74.
253. Poynter JN, Gruber SB, Higgins PDR, Almog R, Bonner JD, Rennert HS, Low M, Greenson JK, Rennert G. Statins and the risk of colorectal cancer. N Engl J Med. 2005; 352: 2184–92. doi: 10.1056/NEJMoa043792.
254. Shao Y-Y, Hsu C-H, Yeh K-H, Chen H-M, Yeh Y-C, Lai C-L, Lin Z-Z, Cheng A-L, Lai M-S. Statin Use Is Associated With Improved Prognosis of Colorectal Cancer in Taiwan. Clin Colorectal Cancer. 2015; 14: 177–184.e4. doi: 10.1016/j.clcc.2015.02.003.
255. Yasuda Y, Shimizu M, Shirakami Y, Sakai H, Kubota M, Hata K, Hirose Y, Tsurumi H, Tanaka T, Moriwaki H. Pitavastatin inhibits azoxymethane-induced colonic preneoplastic lesions in C57BL/KsJ-db/db obese mice. Cancer Sci. 2010; 101: 1701–7. doi: 10.1111/j.1349-7006.2010.01579.x.
256. Malicki S, Winiarski M, Matlok M, Kostarczyk W, Guzdek A, Konturek PC. IL-6 and IL-8 responses of colorectal cancer in vivo and in vitro cancer cells subjected to simvastatin. J Physiol Pharmacol. 2009; 60: 141–6. Available from http://www.ncbi.nlm.nih.gov/pubmed/20065508.
257. Kubota M, Shimizu M, Sakai H, Yasuda Y, Ohno T, Kochi T, Tsurumi H, Tanaka T, Moriwaki H. Renin-angiotensin system inhibitors suppress azoxymethane-induced colonic preneoplastic lesions in C57BL/KsJ-db/db obese mice. Biochem Biophys Res Commun. 2011; 410: 108–13. doi: 10.1016/j.bbrc.2011.05.115.
258. Kochi T, Shimizu M, Ohno T, Baba A, Sumi T, Kubota M, Shirakami Y, Tsurumi H, Tanaka T, Moriwaki H. Preventive effects of the angiotensin-converting enzyme inhibitor, captopril, on the development of azoxymethane-induced colonic preneoplastic lesions in diabetic and hypertensive rats. Oncol Lett. 2014; 8: 223–9. doi: 10.3892/ol.2014.2136.
259. Yoshiji H, Noguchi R, Kaji K, Ikenaka Y, Shirai Y, Namisaki T, Kitade M, Tsujimoto T, Kawaratani H, Fukui H. Attenuation of insulin-resistance-based hepatocarcinogenesis and angiogenesis by combined treatment with branched-chain amino acids and angiotensin-converting enzyme inhibitor in obese diabetic rats. J Gastroenterol. 2010; 45: 443–50. doi: 10.1007/s00535-009-0158-z.
260. Kohan DE, Pritchett Y, Molitch M, Wen S, Garimella T, Audhya P, Andress DL. Addition of atrasentan to renin-angiotensin system blockade reduces albuminuria in diabetic nephropathy. J Am Soc Nephrol. 2011; 22: 763–72. doi: 10.1681/ASN.2010080869.
261. de Zeeuw D, Coll B, Andress D, Brennan JJ, Tang H, Houser M, Correa-Rotter R, Kohan D, Lambers Heerspink HJ, Makino H, Perkovic V, Pritchett Y, Remuzzi G, et al. The endothelin antagonist atrasentan lowers residual albuminuria in patients with type 2 diabetic nephropathy. J Am Soc Nephrol. 2014; 25: 1083–93. doi: 10.1681/ASN.2013080830.
262. Goldkorn A, Ely B, Quinn DI, Tangen CM, Fink LM, Xu T, Twardowski P, Van Veldhuizen PJ, Agarwal N, Carducci MA, Monk JP, Datar RH, Garzotto M, et al. Circulating tumor cell counts are prognostic of overall survival in SWOG S0421: a phase III trial of docetaxel with or without atrasentan for metastatic castration-resistant prostate cancer. J Clin Oncol. 2014; 32: 1136–42. doi: 10.1200/JCO.2013.51.7417.
263. Lara PN, Ely B, Quinn DI, Mack PC, Tangen C, Gertz E, Twardowski PW, Goldkorn A, Hussain M, Vogelzang NJ, Thompson IM, Van Loan MD. Serum biomarkers of bone metabolism in castration-resistant prostate cancer patients with skeletal metastases: results from SWOG 0421. J Natl Cancer Inst. 2014; 106: dju013. doi: 10.1093/jnci/dju013.
264. Quinn DI, Tangen CM, Hussain M, Lara PN, Goldkorn A, Moinpour CM, Garzotto MG, Mack PC, Carducci MA, Monk JP, Twardowski PW, Van Veldhuizen PJ, Agarwal N, et al. Docetaxel and atrasentan versus docetaxel and placebo for men with advanced castration-resistant prostate cancer (SWOG S0421): a randomised phase 3 trial. Lancet Oncol. 2013; 14: 893–900. doi: 10.1016/S1470-2045(13)70294-8.
265. de Zeeuw D, Agarwal R, Amdahl M, Audhya P, Coyne D, Garimella T, Parving H-H, Pritchett Y, Remuzzi G, Ritz E, Andress D. Selective vitamin D receptor activation with paricalcitol for reduction of albuminuria in patients with type 2 diabetes (VITAL study): a randomised controlled trial. Lancet (London, England). 2010; 376: 1543–51. doi: 10.1016/S0140-6736(10)61032-X.
266. Lawrence JA, Akman SA, Melin SA, Case LD, Schwartz GG. Oral paricalcitol (19-nor-1,25-dihydroxyvitamin D2) in women receiving chemotherapy for metastatic breast cancer: a feasibility trial. Cancer Biol Ther. 2013; 14: 476–80. doi: 10.4161/cbt.24350.
267. Pommergaard H-C, Burcharth J, Rosenberg J, Raskov H. Aspirin, Calcitriol, and Calcium Do Not Prevent Adenoma Recurrence in a Randomized Controlled Trial. Gastroenterology. 2016; 150: 114–122.e4. doi: 10.1053/j.gastro.2015.09.010.
268. Baron JA, Barry EL, Mott LA, Rees JR, Sandler RS, Snover DC, Bostick RM, Ivanova A, Cole BF, Ahnen DJ, Beck GJ, Bresalier RS, Burke CA, et al. A Trial of Calcium and Vitamin D for the Prevention of Colorectal Adenomas. N Engl J Med. 2015; 373: 1519–30. doi: 10.1056/NEJMoa1500409.
269. Inoki K, Mori H, Wang J, Suzuki T, Hong S, Yoshida S, Blattner SM, Ikenoue T, Rüegg MA, Hall MN, Kwiatkowski DJ, Rastaldi MP, Huber TB, et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest. 2011; 121: 2181–96. doi: 10.1172/JCI44771.
270. Moreno JA, Moreno S, Rubio-Navarro A, Gómez-Guerrero C, Ortiz A, Egido J. Role of chemokines in proteinuric kidney disorders. Expert Rev Mol Med. 2014; 16: e3. doi: 10.1017/erm.2014.3.
271. Donath MY. Targeting inflammation in the treatment of type 2 diabetes: time to start. Nat Rev Drug Discov. 2014; 13: 465–76. doi: 10.1038/nrd4275.
272. Crusz SM, Balkwill FR. Inflammation and cancer: advances and new agents. Nat Rev Clin Oncol. 2015; 12: 584–96. doi: 10.1038/nrclinonc.2015.105.
273. Perez-Gomez MV, Sanchez-Niño MD, Sanz AB, Zheng B, Martín-Cleary C, Ruiz-Ortega M, Ortiz A, Fernandez-Fernandez B. Targeting inflammation in diabetic kidney disease: early clinical trials. Expert Opin Investig Drugs. 2016; 1–14. doi: 10.1080/13543784.2016.1196184.
274. Chu P-Y, Zatta A, Kiriazis H, Chin-Dusting J, Du X-J, Marshall T, Kaye DM. CXCR4 antagonism attenuates the cardiorenal consequences of mineralocorticoid excess. Circ Heart Fail. 2011; 4: 651–8. doi: 10.1161/CIRCHEARTFAILURE.110.960831.
275. Ding M, Cui S, Li C, Jothy S, Haase V, Steer BM, Marsden PA, Pippin J, Shankland S, Rastaldi MP, Cohen CD, Kretzler M, Quaggin SE. Loss of the tumor suppressor Vhlh leads to upregulation of Cxcr4 and rapidly progressive glomerulonephritis in mice. Nat Med. 2006; 12: 1081–7. doi: 10.1038/nm1460.
276. Wang X, Shaw S, Amiri F, Eaton DC, Marrero MB. Inhibition of the Jak/STAT signaling pathway prevents the high glucose-induced increase in tgf-beta and fibronectin synthesis in mesangial cells. Diabetes. 2002; 51: 3505–9. Available from http://www.ncbi.nlm.nih.gov/pubmed/12453907.
277. Huang JS, Guh JY, Hung WC, Yang ML, Lai YH, Chen HC, Chuang LY. Role of the Janus kinase (JAK)/signal transducters and activators of transcription (STAT) cascade in advanced glycation end-product-induced cellular mitogenesis in NRK-49F cells. Biochem J. 1999; 342:231-8. Available from http://www.ncbi.nlm.nih.gov/pubmed/?term=10432321.
278. Recio C, Oguiza A, Lazaro I, Mallavia B, Egido J, Gomez-Guerrero C. Suppressor of cytokine signaling 1-derived peptide inhibits Janus kinase/signal transducers and activators of transcription pathway and improves inflammation and atherosclerosis in diabetic mice. Arterioscler Thromb Vasc Biol. 2014; 34: 1953–60. doi: 10.1161/ATVBAHA.114.304144.
279. Ortiz-Muñoz G, Lopez-Parra V, Lopez-Franco O, Fernandez-Vizarra P, Mallavia B, Flores C, Sanz A, Blanco J, Mezzano S, Ortiz A, Egido J, Gomez-Guerrero C. Suppressors of cytokine signaling abrogate diabetic nephropathy. J Am Soc Nephrol. 2010; 21: 763–72. doi: 10.1681/ASN.2009060625.
280. Schanstra JP, Zürbig P, Alkhalaf A, Argiles A, Bakker SJL, Beige J, Bilo HJG, Chatzikyrkou C, Dakna M, Dawson J, Delles C, Haller H, Haubitz M, et al. Diagnosis and Prediction of CKD Progression by Assessment of Urinary Peptides. J Am Soc Nephrol. 2015; 26: 1999–2010. doi: 10.1681/ASN.2014050423.
281. Siwy J, Schanstra JP, Argiles A, Bakker SJL, Beige J, Boucek P, Brand K, Delles C, Duranton F, Fernandez-Fernandez B, Jankowski M-L, Al Khatib M, Kunt T, et al. Multicentre prospective validation of a urinary peptidome-based classifier for the diagnosis of type 2 diabetic nephropathy. Nephrol Dial Transplant. 2014; 29: 1563–70. doi: 10.1093/ndt/gfu039.
282. Metzger J, Negm AA, Plentz RR, Weismüller TJ, Wedemeyer J, Karlsen TH, Dakna M, Mullen W, Mischak H, Manns MP, Lankisch TO. Urine proteomic analysis differentiates cholangiocarcinoma from primary sclerosing cholangitis and other benign biliary disorders. Gut. 2013; 62: 122–30. doi: 10.1136/gutjnl-2012-302047.
283. Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis. 2009; 4: 22. doi: 10.1186/1750-1172-4-22.
284. Bisgaard ML, Fenger K, Bülow S, Niebuhr E, Mohr J. Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Hum Mutat. 1994; 3: 121–5. doi: 10.1002/humu.1380030206.
285. Bourke B, Broderick A, Bohane T. Peutz-Jeghers syndrome and management recommendations. Clin Gastroenterol Hepatol. 2006; 4: 1550; author reply 1550. doi: 10.1016/j.cgh.2006.09.028.
286. Shiovitz S, Copeland WK, Passarelli MN, Burnett-Hartman AN, Grady WM, Potter JD, Gallinger S, Buchanan DD, Rosty C, Win AK, Jenkins M, Thibodeau SN, Haile R, et al. Characterisation of familial colorectal cancer Type X, Lynch syndrome, and non-familial colorectal cancer. Br J Cancer. 2014; 111: 598–602. doi: 10.1038/bjc.2014.309.
287. Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997; 386: 623–7. doi: 10.1038/386623a0.
288. Salazar R, Roepman P, Capella G, Moreno V, Simon I, Dreezen C, Lopez-Doriga A, Santos C, Marijnen C, Westerga J, Bruin S, Kerr D, Kuppen P, et al. Gene expression signature to improve prognosis prediction of stage II and III colorectal cancer. J Clin Oncol. 2011; 29: 17–24. doi: 10.1200/JCO.2010.30.1077.
289. Budinska E, Popovici V, Tejpar S, D’Ario G, Lapique N, Sikora KO, Di Narzo AF, Yan P, Hodgson JG, Weinrich S, Bosman F, Roth A, Delorenzi M. Gene expression patterns unveil a new level of molecular heterogeneity in colorectal cancer. J Pathol. 2013; 231: 63–76. doi: 10.1002/path.4212.
290. Oberaigner W, Ebenbichler C, Oberaigner K, Juchum M, Schönherr HR, Lechleitner M. Increased cancer incidence risk in type 2 diabetes mellitus: results from a cohort study in Tyrol/Austria. BMC Public Health. 2014; 14: 1058. doi: 10.1186/1471-2458-14-1058.
291. Flanagan JN, Zheng S, Chiang K-C, Kittaka A, Sakaki T, Nakabayashi S, Zhao X, Spanjaard RA, Persons KS, Mathieu JS, Holick MF, Chen TC. Evaluation of 19-nor-2alpha-(3-hydroxypropyl)-1alpha,25-dihydroxyvitamin D3 as a therapeutic agent for androgen-dependent prostate cancer. Anticancer Res. 2009; 29: 3547–53. Available from http://www.ncbi.nlm.nih.gov/pubmed/19667147.
292. Saleh MA, Boesen EI, Pollock JS, Savin VJ, Pollock DM. Endothelin receptor A-specific stimulation of glomerular inflammation and injury in a streptozotocin-induced rat model of diabetes. Diabetologia. 2011; 54: 979–88. doi: 10.1007/s00125-010-2021-4.
293. Younis IR, George DJ, McManus TJ, Hurwitz H, Creel P, Armstrong AJ, Yu JJ, Bacon K, Hobbs G, Peer CJ, Petros WP. Clinical pharmacology of an atrasentan and docetaxel regimen in men with hormone-refractory prostate cancer. Cancer Chemother Pharmacol. 2014; 73: 991–7. doi: 10.1007/s00280-014-2432-x.
294. Groenewegen G, Walraven M, Vermaat J, de Gast B, Witteveen E, Giles R, Haanen J, Voest E. Targeting the endothelin axis with atrasentan, in combination with IFN-alpha, in metastatic renal cell carcinoma. Br J Cancer. 2012; 106: 284–9. doi: 10.1038/bjc.2011.515.
295. Witteveen PO, van der Mijn KJC, Los M, Kronemeijer RH, Groenewegen G, Voest EE. Phase 1/2 study of atrasentan combined with pegylated liposomal doxorubicin in platinum-resistant recurrent ovarian cancer. Neoplasia. 2010; 12: 941–5. Available from http://www.ncbi.nlm.nih.gov/pubmed/21076619.
296. Armstrong AJ, Creel P, Turnbull J, Moore C, Jaffe TA, Haley S, Petros W, Yenser S, Gockerman JP, Sleep D, Hurwitz H, George DJ. A phase I-II study of docetaxel and atrasentan in men with castration-resistant metastatic prostate cancer. Clin Cancer Res. 2008; 14: 6270–6. doi: 10.1158/1078-0432.CCR-08-1085.
297. Nelson JB, Love W, Chin JL, Saad F, Schulman CC, Sleep DJ, Qian J, Steinberg J, Carducci M, Atrasentan Phase 3 Study Group. Phase 3, randomized, controlled trial of atrasentan in patients with nonmetastatic, hormone-refractory prostate cancer. Cancer. 2008; 113: 2478–87. doi: 10.1002/cncr.23864.
298. Phuphanich S, Carson KA, Grossman SA, Lesser G, Olson J, Mikkelsen T, Desideri S, Fisher JD, New Approaches to Brain Tumor Therapy (NABTT) CNS Consortium. Phase I safety study of escalating doses of atrasentan in adults with recurrent malignant glioma. Neuro Oncol. 2008; 10: 617–23. doi: 10.1215/15228517-2008-013.
299. Chiappori AA, Haura E, Rodriguez FA, Boulware D, Kapoor R, Neuger AM, Lush R, Padilla B, Burton M, Williams C, Simon G, Antonia S, Sullivan DM, et al. Phase I/II study of atrasentan, an endothelin A receptor antagonist, in combination with paclitaxel and carboplatin as first-line therapy in advanced non-small cell lung cancer. Clin Cancer Res. 2008; 14: 1464–9. doi: 10.1158/1078-0432.CCR-07-1508.
300. Carducci MA, Saad F, Abrahamsson P-A, Dearnaley DP, Schulman CC, North SA, Sleep DJ, Isaacson JD, Nelson JB, Atrasentan Phase III Study Group Institutions. A phase 3 randomized controlled trial of the efficacy and safety of atrasentan in men with metastatic hormone-refractory prostate cancer. Cancer. 2007; 110: 1959–66. doi: 10.1002/cncr.22996.
301. Idrisa A, Omigbodun AO, Adeleye JA. Pregnancy in hemoglobin sickle cell patients at the University College Hospital, Ibadan. Int J Gynaecol Obstet. 1992; 38: 83–6. Available from http://www.ncbi.nlm.nih.gov/pubmed/1356849.
302. Finger EC, Cheng C-F, Williams TR, Rankin EB, Bedogni B, Tachiki L, Spong S, Giaccia AJ, Powell MB. CTGF is a therapeutic target for metastatic melanoma. Oncogene. 2014; 33: 1093–100. doi: 10.1038/onc.2013.47.
303. Adler SG, Schwartz S, Williams ME, Arauz-Pacheco C, Bolton WK, Lee T, Li D, Neff TB, Urquilla PR, Sewell KL. Phase 1 study of anti-CTGF monoclonal antibody in patients with diabetes and microalbuminuria. Clin J Am Soc Nephrol. 2010; 5: 1420–8. doi: 10.2215/CJN.09321209.
304. Eccles SA. Metastasis and the Tumor Microenvironment: A Joint Metastasis Research Society-AACR Conference - Research on Metastasis: part 2. IDrugs. 2010; 13: 768–71. Available from http://www.ncbi.nlm.nih.gov/pubmed/21046523.
305. Advani A, Wiggins KJ, Cox AJ, Zhang Y, Gilbert RE, Kelly DJ. Inhibition of the epidermal growth factor receptor preserves podocytes and attenuates albuminuria in experimental diabetic nephropathy. Nephrology (Carlton). 2011; 16: 573–81. doi: 10.1111/j.1440-1797.2011.01451.x.
306. Wassef L, Kelly DJ, Gilbert RE. Epidermal growth factor receptor inhibition attenuates early kidney enlargement in experimental diabetes. Kidney Int. 2004; 66: 1805–14. doi: 10.1111/j.1523-1755.2004.00955.x.