
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a progressive degenerative disease of the central nervous system (CNS) that lead to the progressive death of the motor neurons [1] and death about four years after disease onset [2]. Although various mechanisms, such as neuroinflammation, excitotoxicity, mitochondrial dysfunction, abnormal protein aggregation, and oxidative stress have been proposed to be associated with the pathogenesis of ALS, no single mechanism has been identified as responsible [3]. Cholesterol, a major constituent of the CNS, is synthesized de novo [4]. It is metabolized into diverse types of hydroxycholesterols (OHCs), thereby regulating cholesterol homeostasis [5], cellular apoptosis [6], and signaling [7].
We hypothesized that the synthesis of OHC in the CNS might contribute to the pathogenesis of ALS. The aim of this study was to measure the levels of three major OHCs (24-,25-, and 27-OHCs) in serum (OHCserum) and cerebrospinal fluid (OHCCSF) of patients with ALS and to assess their role in the pathogenesis of ALS experimental models.
RESULTS
Increased levels of OHCs in ALS patients
Patients with ALS who did not receive riluzole treatment (ALS-naive), riluzole-treated patients with ALS (ALS-riluzole), and control groups did not differ significantly in terms of demographic, body mass index, or levels of serum cholesterols (Table 1).
Table 1: Basal characteristics of patients
ALS-naïve |
ALS-riluzole |
Control |
p-values |
|||
|---|---|---|---|---|---|---|
ALS-naïve vs. control |
ALS-naïve vs. ALS-riluzole |
ALS-riluzole vs. control |
||||
Number |
30 |
9 |
33 |
|||
Age (yrs) |
53.8 ± 12.15 (27.7–79.3) |
52.9 ± 6.9 (42.4–66.0) |
50.52 ± 16.51 (20.0–74.7) |
n.s. |
n.s. |
n.s. |
Male |
15 (52%) |
4 (44%) |
16 (48%) |
n.s. |
n.s. |
n.s. |
Total Chol (mg/dL) |
182.11 ± 27.78 (112–210) |
172.54 ± 30.37 (112–210) |
168.45 ± 30.39 (110–225) |
n.s. |
n.s. |
n.s. |
BMI |
21.75 ± 2.88 (17.09–28.23) |
23.14 ± 3.96 (17.24–29.65) |
22.71 ± 4.07 (12.83–33.19) |
n.s. |
n.s. |
n.s. |
Duration of disease (yrs) |
1.00 ± 0.68 (0.25–3.00) |
0.93 ± 0.45 (0.36–1.64) |
n.s. |
|||
ALSFRSr |
38.75 ± 6.52 (24–47) |
38.89 ± 10.67 (12–48) |
n.s. |
|||
Abbreviation: ALSFRSr = amyotrophic lateral sclerosis functional rating scale revised, BMI = body mass index, Chol = cholesterol, CSF = cerebrospinal fluid, OHC = hydroxycholesterol.
The levels of 24-OHCCSF and 25-OHCCSF were significantly higher in the ALS-naïve group (2.03 ± 0.63 ng/mL and 0.14 ± 0.06 ng/mL, respectively) than in the ALS-riluzole group (1.33 ± 0.46 ng/mL, p = 0.006 and 0.07 ± 0.03 ng/mL, p= 0.001) and controls (1.59 ± 0.05 ng/mL, p = 0.018 and 0.09 ± 0.04 ng/mL, p = 0.012). The levels of 27-OHCCSF and 25-OHCserum were also higher in ALS-naïve group (1.05 ± 0.39 ng/mL and 5.39 ± 1.94, ng/mL, respectively) than in controls (0.77 ± 0.32 ng/mL, p = 0.014 and 4.27 ± 1.18 ng/mL, p = 0.017) (Figure 1 and 2).
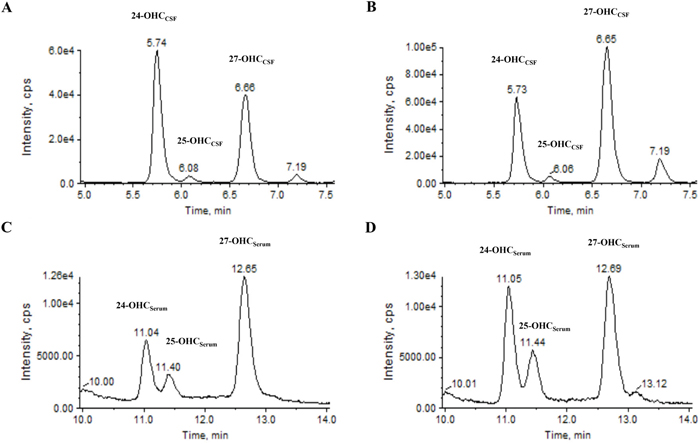
Figure 1: Representative chromatogram of 24-, 25- and 27-OHC in the CSF and serum using ALS patient and control samples. For OHCCSF analysis, chromatogram of A. normal and B. ALS patients were obtained in CSF samples. For OHCserum analysis, chromatogram of C. normal and D. ALS patient were obtained in serum samples. Abbrevations: CSF = cerebrospinal fluid, min = minutes, OHC = hydroxycholesterol.
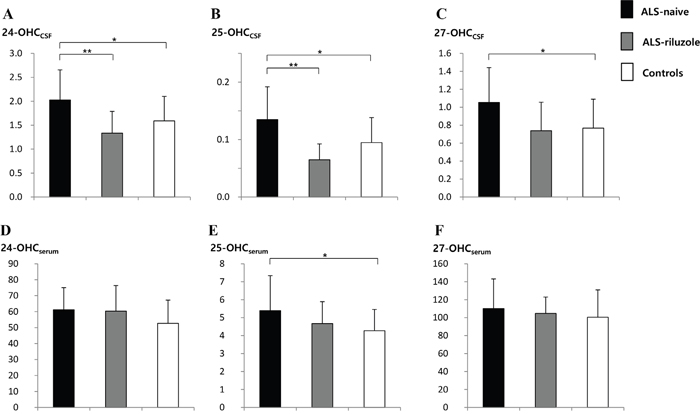
Figure 2: Levels of 24-, 25, and 27-OHC in the CSF and serum of ALS-naïve, ALS-riluzole, and control groups. The level of OHCs in the A-C. CSF and D-F. serum were compared between groups. The levels of (A) 24-OHCCSF and (B) 25-OHCCSF were significantly higher in the ALS-naïve group than both the ALS-riluzole and control groups. The level of (C) 27-OHCCSF and (E) 25-OHCserum were also higher in the ALS-naïve group than in controls. Data for OHCs were expressed in ng/mL. Abbreviations: ALS-naïve = ALS patients without treatment, ALS-riluzole = ALS patients on riluzole treatment, OHC = hydroxycholesterol, OHCCSF = OHC in the cerebrospinal fluid, OHCserum = OHC in the serum. *p < 0.05, **p < 0.01.
Disease severity, measured by the revised ALS functional rating scale (ALSFRSr), was significantly correlated with the levels of 27-OHCCSF(Figure 3C), 24-OHCserum (Figure 3D), and 25-OHCserum (Figure 3E) in all patients with ALS in the study (n = 37). However, multivariate regression analysis for these three OHCs revealed that only 25-OHCserum level was significantly associated with ALSFRSr score (-0.591; 95% CI -5.385, -0.673; p = 0.014).
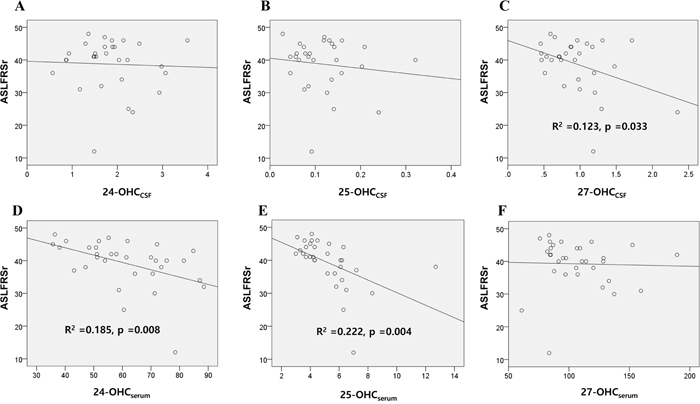
Figure 3: Association of the OHCs with disease severity scores of patients with ALS. The association of the 24, 25, and 27-OHC in the A-C. CSF and D-F. serum of ALS patients with their ALSFRSr were assessed. Univariable linear regression analysis revealed that levels of 27-OHCCSF (C) 24-OHCserum (D) and 25-OHCserum (E) were significantly associated with the ALSFRSr of patients. Data for OHCs were expressed in ng/mL. Abbreviations: ALSFRSr = amyotrophic lateral sclerosis functional rating score revised, OHCCSF = OHC in the cerebrospinal fluid, OHCserum = OHC in the serum.
The progression rate (ΔFS), measured by the change of ALSFRSr per months was significantly correlated with the levels of 27-OHCCSF (Figure 4C) and 25-OHCserum (Figure 4E). Multivariate analysis revealed that only the 25-OHCserum level was significantly associated with ΔFS (0.541; 95% CI 0.105, 0.438; p = 0.002).
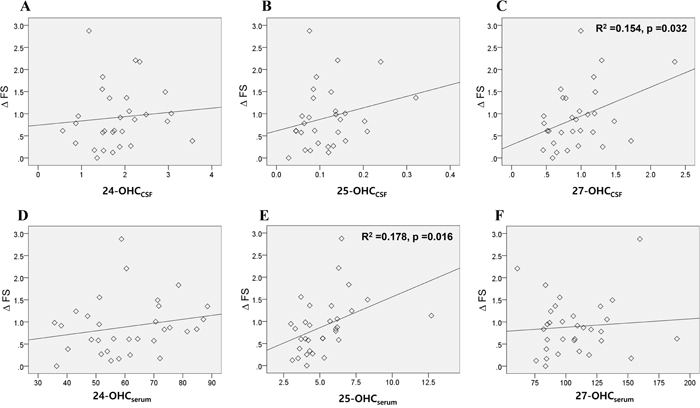
Figure 4: Association of the OHCs with disease progression rate of patients with ALS. The association of the 24, 25, and 27-OHC in the A-C. CSF and D-F. serum of ALS patients with their disease progression rate (ΔFS) were assessed. Univariable linear regression analysis revealed that levels of 27-OHCCSF (C) and 25-OHCserum (E) were significantly associated with the ΔFS of patients. Data for OHCs were expressed in ng/mL. Abbreviations: ΔFS = progression rate in amyotrophic lateral sclerosis functional rating score revised, OHCCSF = OHC in the cerebrospinal fluid, OHCserum = OHC in the serum.
The 25-OHC induced neuronal apoptosis, activated the GSK-3β pathway in ALS in vitro model, which was attenuated by riluzole
Among the three OHCs, the 25-OHC induced most severe motor neuronal death in a concentration of the 2.5uM to 40 uM (Figure 5A) in a motor neuronal cell line stably expressing mutant G93A superoxide dismutase 1 gene (mSOD1-NSC34) cells. It also activated the GSK pathway (Figure 5B) and induced cell apoptosis (Figure 5C). Riluzole reduced 25-OHC-induced apoptosis (Figure 5C), neuronal death (Figure 5D), and activation of GSK-3 (Figure 5E).
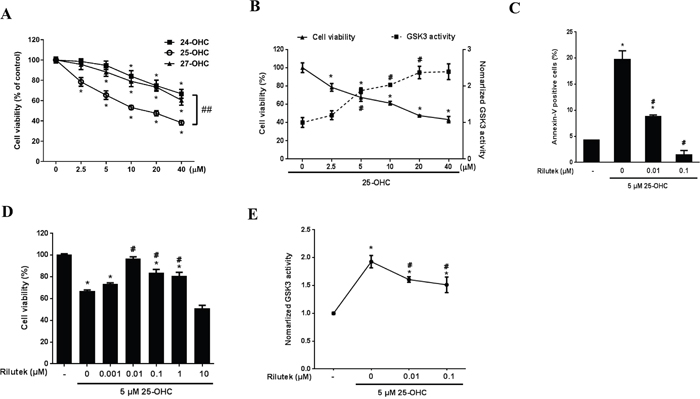
Figure 5: 25-OHC induced mSOD1 motor neuronal cell death and activated the GSK-3 pathway. A. Among the three major OHCS, the 25-OHC induced most severe death of mSOD1-NSC34 cells. B. The 25-OHC activated GSK-3 pathways in association with the cellular death and C. induced apoptosis. This 25-OHC induced processes, including (C) apoptosis, D. cell death, and E. activation of the GSK-3 pathways were inhibited by the riluzole treatment. The data are presented as mean (% of the non-treated group) ± SEM and were compared using Tukey’s test after a one-way ANOVA (n = 5). *p<0.05 when compared with the non-treated group. #p<0.05 when compared with the group that was treated only with 5 μM 25-OHC. ##p<0.05 when compared with the 25-OHC treated group. Abbreviations: mSOD1 = mutant superoxide dismutase 1, OHC = hydroxycholesterol.
Western blot analysis showed that 25-OHC facilitated release of cytochrome c from injured mitochondria, activated caspase-3, and increased the cleavage of poly (ADP-ribose) polymerase (PARP) in mSOD1-NSC34 cells. However, this activation of cell death signaling cascades by 25-OHC was significantly reduced in a dose-dependent manner after treatment with riluzole (Figure 6A-6C). Moreover, immunofluorescence staining revealed that 25-OHC increased the immunoreactivity of cleaved caspase-3 immunoreactivity and Poly ADP ribose polymerase (PARP), which were reduced by treatment with riluzole (Figure 6D).
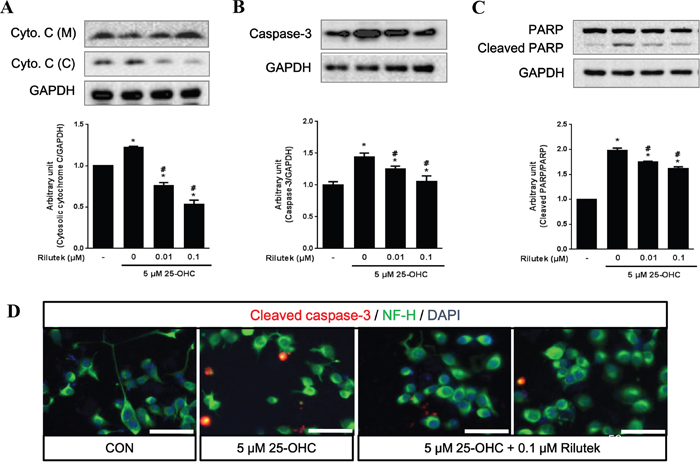
Figure 6: 25-OHC-mediated cell death signaling and the effect of riluzole treatment on mSOD1 motor neuronal cells. The 25-OHC induced cell death elevated signaling proteins, including A. cytosolic cytochrome c, B. caspase-3, C. and cleaved PARP (A-C). These 25-OHC induced cell death signaling was attenuated by treatment with riluzole. D. Immunofluorescence imaging also showed that cleavage of caspase-3 was attenuated by treatment with 0.1uM of riluzole (scale bars = 50 μm). Data were expressed as a ratio of the simultaneously assayed non-treated group's value and were compared using Tukey’s test after a one-way ANOVA (n = 5). *p <0.05 when compared with non-treated group. #p<0.05 when compared with only 5 μM 25-OHC treated group.
Administration of 25-OHC have activated of GSK-3β (Ser9) and phosphorylated tau (Ser396), which were prevented by treatment with riluzole (Figures 7A and 7B). Treatment with either GSK-3 inhibitor (10 μM SB415286) or riluzole reduced the GSK-3β activation induced by 25-OHC, although combined treatment with both GSK-3 inhibitor and riluzole did not produce synthetic effects above and beyond the effects of either on its own (Figure 7C).
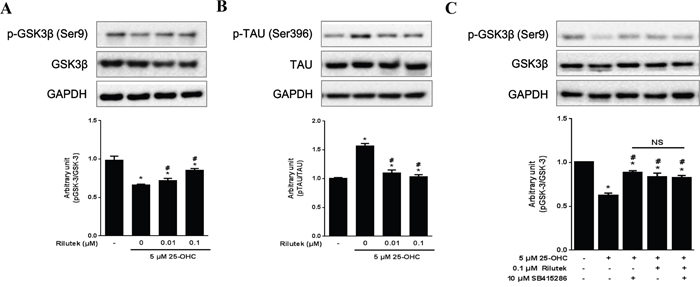
Figure 7: Inhibitory effects of riluzole on the 25-OHC-activated GSK3β pathway in motor neuronal cells. Western blot analysis, performed with antibodies against p-GSK3β (Ser9) and its substrate p-TAU (Ser396) revealed that 25-OHC activated A. GSK3β and B. subsequent tau pathway. C. Both the GSK-3 inhibitor (10 μM SB415286) and riluzole treatments reduced 25-OHC-induced GSK-3β activation. Compounded effects of combined treatment of t GSK-3 inhibitor and riluzole did not differ from the either of these treatments alone (C). Data are expressed as a ratio of the simultaneously assayed non-treated group's value and were compared using Tukey’s test after a one-way ANOVA (n=5). *p<0.05 when compared with non-treated group. #p<0.05 when compared with 5 μM 25-OHC treated group. NS, not significant.
25-OHC induced motor neuronal death via liver X receptor (LXR) signaling
To assess whether the 25-OHC mediate motor neuronal death via LXR signaling, a LXR antagonist, 22(s)-OHC [8], were co-treated with the 25-OHC in mSOD1-NSC34 cells. Treatment of 22(s)-OHC significantly attenuated the 25-OHC induced motor neuronal cells in a dose dependent manner (Figure 8).
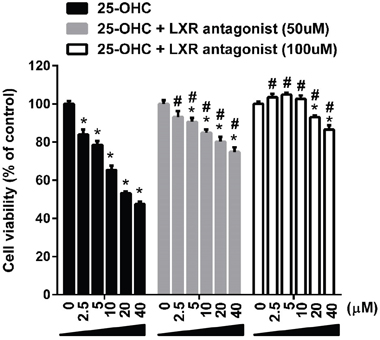
Figure 8: The 25-OHC induced motor neuronal death via LXR signaling. Treatment of LXR antagonist (22(S)-OHC; 50uM, 100uM) significantly attenuated the 25-OHC induced death of the mSOD1-NSC34 cells. *p <0.01, when compared with vehicle control. #p <0.01, when compared with the group treated only with the 25-OHC.
Upregulation of the 25-OHC synthesis in ALS mice
In the CNS, 25-OHC is mostly generated by the cholesterol 25-hydroxylase (CH25H) [9] and, to a lesser extent by the cytochrome P450 3A4 (CYP3A4) [10]. Whereas 25-hydroxycholesterol 7-alpha-hydroxylase (CYP7B1) is involved in the metabolism of 25-OHC [11]. To determine a potential mechanism for the observed increases in 25-OHC in ALS, we examined the mRNA expression of CH25H, CYP3A4, and CYP7B1 in the brain and spinal cord of transgenic mice with the human mutant SOD1 gene containing a glycine 93 (Gly93) to alanine (Ala) substation (mSOD1-G93A mice) at different time points (postnatal day of 60, 90, and 120) (Figure 9). In the asymptomatic stage (60 days), the mRNA expression of these enzymes in mSOD1G93A mice did not differ between ALS and WT mice. However, in the early symptomatic stage (90 days), mRNA expression of the 25-OHC synthesizing enzymes, CH25H and CYP3A4, was increased significantly compared to age-matched WT mice and asymptomatic mSOD1-G93A mice; these effects were primarily observed in brain tissue. The 25-OHC metabolizing enzyme, CYP7B1, was also moderately increased in the early symptomatic SOD1G93A mice, both in the brain and the spinal cord. Interestingly, these high levels of mRNA expressions of 25-OHC associated enzymes were only observed in the early symptomatic stage but not in the late symptomatic stage in mSOD1G93A mice.
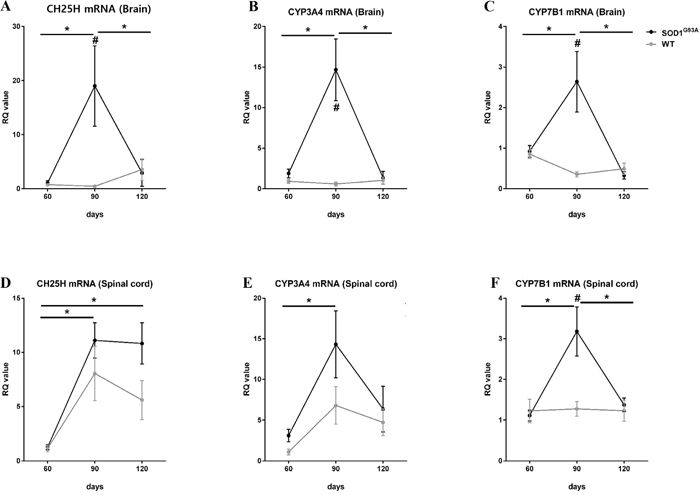
Figure 9: Increased activities of 25-OHC synthesis and metabolism in the early symptomatic stage of SOD1G93A mice. The qRT-PCR reactions were performed with mRNA with primers for the 25-OHC synthesizing enzyme (CH25H and CYP3A4) and for the 25-OHC metabolizing enzyme (CYP7B1) using A-C. brain and D-F. spinal cord at different disease stages (60, 90, and 120 days) of SOD1G93A mice and age-matched WT controls. (A). The expression of CH25H and (B) CYP3A4 mRNA were significantly increased in the brain tissues of early symptomatic stage (90 days) SOD1G93A mice compared to age-matched WT controls and asymptomatic SOD1G93A mice. (C and F). The expression of CYP7B1 was also modestly increased in the brain and spinal cord of SOD1G93A mice in early symptomatic stages. These high expression levels of 25-OHC-associated enzymes were not observed in the late symptomatic stage of SOD1G93A mice. Group means ± SEM are plotted; #p <0.05; SOD1G93A mice compared to WT mice on the same day. *p <0.05; SOD1G93A mice compared at different time points. Differences between groups were analyzed with two-tailed Student’s t-tests. Abbreviations: CH25H = cholesterol 25-hydroxylase, CYP3A4= Cytochrome P450 3A4, CYP7B1 = 25-hydroxycholesterol 7-alpha-hydroxylase, SOD1 = superoxide dismutase 1, WT = wild type.
DISCUSSION
This study showed that the 25-OHC is involved in the pathogenesis of ALS. The levels of 25-OHCCSF and 25-OHCserum were higher in riluzole-naive ALS patients than in controls and/or riluzole-treated ALS patients. Moreover the level of 25-OHCserum in ALS patients were significantly associated with their disease severity and rate of progressions. The 25-OHC also induced cellular apoptosis and activated the GSK-3β/ LXR pathways in a cellular model of ALS. Lastly the expression of enzymes important for synthesis and metabolism of 25-OHC was increased in an animal model of ALS, in particular, at the early symptomatic stage of the disease.
Our observations are in line with several evidences from the existing literature. Specifically, the expression of the 25-hydroxylase gene was shown to be increased in autopsied tissues of patients with ALS [12]. The inactivation of liver X receptor beta (LXRß), which promotes cholesterol transport in the CNS, has induced the pathogenesis of ALS pathogenesis in a mouse model [13]. The mutation in the CYP7B1 gene, responsible for 25-OHC metabolism, caused the hereditary degenerative disease involving motor neurons [14]. The metabolism for lipid were dysregulated in both mouse models of ALS and patients with ALS [15, 16]. Finally, 25-OHC can induce mitochondria-dependent cell apoptosis via activation of glycogen synthesis kinase–3β (GSK-3β) [17], an enzyme considered to be involved in the disease progression of ALS [18].
In a recent study of OHCs in ALS patients, levels of 25-OHCserum tended to be higher in ALS patients than controls, but this difference did not reach statistical significance [19], in contrast to our observations. The reason for this discrepancy may be that our patient inclusion criteria were stringent, such that possible or probable cases of ALS according to the El Escorial criteria were excluded [20]. Moreover, we have controlled the compounding effects of riluzole treatment on OHC level, measured the severity of patients by ALSFRSr, and analyzed the OHCCSF, as well as the OHCserum, which could have contributed the more robust and readily observable results in our study.
Among the three major OHCs, the 25-OHC induced most severe motor neuronal death in vitro model of ALS compared to the 24- and 27-OHC (Figure 5A). The 25-OHC is produced in the macrophage and/or glia by the enzyme CH25H [21, 22], in response to Toll-like receptor-4 (TLR-4) signaling [23] Moreover patients with ALS had increased expression of the TLR-4 in their glia of spinal cord. [24]. We speculate that this 25-OHC could mediate the motor neuronal death of ALS, via GSK3-ß and LXR pathway, in response to the upregulated the inflammatory signals of the glia (Figure 10).
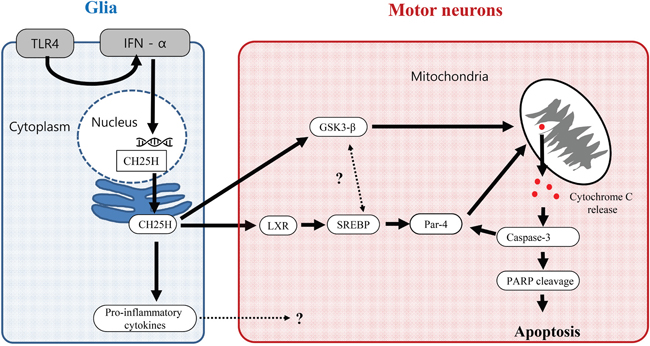
Figure 10: Proposed 25-OHC dependent signaling pathway contributing the motor neuronal death in ALS. Abbreviations: CH25H = cholesterol 25-hydroxylase, GSK-3β = glycogen synthesis kinase–3β, IFN = interferon, LXR = liver X receptor, PARP = poly (ADP-ribose) polymerase, PAR-4 = Prostate Apoptosis Response 4, SREBP = sterol regulatory element-binding protein, TLR4 = Toll-like receptor-4.
The OHC engaged in diverse biological activities, including cell signaling, cellular cholesterol synthesis, and cytotoxicity [25, 26]. Among these metabolites, the level of 24-OHC has been shown to increase in patients with Alzheimer’s disease and is associated with disease severity [27]. The metabolite 27-OHC is proposed to reflect the effect of the peripheral hypercholesterolemia to the CNS via its net flux from peripheral circulation to the CNS [28]. Recently, 25-OHC has drawn attention for its role as both an anti-inflammatory and pro-inflammatory mediator [11]. In addition to these important roles of OHCs, we speculated that 25-OHC might play a pathogenic role in ALS by mediating GSK-3ß activation, LXR signaling, and neuronal apoptosis.
Although disease severity in patients with ALS patients was correlated with levels of 25-OHCserum, it was not correlated with levels of 25-OHCCSF. This may explained by our finding that the expression of 25-OHC synthesizing enzyme was only transiently increased in the CNS of ALS mice during the early symptomatic stage of the disease. It may be that this temporarily high expression of 25-OHC-synthesizing enzymes may have interfered with the production of 25-OHC in the CNS of patients who are in the advanced stages of ALS.
The precise cause of decreased 25-OHCCSF in ALS-riluzole patients remains unclear. However, in light of a previous study showing that glutamate excitotoxicity increased both cholesterol and cholesterol oxidation products in the brain [14], these results may be attributable to anti-glutamatergic effects of riluzole [29].
Our study has several limitations. First, the number of patients were relatively small. Second, the 25-OHC may not be solely responsible for the whole pathogenesis of ALS. Because the ALS-riluzole group, who had relatively low levels of 25-OHCCSF, continued to experience the disease progression [30]. Moreover, the 25-OHC might also mediate diverse disease conditions, including viral infection [31], X-linked adrenoleukodystrophy [8], multiple sclerosis [32], and neuromyelitis optica [33], other than the ALS [23]. Therefore, it seems that the 25-OHC might be one of the diverse inflammatory mediator rather than a specific marker for ALS. Second, we have used the serum and CSF from patients as controls, rather than those from healthy volunteers. This is for the ethical issues of performing the lumbar punctures on the healthy subject. However, as we have excluded both the degenerative or inflammatory diseases of the CNS from the controls, we speculate that this issue would not significantly affect the result of our study. Lastly, the level of OHCs in the CSF and serum of ALS animal could also have been another interesting point, which were not performed in our study because the concentrations of the OHC were expected to be extremely low in mice.
Our data showed that the 25-OHC could be an important mediator of the ALS pathogenesis involving in the GSK3-ß activation and neuronal apoptosis, which could eventually trigger the onset or accelerating the progression of ALS symptoms. Our results may reveal a novel ALS treatment target and provide insight into the involvement in of CNS cholesterol metabolism in ALS pathogenesis.
MATERIALS AND METHODS
Patients
Patients who conformed to the diagnosis of definite or probable ALS according to the revised El Escorial criteria [20] were recruited from the ALS Clinic at the Seoul National University Hospital between December 2009 and May 2012. The following patients were excluded: those diagnosed more than 3 years before study commencement, those aged < 20 years or >80 years, those with respiratory support (tracheostomy, non-invasive ventilation, or oxygen inhalation), those fed via levin tube or gastrostomy tube (percutaneous endoscopic gastrostomy, PEG), those with presence of an acute systemic illness (per se, aspiration pneumonia), previous enrollment in the clinical drug or stem cell trial for ALS, or absence of written consent. ALS patients were also dichotomized according to the use of riluzole, as ALS-naive group and ALS-riluzole group [30].
The control group was comprised of patients who visited the Neurology Clinic at the Seoul National University Hospital. Patients were excluded from the control group if they had been diagnosed with a either neurodegenerative [20] or neuroinflammatory disease [34] of the central nervous system, any clinical or electrophysiological evidence of motor neuron disease [20], or if they had used an oral or intravenous steroid that might have affected cholesterol metabolism.
Clinical parameter and sampling protocols
Age, gender, level of serum total cholesterol, body weight, and height were assessed in patients with ALS and non-ALS controls. The ALSFRSr [35] and duration of disease were also assessed in patients with ALS. The progression rate (ΔFS) was calculated with minor modification of the previous study as follows; Δ FS = (48 - ALSFRSr at “time of sampling”)/duration from onset to diagnosis (month) [36]. All samples of CSF and serum were immediately centrifuged and stored at – 80oC after collection, according to standard protocols [37].
Measurement for the CSF and serum level of 24-, 25-, and 27-hydroxycholesterols
Liquid chromatography-tandem mass spectrometry (LC–MS/MS) analyses were performed at Korea Institute of Science and Technology (KIST), according to our recent methods [33]. Briefly, an API 4000 triple-quadrupole mass spectrometer (AB Sciex, Toronto, Canada) with an electrospray ionization source, in the positive ionization mode was used. The electrospray source was coupled online with a Shimadzu ultra-fast LC system (Shimadzu Corporation, Kyoto, Japan).
Standard protocol approval, registration, and patient consent
This study was approved by the Seoul National University Hospital Institutional Review Board (IRB number: H-0904-057-279). All patients provided written informed consent prior to participation. This study was approved by the IACUC (Institutional Animal Care and Use Committee) of Hanyang University.
Cell culture, treatment, and the GSK-3 activity
The NSC-34 mouse motor neuron cell line [38] was purchased from CELLutions (CELLutions Biosystems Inc., Ontario, Canada), and the cells were grown in the high glucose formulation of DMEM (D5796; Sigma, USA) supplemented with 10% fetal bovine serum (FBS) and 1% PenStrp. To induce differentiation of the NSC-34 cells (dNSC-34), they were grown to confluence, and the medium was replaced with a fresh medium consisting of a 1:1 mixture of DMEM with Ham's F12 containing 1% FBS, 1% PenStrp, and 1% modified Eagle's medium non-essential amino acids, and were maintained for 3 days until use [39]. All media were purchased from GIBCO (GIBCO, USA). We stably overexpressed wild and mutant G93A superoxide dismutase 1 (SOD1, NM_000454) in NSC-34 cell lines that were selected using 100 μg/mL geneticin [40]. Exogenous 22(S)-, 24-,25-, and 27-OHC (Sigma, USA) was dissolved in ethanol [41]. The final concentration of ethanol in media did not exceed 0.1%. Riluzole was provided by Sanofi-aventis (Korea). Cells were pre-treated with several concentrations of 25-OHC (0, 2.5, 5, 10, 20 μM, and 40 μM) alone for 24 h, then treated with riluzole (0, 0.01, 0.1, 1, 10 μM, and 100 μM) for 24 h, and then washed carefully several times with phosphate-buffered saline (PBS). Cell viability was assessed by the MTT assay [42]. GSK-3 activity was evaluated using GSK-3 substrate phosphoglycogen synthase peptide-2 (Upstate, USA) as described previously [43].
Immunoblot analysis
After chemical treatment, cells were lysed with RIPA buffer supplemented with phosphatase inhibitor. Cell lysates were subjected to immunoblotting with antibodies; p-Akt (Ser473) (1:500, Cell Signaling Technology, Danvers, MA, USA), Akt (1:1000, Cell Signaling), p-GSK-3β (Ser9) and GSK-3β (1:1000, Santa Cruz Biotechnology, Dallas, Texas, USA), cytochrome c (1:500, Santa Cruz Biotech), PARP (1:1000, Santa Cruz Biotech), caspase-3 (1:1000, Cell Signaling), p-Tau (Ser396) and Tau (1:1000, Invitrogen), and GAPDH (1:1000, Santa Cruz Biotechnology). To evaluate cytosolic cytochrome c levels, cell pellets were fractionized using the Qproteome cell compartment kit (Qiagen, Germantown, MD, USA). The reactive bands were detected by ECL (Amersham Pharmacia Biotech, Piscataway, NJ, USA) and quantified with an image analyzer (Bio-Rad, Quantity One-4, 2, 0). The same membranes were probed for GAPDH as an internal control [44].
Immunocytochemistry
Briefly, the cells were washed with PBS, fixed in 4% formaldehyde, and permeabilized with 0.1% Triton X-100 for 5 min. Primary antibodies used were as follows: NF-H (neurofilament heavy chain, 1:2000, Abcam, Cambridge, MA, USA) and cleaved caspase-3 (1:200, Cell Signaling). For detection, cells were labeled with fluorescent secondary AlexaFluor 488-labelled anti-chicken and TRITC-conjugated anti-rabbit antibodies (1:500, Molecular Probes, Invitrogen, USA) for visualization. Slides were cover-slipped with a drop of DAPI medium (Sigma, Saint Louis, MO, USA). Cells without the primary antibody served as negative controls.
Evaluation of apoptotic cells
To evaluate apoptosis, cells were stained by annexin V-FITC apoptosis detection kit (Sigma) following the manufacturer’s instructions. Cells were washed and briefly trypsinized, and then washed twice with cold PBS. Cells were pelleted by centrifugation, resuspended in 1X binding buffer, and incubated with a staining solution (annexin V-FITC and PI) for 15 min. in the dark at 4°C. The cells were then resuspended in 1X binding buffer. Samples were kept on ice during the entire procedure and analyzed immediately by flow cytometry. Ten thousand cells from each sample were scanned and analyzed by FACS Canto (Becton Dickinson) using the standard configuration and parameters. Data acquisition and analysis was performed using the FACS DIVA software (BD Biosciences, San Jose, California, USA). Necrosis and apoptosis were determined by PI (FL2) and annexin V-FITC (FL1) fluorescence, respectively.
Transgenic SOD1G93A mice
B6SJL-Tg (SOD1-G93A) 1Gur/J mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). The mice were housed under a 12-h light/dark cycle and bred following the supplier’s protocol. The presence of the human G93A transgene was confirmed by polymerase chain reaction (PCR) and evaluated for transgene copy numbers [45]. In this strain, the first symptoms and end-stage symptoms of ALS appear at approximately 77 and 136 days of age [46]. Thus, we divided the SOD1G93A mice into three groups that were evaluated at the following time points: 60 (asymptomatic), 90 (early symptomatic), and 120 days of age (late symptomatic) [47]. Wild-type (WT) mice (B6SJLF1/J) obtained from the Jackson Laboratory were used as controls. All animal experiments were approved by the Institutional Animal Care and Use Committee of Hanyang University.
Real-time polymerase chain reaction (RT-PCR)
We quantified mRNA expression of CH25H and CYP3A4, which synthesize 25-OHC, and the CYP7B1, which metabolizes 25-OHC, using the mSOD1G93A mice (n = 30) and age-matched WT mice (n = 30). Mice were randomly assigned into three groups that were evaluated at each time points: n = 10. RNA was isolated from the lumbar spinal cord and cortical brain regions of mice and extracted using RNeasy Mini Prep (Qiagen, Germany). The quantity and integrity of RNA was evaluated by measuring the absorbance at 260 nm via Nanodrop (Thermo Scientific, ND-2000). The cDNA was synthesized from 2.5 μg of RNA using Vilo cDNA kit (Invitrogen, USA) per the manufacturer’s protocol. Primers for quantitative polymerase chain reaction (PCR) were purchased from Qiagen. We analyzed CYP3A4 (Qiagen, Germany, PPM03943B), CYP7B1 (Qiagen, Germany, PPM03979F), CH25H (Qiagen, Germany, PPM27829A), and GAPDH (Qiagen, Germany, PPM02946E). First-strand cDNA was amplified using Power SYBR Green PCR master mix (Applied Biosystems, USA) with primers. Real-time RT-PCR was conducted with Applied Biosystems StepOnePlusTM (Carlsbad, USA) at 95°C for 10 min, followed by 40 cycles of 15 sec at 95°C and 1 min at 60°C. A melting curve was performed to check the specificity of amplification. The relative quantity (RQ) was calculated by 2-ΔΔCt, taking GAPDH as the interval standard control. Each sample was analyzed in triplicate.
Statistical analyses
Clinical data are presented as means ± standard deviation. Experimental data are presented as means ± SEM of five or more independent experiments. Differences between groups were analyzed by Student’s t-test or one-way ANOVA followed by Tukey’s post hoc comparisons. Two-tailed p-values < 0.05 were considered statistically significant. Linear regression analyses were used to test for associations between values. All statistical analyses were performed using the SPSS 17.0 software package for Windows (SPSS, Seoul, Korea).
Abbreviations
ALS: amyotrophic lateral sclerosis; ALSFRSr: amyotrophic lateral sclerosis functional rating scale revised; CH25H: cholesterol 25-hydroxylase; CNS: central nervous system; CSF: cerebrospinal fluid; CYP3A4: cytochrome P450 3A4; CYP7B1: 25-hydroxycholesterol 7-alpha-hydroxylase; GSK: glycogen synthase kinase; HSP: hereditary spastic paraplegia; OHC: hydroxycholesterol; PARP: poly (ADP-ribose) polymerase; PBS: phosphate-buffered saline; SOD1: superoxide dismutase 1.
ACKNOWLEDGMENTS
This study was supported by the grant of National Research Foundation fund NRF-2014R1A1A2055741, Korea Health Industry Development Fund - HI15C0876, and partially supported by grant of Sanofi-Korea.
CONFLICTS OF INTEREST
All authors declare that there is no conflicts of interest
Author contributions
Sung-Min Kim, contributed to the conception and design of the work, interpretation of data and writing the manuscript. Min-Young Noh performed an in vitro experiment and wrote some of the manucript. Heejaung Kim performed an in vivo experiment. So-young Cheon contributed to an in vitro experiment. Kang Mi Lee, Jaeick Lee, and Eunju Cha, performed a mass spectrometry analysis for the serum and CSF of patients. Kyung Seok Park, Kwang-Woo Lee, and Jung-Joon Sung, M.D contributed to the acquisition of data. Seung Hyun Kim contributed to the acquisition of data, design of the work and also interpretation of the data.
REFERENCES
1. Haverkamp LJ, Appel V, Appel SH. Natural history of amyotrophic lateral sclerosis in a database population. Validation of a scoring system and a model for survival prediction. Brain. 1995; 118:707-719.
2. Magnus T, Beck M, Giess R, Puls I, Naumann M, Toyka K. Disease progression in amyotrophic lateral sclerosis: predictors of survival. Muscle & nerve. 2002; 25:709.
3. Kiernan M, Vucic S, Cheah B, Turner M, Eisen A, Hardiman O, Burrell J, Zoing M. Amyotrophic lateral sclerosis. Lancet. 2011; 377:942.
4. Björkhem I, Cedazo-Minguez A, Leoni V, Meaney S. Oxysterols and neurodegenerative diseases. Molecular aspects of medicine. 2009; 30:171-179.
5. Schroepfer GJ. Oxysterols: modulators of cholesterol metabolism and other processes. Physiological Reviews. 2000; 80:361-554.
6. Björkhem I. Crossing the barrier: oxysterols as cholesterol transporters and metabolic modulators in the brain. Journal of internal medicine. 2006; 260493-508.
7. Lappano R, Recchia AG, De Francesco EM, Angelone T, Cerra MC, Picard D, Maggiolini M. The cholesterol metabolite 25-hydroxycholesterol activates estrogen receptor α-mediated signaling in cancer cells and in cardiomyocytes. PloS one. 2011; 6:e16631.
8. Jang J, Park S, Jin Hur H, Cho HJ, Hwang I, Pyo Kang Y, Im I, Lee H, Lee E, Yang W, Kang HC, Won Kwon S, Yu JW, Kim DW. 25-hydroxycholesterol contributes to cerebral inflammation of X-linked adrenoleukodystrophy through activation of the NLRP3 inflammasome. Nat Commun. 2016; 7:13129.
9. Lund E, Kerr T, Sakai J, Li W, Russell D. cDNA cloning of mouse and human cholesterol 25-hydroxylases, polytopic membrane proteins that synthesize a potent oxysterol regulator of lipid metabolism. The Journal of biological chemistry. 1998; 273:34316.
10. Honda A, Miyazaki T, Ikegami T, Iwamoto J, Maeda T, Hirayama T, Saito Y, Teramoto T, Matsuzaki Y. Cholesterol 25-hydroxylation activity of CYP3A. Journal of lipid research. 2011; 52:1509.
11. Cyster J, Dang E, Reboldi A, Yi T. 25-Hydroxycholesterols in innate and adaptive immunity. Nature reviews Immunology. 2014; 14:731.
12. Malaspina A, Kaushik N, De Belleroche J. Differential expression of 14 genes in amyotrophic lateral sclerosis spinal cord detected using gridded cDNA arrays. Journal of neurochemistry. 2001; 77:132-145.
13. Andersson S, Gustafsson N, Warner M, Gustafsson JA. Inactivation of liver X receptor beta leads to adult-onset motor neuron degeneration in male mice. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102:3857-3862.
14. Tsaousidou MK, Ouahchi K, Warner TT, Yang Y, Simpson MA, Laing NG, Wilkinson PA, Madrid RE, Patel H, Hentati F. Sequence Alterations within CYP7B1 Implicate Defective Cholesterol Homeostasis in Motor-Neuron Degeneration. American journal of human genetics. 2008; 82:510.
15. Dupuis L, Oudart H, René F, Gonzalez dAJ, Loeffler J. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101:11159.
16. Yang JW, Kim SM, Kim HJ, Kim JE, Park KS, Kim SH, Lee KW, Sung JJ. Hypolipidemia in patients with amyotrophic lateral sclerosis: a possible gender difference? J Clin Neurol. 2013; 9:125-129.
17. Choi Y, Kim Y, Choi I, Kim S-W, Kim W-K. 25-Hydroxycholesterol induces mitochondria-dependent apoptosis via activation of glycogen synthase kinase-3 β in PC12 cells. Free radical research. 2008; 42:544-553.
18. Koh S-H, Kim Y, Kim HY, Hwang S, Lee CH, Kim SH. Inhibition of glycogen synthase kinase-3 suppresses the onset of symptoms and disease progression of G93A-SOD1 mouse model of ALS. Experimental neurology. 2007; 205:336-346.
19. Wuolikainen A, Acimovic J, Lövgren-Sandblom A, Parini P, Andersen PM, Björkhem I. Cholesterol, Oxysterol, Triglyceride, and Coenzyme Q Homeostasis in ALS. Evidence against the Hypothesis That Elevated 27-Hydroxycholesterol Is a Pathogenic Factor. PloS one. 2014; 9.
20. Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis. 2000; 1:293-299.
21. Olah M, Amor S, Brouwer N, Vinet J, Eggen B, Biber K, Boddeke H. Identification of a microglia phenotype supportive of remyelination. Glia. 2012; 60:306.
22. Zhang J, Akwa Y, el-Etr M, Baulieu E, Sjövall J. Metabolism of 27-, 25-and 24-hydroxycholesterol in rat glial cells and neurons. The Biochemical journal. 1997; 322:175.
23. Diczfalusy U, Olofsson KE, Carlsson A-M, Gong M, Golenbock DT, Rooyackers O, Fläring U, Björkbacka H. Marked upregulation of cholesterol 25-hydroxylase expression by lipopolysaccharide. Journal of lipid research. 2009; 50:2258-2264.
24. Casula M, Iyer A, Spliet W, Anink J, Steentjes K, Sta M, Troost D, Aronica E. Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience. 2011; 179:233.
25. Vejux A, Malvitte L, Lizard G. Side effects of oxysterols: cytotoxicity, oxidation, inflammation, and phospholipidosis. Brazilian journal of medical and biological research. 2008; 41:545-556.
26. Torocsik D, Szanto A, Nagy L. Oxysterol signaling links cholesterol metabolism and inflammation via the liver X receptor in macrophages. Molecular aspects of medicine. 2009; 30:134-152.
27. Lütjohann D, Papassotiropoulos A, Björkhem I, Locatelli S, Bagli M, Oehring RD, Schlegel U, Jessen F, Rao ML, von Bergmann K. Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. Journal of lipid research. 2000; 41:195-198.
28. Heverin M, Meaney S, Lütjohann D, Diczfalusy U, Wahren J, Björkhem I. Crossing the barrier: net flux of 27-hydroxycholesterol into the human brain. Journal of lipid research. 2005; 46:1047.
29. Chéramy A, Barbeito L, Godeheu G, Glowinski J. Riluzole inhibits the release of glutamate in the caudate nucleus of the cat in vivo. Neuroscience letters. 1992; 147:209.
30. Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. New England Journal of Medicine. 1994; 330:585-591.
31. Gold ES, Diercks AH, Podolsky I, Podyminogin RL, Askovich PS, Treuting PM, Aderem A. 25-Hydroxycholesterol acts as an amplifier of inflammatory signaling. Proceedings of the National Academy of Sciences of the United States of America. 2014; 111:10666.
32. Reboldi A, Dang EV, McDonald JG, Liang G, Russell DW, Cyster JG. 25-hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science (New York, NY). 2014; 345:679.
33. Cha E, Lee KM, Park, KD, Park KS, Lee K-W, Kim S-M, Lee J. Hydroxycholesterol Levels in the Serum and Cerebrospinal Fluid of Patients with Neuromyelitis Optica Revealed by LC-Ag+CIS/MS/MS and LC-ESI/MS/MS with Picolinic Derivatization: Increased Levels and Association with Disability during Acute Attack PloS one. 2016; 11:e0167819.
34. Leoni V, Masterman T, Patel P, Meaney S, Diczfalusy U, Björkhem I. Side chain oxidized oxysterols in cerebrospinal fluid and the integrity of blood-brain and blood-cerebrospinal fluid barriers. Journal of lipid research. 2003; 44:793-799.
35. Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, Nakanishi A. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. Journal of the neurological sciences. 1999; 169:13-21.
36. Kimura F, Fujimura C, Ishida S, Nakajima H, Furutama D, Uehara H, Shinoda K, Sugino M, Hanafusa T. Progression rate of ALSFRS-R at time of diagnosis predicts survival time in ALS. Neurology. 2006; 66:265.
37. Teunissen C, Petzold A, Bennett J, Berven F, Brundin L, Comabella M, Franciotta D, Frederiksen J, Fleming J, Furlan R. A consensus protocol for the standardization of cerebrospinal fluid collection and biobanking. Neurology. 2009; 73:1914-1922.
38. Cashman NR, Durham HD, Blusztajn JK, Oda K, Tabira T, Shaw IT, Dahrouge S, Antel JP. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Developmental dynamics. 1992; 194:209-221.
39. Eggett CJ, Crosier S, Manning P, Cookson MR, Menzies FM, McNeil CJ, Shaw PJ. Development and characterisation of a glutamate-sensitive motor neurone cell line. J Neurochem. 2000; 74:1895-1902.
40. Cho GW, Kim GY, Baek S, Kim H, Kim T, Kim HJ, Kim SH. Recombinant human erythropoietin reduces aggregation of mutant Cu/Zn-binding superoxide dismutase (SOD1) in NSC-34 cells. Neuroscience letters. 2011; 504:107-111.
41. Ning Y, Chen S, Li X, Ma Y, Zhao F, Yin L. Cholesterol, LDL, and 25-hydroxycholesterol regulate expression of the steroidogenic acute regulatory protein in microvascular endothelial cell line (bEnd.3). Biochem Biophys Res Commun. 2006; 342:1249-1256.
42. Lee YJ, Park HH, Koh SH, Choi NY, Lee KY. Amlodipine besylate and amlodipine camsylate prevent cortical neuronal cell death induced by oxidative stress. Journal of neurochemistry. 2011; 119:1262-1270.
43. Noh MY, Koh SH, Kim Y, Kim HY, Cho GW, Kim SH. Neuroprotective effects of donepezil through inhibition of GSK-3 activity in amyloid-beta-induced neuronal cell death. Journal of neurochemistry. 2009; 108:1116-1125.
44. Noh MY, Koh SH, Kim SM, Maurice T, Ku SK, Kim SH. Neuroprotective effects of donepezil against Abeta42-induced neuronal toxicity are mediated through not only enhancing PP2A activity, but also regulating GSK-3beta and nAChRs activity. Journal of neurochemistry. 2013.
45. Alexander GM, Erwin KL, Byers N, Deitch JS, Augelli BJ, Blankenhorn EP, Heiman-Patterson TD. Effect of transgene copy number on survival in the G93A SOD1 transgenic mouse model of ALS. Brain Res Mol Brain Res. 2004; 130:7-15.
46. Kieran D, Woods I, Villunger A, Strasser A, Prehn JH. Deletion of the BH3-only protein puma protects motoneurons from ER stress-induced apoptosis and delays motoneuron loss in ALS mice. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104:20606-20611.
47. Koh SH, Kim Y, Kim HY, Cho GW, Kim KS, Kim SH. Recombinant human erythropoietin suppresses symptom onset and progression of G93A-SOD1 mouse model of ALS by preventing motor neuron death and inflammation. Eur J Neurosci. 2007; 25:1923-1930.