
Introduction
Neuroblastoma(NB), a childhood cancer of the developing sympathetic nervous system, is the most common pediatric solid tumor, accounting for approximately 7% of childhood malignancies and 15% of pediatric oncology deaths [1]. According to the Cancer Statistics Review of Surveillance, Epidemiology, and End Results Program (SEER) conducted by the United States, more than 650 cases are diagnosed each year in North America [2, 3]. The incidence is about 10.54 cases per 1 million per year in children younger than 15 years [4, 5]. Prognosis of NB is associated with a number of factors, including International Neuroblastoma Risk Group (INRG) staging, age at diagnosis, histopathological classification, degree of tumor differentiation, amplification of N-MYC, loss of heterozygosity of 11q and DNA ploidy [6]. Based on the above factors, neuroblastoma patients can be classified into 4 groups: extremely lowrisk , lowrisk , moderaterisk , and highrisk [6]. Patients with low or intermediate risk can achieve an overall survival (OS) rate greater than 95% with surgery alone [7]. Furthermore, some studies have shown that infants with localized tumors can be cured without any treatment, including surgery [8, 9]. Approximately 50% of cases are high-risk with overall survival rates less than 40% [10]. High-risk patients often go through rigorous treatment consisting of 3 treatment blocks: Induction (chemotherapy and tumor resection), consolidation (high-dose chemotherapy with autologous stem-cell rescue and external-beam radiotherapy) and post-consolidation (anti-ganglioside 2 immunotherapy with cytokines and cis-retinoic acid) [7]. Heterogeneity is a clinical hallmark of NB, represented by its wide range of clinical behaviors and diverse response to treatments [11]. A subset of tumor will undergo spontaneous regression; while others will progress relentlessly into high-risk metastatic disease with poor prognosis despite the use of multimodal intensive treatment. Such diversity can be attributed to molecular differences [11]. Currently, molecular signature only constitutes a small portion of parameters used for prognostic evaluation, including MYCN amplification, 11q absence and DNA ploidy. However other molecular oriented parameters, such as mutations in ATRX, ALK, and variations of chromosome fragments, such as 1p and 17q, have not yet been included in the evaluation systems of NB. The most likely reason is that neuroblastomas show some mutations (mentioned below), but the most lack mutations in genes that are frequently mutated [12]. Surprisingly, some studies have shown high-risk NB was associated with fewer recurrent somatic mutations [13]. And compared with adult cancers, neuroblastomas show lower number of activating mutations affect protein functions [12].
This review primarily focuses on both germline mutations predisposing children to the development of neuroblastoma and somatic events associated with neuroblastoma pathogenesis and clinical phenotypes. Through the comprehensive summary, it may provide some tips in the studies of genetic studies or targeted therapies for neuroblastoma.
Germline Mutations of NB
Recent studies have shown that germline mutations of NB can be classified into two types, namely familial, and sporadic genetic susceptible genes.
Familial NB
Familial NB, primarily featuring rare mutations of certain genes, merely accounts for 1 percent of all the NB patients [14]. Familial NB is incomplete explicit autosomal dominant inheritance. In contrast to sporadic cases, familial cases occur at a younger age, related to multifocal primary tumors [15]. According to the literatures, germline mutations in familial NB mainly occur in two genes, namely paired-like homeobox 2B (PHOX2B) and anaplastic lymphoma kinase (ALK).
PHOX2B, the first identified predisposition gene related to NB [16], locates in 4p12 area of human chromosome, which encodes and regulates transcription factor of neural crest development. Most of patients harboring PHOX2B mutations are accompanied by complications, including congenital centrum hypopnea syndrome, congenital megacolon, multiple neurofibromatosis and pheochromocytoma [17, 18]. Several mutations in the PHOX2B have been identified in sporadic and familial neuroblastoma [17]. These mutations are believed to interfere with the PHOX2B protein’s role in promoting nerve cell differentiation. However, germline mutations of PHOX2B accounted for merely 6.4 % of hereditary neuroblastoma cases and were rarely detected in more common sporadic cases of the disease, indicating that the gene was not the major pathogenic gene [17, 19].
ALK gene mutations are more common than PHOX2B gene mutations in familial NB. ALK gene lies in human chromosome 2p23, encoding tyrosine kinase receptor that belongs to insulin receptor superfamily. It plays an important role in brain and specific neuron development. Amplification or mutation of ALK promotes phosphorylation of ALK, leading to increased kinase activity and ultimately results in tumorigenesis [20, 21]. In familial NB, ALK mutations often occur in coding areas, such as hotspots F1174, F1245 and R1275. Among these loci, the R1275Q mutation is the most common germline ALK mutation, occurring in about 45% of hereditary neuroblastomas [22]. These mutations also occur in the somatic cells, the most common somatic mutation in neuroblastoma, accounting for 6%-12% of sporadic neuroblastoma [12, 20, 23-28]. Among these mutations, ALK R1275Q is also the most common somatic ALK alteration [29, 30]. In addition, F1174L mutants are observed in a higher frequency in MYCN-amplified tumors [31], suggesting their pathogenic association [32]. ALK interacts with MYCN via multiple mechanisms: ALK was found to induce the expression of MYCN by promotor activation [33] and activation through ERK5 [34]; In addition, phosphoinositide 3-kinase/AKT further activates the ALK downstream signaling controlling glycogen synthase kinase 3 beta activity and MYCN protein stabilization. The observed synergistic effect of that mutant ALK accelerated tumor formation in MYCN transgenic mice [32, 35, 36] could be explained by the above mechanism.
With the knowledge of familial NB, recent years some researchers have proposed that screening of germline mutation of ALK and PHOX2B for sick children with NB family history or two-sided adrenal gland goiter should be performed. If ALK or PHOX2B gene was mutated, the sick children should undergo abdomen ultrasound and urine catecholamine levels detection every three months until the sick children reach 5 years old even though they are asymptomatic [37]. However, A recent study from pediatric cancer genome project suggested that less than 8.5% of pediatric cancer patients including neuroblastoma could be detected germline mutated predisposition genes. In addition, only 40% of their patients with germline mutations were pathogenic or probably pathogenic and that could be evaluated had a family history of cancer [38]. Derived from these, family history did not predict the presence of an underlying predisposition syndrome in most patients. This conclusion challenges the conventional belief for family history of NB.Thus, due to lack of enough evidences, screening of sick children with familial NB has yet to be further improved.
Sporadic NB
Sporadic neuroblastoma is more common. Through using genome-wide association studies (GWAS), several common genomic variables (single nucleotide polymorphisms [SNPs] and copy number variations [CNVs]) associated with sporadic neuroblastoma have been identfied.These genomic variables could be classified into three categories, among which two categories were related to NB risks. The first category was found in NB patients with high risk, including CASC15/14, BARD1, LMO1, LIN28B and HACE1 [37, 39]. The second category occurred in NB patients with low risk, including genes DUSP12、DDX、IL31RA and HSD17B12 [40]. The third category was variation of germline copy numbers, for example, gene NBPF23 [41].
Maris et al. identified common SNPs at 6p22 within CASC15 and CASC14 genes associated with neuroblastoma risk. Homozygosity for the at-risk G allele of rs6939340, the most significantly associated SNP, has an increased risk of developing neuroblastoma with odds ratio of 1.97. These gene variations were more likely to occur among stage 4 NB patients and individuals who carried MYCN amplification [42].
Several SNPs in BRCA1 associated RING domain 1 (BARD1) located in the area 2q35 of human chromosome were related with invasive NB. The function of the protein encoded by gene BARD1 is to form heterodimer in conjunction with BRCA1 protein. Stable formation of the heterodimer may be critical for BRCA1 exerting cancer inhibition [43]. A study comparing 397 high-risk cases and 2,043 controls revealed six new SNPs at 2q35 within the BARD1 gene locus significantly associated with NB [43]. They show that common variation in BARD1 associates with the risk of the aggressive and most clinically corresponding subtype of human neuroblastoma. Furthermore, Pugh et al. tested tumor tissue DNA and the matching peripheral blood DNA by using the next sequence, discovering gene BARD1 could undergo germline mutations (c.334C > T, c.1921C > T) [12].
The LMO1 risk alleles and copy-number gains are associated with increased LMO1 expression in NB primary tumors and cell lines, consistent with a gain of functional role in tumorigenesis [44]. The protein coded by gene LMO1 was transcription factor, including two LIM areas rich in cysteine, which played a role in protein interactions. Previous studies also indicate that LMO1 participates in regulation of nerve system development [45]. In the first GWAS performed on Chinese children, Wang et al. found that LMO1 on chromosome 11p15.4 was related to susceptibility of NB. They conducted NB GWAS including 549 cases (244 NB patients and 305 healthy controls). Eleven SNPs located within LMO1 were found significantly associated with NB, and rs204926 was confirmed to have the closest relationship [39]. At the somatic level, the LMO1 is mutated in 12% of NB, primarily through duplication, and this is associated with disease progression and poor survival [44].
LIN28B
Given the central role of MYCN in neuroblastoma biology, understanding its upstream regulators is also important. LIN28B has been shown to positively regulate MYCN levels through let-7 binding [2]. MYCN indirectly affects the expression of LIN28B through regulating miR-26a-5p( MiR-26a-5p and miR-26b-5p regulate LIN28B expression), and directly regulates LIN28B expression through a binding site within LIN28B promoter [46]. Taken together, these data points to a complex reciprocal regulatory relationship between the two genes [47]. LIN28B-let-7-MYCN regulation and control system blocked the differentiation of normal neuroblasts cells [48].
Besides common SNP variations, neuroblastoma was the first tumor that germline copy number variation was found to contribute to its susceptibility [41]. NBPF23 (Neuroblastoma breakpoint family, member 23) is located on chromosome 1q21.1 harboring a region where varies in copy number among population. Furthermore hemizygous deletion of this region is significantly associated with neuroblastoma [37]. The neuroblastoma breakpoint family (NBPF) has been found to play regulatory roles in neuroblastoma development and human evolution. However, the mechanism for the regulation and function of this family is still unknown. NBPF may function as DNA-binding transcription factor in nucleus, which provides important new insights into the functions of NBPF genes in the human cells [49].
Other germline mutations related to NB
DNA repair genes. DNA repair genes have been reported in oncogenesis of multiple cancers including neuroblastoma, such as BRCA1/2, PALB2, FANCD2 and CHEK2 et al. [50-55]. Pugh et al. performed whole genome sequencing of peripheral blood DNA samples on 240 cases of NB patients, and found a few germline mutations including CHEK2 (c.433C > T, c.542G > A and c.539G > A), PINK1 (c.1040T > C and c.836G > A), BARD1(c.334C > T and c.1921C > T) and PALB2 (c.1684+1C > A) [12]. Brooks et al. reported 20 cases of pediatric cancer including neuroblastoma among 379 families, uncovering mutations in either BRCA1 or BRCA2 [54]. Other studies also identified a deletion/insertion in the FANCD2 gene in nephro- and neuroblastomas [55]. Particularly, germline mutations in BRCA2 and PALB2, that PALB2 binds to the N terminus of BRCA2 and has a key role in localization and stabilization of BRCA2, have been detected that they were associated with the development of neuroblastoma [51, 52].
STK11
Papillary thyroid carcinoma (PTC), known as a secondary malignancy after treatment for neuroblastoma (NB), is rare in children. Targeted next-generation sequencing analysis for a 46 cancer-gene profile was performed on both tumors and peripheral blood DNA. A heterozygous missense mutation in STK11 (F354L) was identified in both NB and PTC. This mutation was also detected in peripheral blood cells [56]. However, the role of this gene in NBs requires further study.
Somatic Mutations in NB
Somatic events of NB can be classified as copy number variations and somatic gene mutations.
Copy number variations
Copy number variant (CNV) is an important and major source of variation in the human genome, comprising of large insertions and deletions that lead to gain or lose segments of chromosomes. Traditional genome-wide approaches to detect CNVs make use of single nucleotide polymorphism (SNP) array data or array comparative genome hybridization (aCGH) [57-59]. The minimum detectable size and breakpoint resolution are limited, for all results relying on the density of probes on the array. NGS offers higher sensitivity, and is a cost-effective alternative for CNV testing [60-62]. Copy number variation is relative to the structure variation of genome, including MYCN and other genes amplifications and chromosomal gains and losses. Comparing to adult tumors, the number of genetic somatic mutations in neuroblastoma was low, but the frequency of recurrent copy number variations was relatively high. Therefore CNVs could be used as biomarkers for neuroblastoma [26].
Somatic DNA amplification
DNA amplification plays a critical role in the development of solid tumors, potentially by causing overexpression of oncogenes. MYCN was the first proto-oncogene found to be amplified with significant clinical relevance, and its status was routinely used to direct treatment. As mentioned above, genes like LIN28B and ALK have been shown to regulate MYCN levels through certain mechanisms, bringing the old enemy into the focus of current and future targeted drug efforts.
MYCN, amplification of c-MYC homolog, has been shown to strongly correlate with poor prognosis [32, 63], considered as the best-characterized biomarker of risk for NB [63, 64]. It is located at chromosome 2p24 [65] and plays an important role in shortening cell cycle, promoting cell proliferation, inhibiting cell differentiation and apoptosis. Amplification of MYCN (no less than 10 times duplication of diploid genome or greater than 4 times duplication of number 2 chromosome related to signal) accounts for about 22% of gross occurrence of NB, mostly co-existing with other perilous factors [66].
Other regional amplifications. Previous studies have also reported other amplifications, such as ALK, DDX1 and OCD1 amplifications, associated with low recurrence and often co-occurring with MYNC amplification. Apart from MYCN, ALK is the most frequently amplified gene, accounting for 4% of NBs [23-25], followed by DDX1, located in close vicinity of MYCN [67]. ODC1 amplification at band 2p25, somatic amplification at 12q13-15 locus containing CDK4 (12q13_14) and MDM2 (12q15) were always found to be co-amplified with MYCN [26, 68, 69]. The NAG gene mapped in close vicinity to MYCN on chromosome band 2p24 was also found to be co-amplified with MYCN [70]. However, in the cases with absence of MYCN amplification, the amplicons at 12q13-14 and 12q13-15 , and CDK6 gene at 7q21, CCND1 gene at 11q13 were also detected [69]. Surprisingly, most of these genes are involved in cell cycle. This suggests that cell cycle regulators could play a role in neuroblastoma tumorigenesis.
Somatically chromosomal gains and losses
Somatically chromosomal imbalance is a key feature of NB: they occur as genomic amplifications or deletions as well as whole or segmental chromosome imbalances. Many chromosome alterations (whole chromosome gains or losses), resulting in hyperdiploidy, are associated with low-risk disease and favorable outcomes, while segmental chromosomal alterations (deletions of chromosome arms 1p, 3p, 4p, 9p, 11q and gains of chromosome arms 1q, 2p, 17q) are associated with more aggressive diseases [16, 71].
Loss of 1p and 11q
Loss of heterozygosity at chromosome 1p36, occurring in 23%-35% patients, is associated with other high-risk clinical and genomic features, such as older age, MYCN amplification, and metastatic disease [72-75]. Compared to the tumors without MYCN amplification, tumors with MYCN amplification generally had larger 1p deletions (the median size of deletion for MYCN-amplified tumors was 84 Mb and for non-amplified 46 Mb) [76]. Genes located at chromosome 1p36 including CHD5, CAMTA1 and PIK3CD are mostly likely to be deleted in the tumor [77-79]. CHD5 was first identified as a candidate tumor suppressor gene(TSG) which was frequently deleted in 1p36.31 of NBs [80]. High CHD5 is strongly related to favorable clinical, biological features and outcome. On the contrary, low/absent expression is associated with older age, higher stage, MYCN amplification and a poor outcome [81, 82]. Loss of chromosome 11q, most commonly seen in tumors without MYCN amplification, occurs in approximately 33% of neuroblastomas and is associated with a poor prognosis [72]. The related genes located at chromosome 11q include CADM1 and ATM [83, 84]. CAMTA1 qualifies as a TSG in NB. Low CAMTA1 expression is associated with unfavorable features (advanced stage, MYCN amplification) and poor outcome [79, 85].
17q gain
The most frequently identified genomic alteration of neuroblastoma cells is somatic gain of the distal portion of chromosome 17q, which occurs in at least half of primary tumors, predicting an overall poor prognosis, and frequently associated with other parameters of aggressive disease such as older age, MYCN amplification, and chromosome 1p deletion [86, 87]. At least two genes, survivin/BIRC5 and nm23/NME1, mapping to 17q gain regions, have been implicated contributing to the aggressive phenotype of neuroblastomas. Survivin/BIRC5 is an anti-apoptotic protein, and its expression is associated with poor prognosis, and nm23/NME1 encodes a nucleoside diphosphate kinases, involved in cell proliferation and differentiation. Overexpression of NME1 is associated with unfavorable outcome and aggressive features [79, 88, 89].
Loss of 9p and 3p
Caren et al. employed SNPs chip to analyze NB samples, and found chromosome 9p might undergo homozygous or heterozygosis loss. The genes involved included CDKN2A and CDKN2B. A region of homozygous deletion was discovered in one NB tumor sample, located in chromosome 3p24.1, harboring the gene RBMS3. They also detected two homozygous deletions in a NB cell line Kelly, one in chromosome 3p, covering the gene LSAMP, the other in gene PTPRD in chromosome 9p [76].
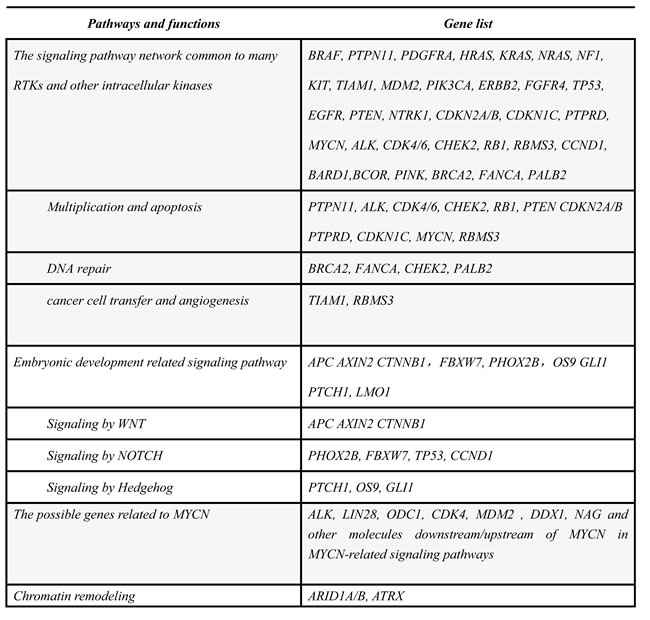
Table 1: Classification of functions of candidate genes relating to NB
Somatic mutations of NB
Compared with adult tumors, mutation frequencies of tumor cell of children are relatively lower [90], but activating mutations frequently affect specific biological processes in aggressive neuroblastoma [91].
ATRX, a gene plays an important role in epigenetic regulation, was found to be mutated in approximately 50% of adolescent and young adults with neuroblastoma [28, 92]. Although located in chromosome X, ATRX mutations were found in both males and females, consistent with previous reports [28]. Putative genetic loss-of-function alterations in the ATRX gene have been identified in nearly 10% of neuroblastomas [12, 27], and enriched in older patients [28]. In infant tumors or in tumors with MYCN amplification, no ATRX mutations are found, indicating ATRX alterations occur in a subtype of NB. Although the mechanism is poorly understood, neuroblastoma tumors with ATRX loss of function mutations were found to have lengthened telomeres. Nevertheless, these findings suggest that anti-telomerase-based therapies might benefit neuroblastoma patients with ATRX mutations [21, 93].
Other genes involved in chromatin regulation. Protein products encoded by gene ARID1A located at chromosome 1p35.3 and gene ARID1B located at chromosome 6q25.1 are SWI/SNF family members [94]. They regulate gene expression by modifying chromatin structure. Mark Sausen et al. [26] detected gene ARID1A could undergo nonsense, missense, disconnection mutation, and heterozygosity deficiency of somatic cell; ARID1B could suffer heterozygosity deficiency and point mutation of somatic cell. They contributed to failures of early stage treatment for NB patients and low survival rate to mutations of the two genes, suggesting ARID1A and ARID1B as contributors to neuroblastoma oncogenesis [26]. Interestingly, several sequence alterations in other genes involved in chromatin regulation in neuroblastoma have been found, including EP300, CREBBP, TTF2, KDM5A, CHD9, and gene IKZF1 [91], which might undergo somatic mutations and promote NB occurrence [44]. Thus, Chromatin remodeling may play an important role in the occurrence of NB.
PTPN11, locates at chromosome 12q24, encoding protein SHP-2, has an important role in signal transduction downstream of growth factor receptor signalling. Activating mutations of PTPN11 have been associated with developmental pathologies in neuroblastoma. Through activation of the RAS-ERK signalling pathway, SHP2 is ubiquitously expressed and regulates cell survival and proliferation. Therefore, reduction of SHP2 activity to suppress tumour cell growth is a potential target of cancer therapy [95-97].
NTRK1 encodes TrkA, which is a high affinity nerve growth factor (NGF) receptor. TrkA is involved in neural crest cell differentiation, and its expression has been reported to be associated with a favourable prognosis in neuroblastoma [98, 99]. NTRK1 has been detected in NB to undertake c.1810C > T mutation, which is common in NB for baby younger than 18 months without MYCN multiplication [100].
Promising Therapeutic Targets
With the efforts to collect large numbers of clinically annotated specimens and the advancements in technologies, researchers have discovered numerous therapeutic targets and subsequently developed agents to target them. Although the most frequent alteration in neuroblastoma is the amplification of MYCN, it’s difficult to target MYCN directly. As an alternative to target MYCN, we could target effector molecules downstream/upstream of MYCN in MYCN-related signaling pathways, such as the p53/MDM2/p14 (ARF) pathway, WNT/β-catenin pathway, and PI3K/mTOR pathway [64, 79, 101-107]. Some studies have shown that PI3 or Aurora A kinase inhibition can destabilize MYCN protein. Currently, an Aurora A kinase inhibitor is being evaluated in combination with irinotecan and temozolomide in a phase I clinical trial [7]. ALK alterations may arise in relapsed patients [108], occupying the position after the MYCN in the NB candidate genes. Crizotinib is a dual ALK/MET inhibitor, approved by the US food and drug administration (FDA) for patients with NSCLC harboring ALK rearrangements. However, a phase I clinical trial evaluating the efficacy of crizotinib as a single-agent revealed unfavorable results in neuroblastoma patients [109]. Therefore, the proportion of ALK-positive neuroblastoma patients benefits from Crizotinib is limited [110-112]. ATRX was found to be mutated in approximately 50% of adolescent and young adults with neuroblastoma [28, 92]. Preclinical and clinical investigations are required to prove the efficacy of ATRX inhibitors in this group of patients. The identification of carcinogenic mutations and an increased understanding of how these mutations trigger tumorigenesis enable researchers to develop agents to target such alteration.
Conclusions and Perspectives
In summary, the research on the pathogenesis of NB is facing a great challenge for its extremaly low mutation frequency. Occurrence of NB may be a multi-stage process with the simultaneous activation of multiple carcinogenic signalling routes, suggesting that growing interest should be stimulated in identifying disrupted pathways, instead of single mutated genes, thereby unearthing biological systems perturbed in neuroblastoma. There are a number of proteins and pathways that are relevant to NB pathogenesis. Major genes and pathways include (Table 1): a) The signaling pathway network common to many RTKs and other intracellular kinases, represented by RAS, RAF, PI3K, MAPK, MEK, AKT and mTOR [113]. These signal routes lead to cancer via mechanisms such as apoptosis, multiplication, DNA repair, cancer cell transfer and angiogenesis. The RAS pathway is among the most frequently mutated pathways in human cancer. Studies showed that RAS mutations could be predictive markers for treatment in neuroblastomas [114]; b) Embryonic development-related signaling pathways, such as Wnt, Notch and Hedgehog [115]. Taking NOTCH signal as an example, inhibiting Notch signal leads to NB cells differentiate more maturely [116]. At the early stage of nerve development, Notch signalling route may regulate multiplication, differentiation and apoptosis of cells, which may take part in generation of tumor angiogenesis through connecting to signal PI3K-AKT [117]. The Notch pathway may be activated by the homeobox transcription factor PHOX2B [118].
Since 2005, GWAS has been frequently used for tumor studies by means of comparison between different frequencies of SNP in cases and controls to seek risk mutations. However , GWAS is not a powerful approach for studying rare or uncommon SNPs, because GWAS results tend to have problems such as false positive, false negative, only a few single nucleotide polymorphisms detected locate in functional areas, and insensibility to rare mutation and structure mutation, leading to limitation of its application [119]. The paradigm of DNA next generation sequencing(NGS) could process millions of DNA templates in parallel, resulting in a low cost per base and a throughput on the gigabase (Gb) scale [120]. Progress of sequencing technique of new generation promotes fast development of whole genome sequencing, whole genome exon sequencing and depth sequencing of target genes, which is possible to solve the above-mentioned problems. Moreover, according to the characteristics of gene variants in neuroblastoma, perhaps the main power of NGS was the possibility to combine both mutation and copy-number events, to generate broad cataloging of genetic variations in neuroblastomas. However, the biggest challenge for us is that we must be able to distinguish functionally relevant mutations from nonpathogenic variants. This distinction is critical not only to correlate these with oncogenic potency, but also to deliver to the research of pathogenesis of NB. Thus, deep resequencing of target genes captured is a key method to locate potential pathopoiesia mutation of neuroblastoma, featuring high degree of specificity and feasibility, which provides potential new target spots for therapy intervening measure.
Acknowledgments
Thanks are due to the National Key Clinical Specialist Construction Programs of China (No.2013-544) and Medical University of Tianjin Science Foundation(2014KYM07) for funding support, and to Professor Kexin Chen and Fengju Song, Dr. Hanhan Zhang and partner Jiayi Liu for valuable discussion.
Conflicts of Interest
The authors declared no potential financial conflicts of interest.
References
1. Miller RW, Young JL, Jr. and Novakovic B. Childhood cancer. Cancer. 1995; 75:395-405.
2. Speleman F, Park JR and Henderson TO. Neuroblastoma: A Tough Nut to Crack. American Society of Clinical Oncology educational book American Society of Clinical Oncology Meeting. 2016; 35:e548-557.
3. Howlader N, Noone AM, Krapcho M, Miller D, Bishop K, Altekruse SF, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, et al. SEER Cancer Statistics Review, 1975-2013, National Cancer Institute. Bethesda, MD, http://seer.cancer.gov/csr/1975_2013/, based on November 2015 SEER data submission, posted to the SEER web site, April 2016.
4. London WB, Castleberry RP, Matthay KK, Look AT, Seeger RC, Shimada H, Thorner P, Brodeur G, Maris JM, Reynolds CP and Cohn SL. Evidence for an age cutoff greater than 365 days for neuroblastoma risk group stratification in the Children’s Oncology Group. Journal of clinical oncology. 2005; 23:6459-6465.
5. Gurney JG, Ross JA, Wall DA, Bleyer WA, Severson RK and Robison LL. Infant cancer in the U.S.: histology-specific incidence and trends, 1973 to 1992. J Pediatr Hematol Oncol. 1997; 19:428-432.
6. Cohn SL, Pearson AD, London WB, Monclair T, Ambros PF, Brodeur GM, Faldum A, Hero B, Iehara T, Machin D, Mosseri V, Simon T, Garaventa A, et al. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. Journal of clinical oncology. 2009; 27:289-297.
7. Pinto NR, Applebaum MA, Volchenboum SL, Matthay KK, London WB, Ambros PF, Nakagawara A, Berthold F, Schleiermacher G, Park JR, Valteau-Couanet D, Pearson AD and Cohn SL. Advances in Risk Classification and Treatment Strategies for Neuroblastoma. Journal of clinical oncology. 2015; 33:3008-3017.
8. Nuchtern JG, London WB, Barnewolt CE, Naranjo A, McGrady PW, Geiger JD, Diller L, Schmidt ML, Maris JM, Cohn SL and Shamberger RC. A prospective study of expectant observation as primary therapy for neuroblastoma in young infants: a Children’s Oncology Group study. Annals of surgery. 2012; 256:573-580.
9. Simon T, Hero B, Benz-Bohm G, von Schweinitz D and Berthold F. Review of image defined risk factors in localized neuroblastoma patients: Results of the GPOH NB97 trial. Pediatric blood & cancer. 2008; 50:965-969.
10. Levy D, Aerts I, Michon J, Lumbroso-Le Rouic L, Cellier C and Orbach D. Childhood cancer: progress but prognosis still very unequal. Example of Retinoblastoma and high-risk Neuroblastoma. Bulletin du cancer. 2014; 101:250-257.
11. Matthay KK, George RE and Yu AL. Promising therapeutic targets in neuroblastoma. Clin Cancer Res. 2012; 18:2740-2753.
12. Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, Kim J, Lawrence MS, Lichenstein L, et al. The genetic landscape of high-risk neuroblastoma. Nature genetics. 2013; 45:279-284.
13. Matas-Rico E, van Veen M, Leyton-Puig D, van den Berg J, Koster J, Kedziora KM, Molenaar B, Weerts MJ, de Rink I, Medema RH, Giepmans BN, Perrakis A, Jalink K, et al. Glycerophosphodiesterase GDE2 Promotes Neuroblastoma Differentiation through Glypican Release and Is a Marker of Clinical Outcome. Cancer cell. 2016; 30:548-562.
14. Maris JM, Hogarty MD, Bagatell R and Cohn SL. Neuroblastoma. Lancet (London, England). 2007; 369:2106-2120.
15. Knudson AG, Jr. and Strong LC. Mutation and cancer: neuroblastoma and pheochromocytoma. American journal of human genetics. 1972; 24:514-532.
16. Deyell RJ and Attiyeh EF. Advances in the understanding of constitutional and somatic genomic alterations in neuroblastoma. Cancer genetics. 2011; 204:113-121.
17. Raabe EH, Laudenslager M, Winter C, Wasserman N, Cole K, LaQuaglia M, Maris DJ, Mosse YP and Maris JM. Prevalence and functional consequence of PHOX2B mutations in neuroblastoma. Oncogene. 2008; 27:469-476.
18. Amiel J, Laudier B, Attie-Bitach T, Trang H, de Pontual L, Gener B, Trochet D, Etchevers H, Ray P, Simonneau M, Vekemans M, Munnich A, Gaultier C, et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nature genetics. 2003; 33:459-461.
19. Serra A, Haberle B, Konig IR, Kappler R, Suttorp M, Schackert HK, Roesner D and Fitze G. Rare occurrence of PHOX2b mutations in sporadic neuroblastomas. J Pediatr Hematol Oncol. 2008; 30:728-732.
20. Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, Laureys G, Speleman F, Kim C, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008; 455:930-935.
21. Sridhar S, Al-Moallem B, Kamal H, Terrile M and Stallings RL. New insights into the genetics of neuroblastoma. Molecular diagnosis & therapy. 2013; 17:63-69.
22. Devoto M, Specchia C, Laudenslager M, Longo L, Hakonarson H, Maris J and Mosse Y. Genome-wide linkage analysis to identify genetic modifiers of ALK mutation penetrance in familial neuroblastoma. Human heredity. 2011; 71:135-139.
23. Ogawa S, Takita J, Sanada M and Hayashi Y. Oncogenic mutations of ALK in neuroblastoma. Cancer science. 2011; 102:302-308.
24. George RE, Sanda T, Hanna M, Frohling S, Luther W, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, Xue L, Zozulya S, Gregor VE, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008; 455:975-978.
25. Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, Valteau-Couanet D, Frebourg T, Michon J, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008; 455:967-970.
26. Sausen M, Leary RJ, Jones S, Wu J, Reynolds CP, Liu X, Blackford A, Parmigiani G, Diaz LA, Jr., Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE, et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nature genetics. 2013; 45:12-17.
27. Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J, Westerman BA, van Arkel J, Ebus ME, Haneveld F, Lakeman A, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012; 483:589-593.
28. Cheung NK, Zhang J, Lu C, Parker M, Bahrami A, Tickoo SK, Heguy A, Pappo AS, Federico S, Dalton J, Cheung IY, Ding L, Fulton R, Wang J, et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. Jama. 2012; 307:1062-1071.
29. Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, Ryles H, Laudenslager M, Rappaport EF, Wood AC, McGrady PW, Hogarty MD, London WB, Radhakrishnan R, et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer cell. 2014; 26:682-694.
30. de Pontual L, Kettaneh D, Gordon CT, Oufadem M, Boddaert N, Lees M, Balu L, Lachassinne E, Petros A, Mollet J, Wilson LC, Munnich A, Brugiere L, et al. Germline gain-of-function mutations of ALK disrupt central nervous system development. Human mutation. 2011; 32:272-276.
31. De Brouwer S, De Preter K, Kumps C, Zabrocki P, Porcu M, Westerhout EM, Lakeman A, Vandesompele J, Hoebeeck J, Van Maerken T, De Paepe A, Laureys G, Schulte JH, et al. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin Cancer Res. 2010; 16:4353-4362.
32. Zhu S, Lee JS, Guo F, Shin J, Perez-Atayde AR, Kutok JL, Rodig SJ, Neuberg DS, Helman D, Feng H, Stewart RA, Wang W, George RE, et al. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer cell. 2012; 21:362-373.
33. Schonherr C, Ruuth K, Kamaraj S, Wang CL, Yang HL, Combaret V, Djos A, Martinsson T, Christensen JG, Palmer RH and Hallberg B. Anaplastic Lymphoma Kinase (ALK) regulates initiation of transcription of MYCN in neuroblastoma cells. Oncogene. 2012; 31:5193-5200.
34. Umapathy G, El Wakil A, Witek B, Chesler L, Danielson L, Deng X, Gray NS, Johansson M, Kvarnbrink S, Ruuth K, Schonherr C, Palmer RH and Hallberg B. The kinase ALK stimulates the kinase ERK5 to promote the expression of the oncogene MYCN in neuroblastoma. Science signaling. 2014; 7:ra102.
35. Heukamp LC, Thor T, Schramm A, De Preter K, Kumps C, De Wilde B, Odersky A, Peifer M, Lindner S, Spruessel A, Pattyn F, Mestdagh P, Menten B, et al. Targeted expression of mutated ALK induces neuroblastoma in transgenic mice. Science translational medicine. 2012; 4:141ra191.
36. Berry T, Luther W, Bhatnagar N, Jamin Y, Poon E, Sanda T, Pei D, Sharma B, Vetharoy WR, Hallsworth A, Ahmad Z, Barker K, Moreau L, et al. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer cell. 2012; 22:117-130.
37. Bosse KR and Maris JM. Advances in the translational genomics of neuroblastoma: From improving risk stratification and revealing novel biology to identifying actionable genomic alterations. Cancer. 2016; 122:20-33.
38. Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, Hedges D, Ma X, Zhou X, Yergeau DA, Wilkinson MR, Vadodaria B, Chen X, et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. The New England journal of medicine. 2015; 373:2336-2346.
39. Lu J, Chu P, Wang H, Jin Y, Han S, Han W, Tai J, Guo Y and Ni X. Candidate Gene Association Analysis of Neuroblastoma in Chinese Children Strengthens the Role of LMO1. PloS one. 2015; 10:e0127856.
40. Nguyen le B, Diskin SJ, Capasso M, Wang K, Diamond MA, Glessner J, Kim C, Attiyeh EF, Mosse YP, Cole K, Iolascon A, Devoto M, Hakonarson H, et al. Phenotype restricted genome-wide association study using a gene-centric approach identifies three low-risk neuroblastoma susceptibility Loci. PLoS genetics. 2011; 7:e1002026.
41. Diskin SJ, Hou C, Glessner JT, Attiyeh EF, Laudenslager M, Bosse K, Cole K, Mosse YP, Wood A, Lynch JE, Pecor K, Diamond M, Winter C, et al. Copy number variation at 1q21.1 associated with neuroblastoma. Nature. 2009; 459:987-991.
42. Maris JM, Mosse YP, Bradfield JP, Hou C, Monni S, Scott RH, Asgharzadeh S, Attiyeh EF, Diskin SJ, Laudenslager M, Winter C, Cole KA, Glessner JT, et al. Chromosome 6p22 locus associated with clinically aggressive neuroblastoma. The New England journal of medicine. 2008; 358:2585-2593.
43. Capasso M, Devoto M, Hou C, Asgharzadeh S, Glessner JT, Attiyeh EF, Mosse YP, Kim C, Diskin SJ, Cole KA, Bosse K, Diamond M, Laudenslager M, et al. Common variations in BARD1 influence susceptibility to high-risk neuroblastoma. Nature genetics. 2009; 41:718-723.
44. Schleiermacher G, Janoueix-Lerosey I and Delattre O. Recent insights into the biology of neuroblastoma. International journal of cancer. 2014; 135:2249-2261.
45. Wang K, Diskin SJ, Zhang H, Attiyeh EF, Winter C, Hou C, Schnepp RW, Diamond M, Bosse K, Mayes PA, Glessner J, Kim C, Frackelton E, et al. Integrative genomics identifies LMO1 as a neuroblastoma oncogene. Nature. 2011; 469:216-220.
46. Beckers A, Van Peer G, Carter DR, Mets E, Althoff K, Cheung BB, Schulte JH, Mestdagh P, Vandesompele J, Marshall GM, De Preter K and Speleman F. MYCN-targeting miRNAs are predominantly downregulated during MYCNdriven neuroblastoma tumor formation. Oncotarget. 2015; 6:5204-5216. doi: 10.18632/oncotarget.2477.
47. Cotterman R and Knoepfler PS. N-Myc regulates expression of pluripotency genes in neuroblastoma including lif, klf2, klf4, and lin28b. PloS one. 2009; 4:e5799.
48. Molenaar JJ, Domingo-Fernandez R, Ebus ME, Lindner S, Koster J, Drabek K, Mestdagh P, van Sluis P, Valentijn LJ, van Nes J, Broekmans M, Haneveld F, Volckmann R, et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nature genetics. 2012; 44:1199-1206.
49. Zhou F, Xing Y, Xu X, Yang Y, Zhang J, Ma Z and Wang J. NBPF is a potential DNA-binding transcription factor that is directly regulated by NF-kappaB. The international journal of biochemistry & cell biology. 2013; 45:2479-2490.
50. Lahtz C and Pfeifer GP. Epigenetic changes of DNA repair genes in cancer. Journal of molecular cell biology. 2011; 3:51-58.
51. Howlett NG. Fanconi anemia: Fanconi anemia, breast and embryonal cancer risk revisited. European journal of human genetics. 2007; 15:715-717.
52. Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ and Livingston DM. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Molecular cell. 2006; 22:719-729.
53. Magnusson S, Borg A, Kristoffersson U, Nilbert M, Wiebe T and Olsson H. Higher occurrence of childhood cancer in families with germline mutations in BRCA2, MMR and CDKN2A genes. Familial cancer. 2008; 7:331-337.
54. Brooks GA, Stopfer JE, Erlichman J, Davidson R, Nathanson KL and Domchek SM. Childhood cancer in families with and without BRCA1 or BRCA2 mutations ascertained at a high-risk breast cancer clinic. Cancer biology & therapy. 2006; 5:1098-1102.
55. Theruvath J, Russo A, Kron B, Paret C, Wingerter A, El Malki K, Neu MA, Alt F, Staatz G, Stein R, Seidmann L, Prawitt D and Faber J. Next-generation sequencing reveals germline mutations in an infant with synchronous occurrence of nephro- and neuroblastoma. Pediatric hematology and oncology. 2016; 33:264-275.
56. Buryk MA, Picarsic JL, Creary SE, Shaw PH, Simons JP, Deutsch M, Monaco SE, Nikiforov YE and Witchel SF. Identification of Unique, Heterozygous Germline Mutation, STK11 (p.F354L), in a Child with an Encapsulated Follicular Variant of Papillary Thyroid Carcinoma within Six Months of Completing Treatment for Neuroblastoma. Pediatric and developmental pathology. 2015; 18:318-323.
57. Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, Collins C, Kuo WL, Chen C, Zhai Y, Dairkee SH, Ljung BM, Gray JW, et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nature genetics. 1998; 20:207-211.
58. Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, Hakonarson H and Bucan M. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome research. 2007; 17:1665-1674.
59. Carter NP. Methods and strategies for analyzing copy number variation using DNA microarrays. Nature genetics. 2007; 39:S16-21.
60. Chiang DY, Getz G, Jaffe DB, O’Kelly MJ, Zhao X, Carter SL, Russ C, Nusbaum C, Meyerson M and Lander ES. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nature methods. 2009; 6:99-103.
61. Yoon S, Xuan Z, Makarov V, Ye K and Sebat J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome research. 2009; 19:1586-1592.
62. Jiang Y, Oldridge DA, Diskin SJ and Zhang NR. CODEX: a normalization and copy number variation detection method for whole exome sequencing. Nucleic acids research. 2015; 43:e39.
63. Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY and Hammond D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. The New England journal of medicine. 1985; 313:1111-1116.
64. Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P, Garnett MJ, McDermott U, Benes CH, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer discovery. 2013; 3:308-323.
65. Brodeur GM, Seeger RC, Schwab M, Varmus HE and Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science (New York, NY). 1984; 224:1121-1124.
66. Jeison M, Yaniv I and Ash S. Genetic stratification of neuroblastoma for treatment tailoring. Future oncology (London, England). 2011; 7:1087-1099.
67. Fix A, Lucchesi C, Ribeiro A, Lequin D, Pierron G, Schleiermacher G, Delattre O and Janoueix-Lerosey I. Characterization of amplicons in neuroblastoma: high-resolution mapping using DNA microarrays, relationship with outcome, and identification of overexpressed genes. Genes, chromosomes & cancer. 2008; 47:819-834.
68. Spitz R, Oberthuer A, Zapatka M, Brors B, Hero B, Ernestus K, Oestreich J, Fischer M, Simon T and Berthold F. Oligonucleotide array-based comparative genomic hybridization (aCGH) of 90 neuroblastomas reveals aberration patterns closely associated with relapse pattern and outcome. Genes, chromosomes & cancer. 2006; 45:1130-1142.
69. Guimier A, Ferrand S, Pierron G, Couturier J, Janoueix-Lerosey I, Combaret V, Mosseri V, Thebaud E, Gambart M, Plantaz D, Marabelle A, Coze C, Rialland X, et al. Clinical characteristics and outcome of patients with neuroblastoma presenting genomic amplification of loci other than MYCN. PloS one. 2014; 9:e101990.
70. Wimmer K, Zhu XX, Lamb BJ, Kuick R, Ambros PF, Kovar H, Thoraval D, Motyka S, Alberts JR and Hanash SM. Co-amplification of a novel gene, NAG, with the N-myc gene in neuroblastoma. Oncogene. 1999; 18:233-238.
71. Janoueix-Lerosey I, Schleiermacher G, Michels E, Mosseri V, Ribeiro A, Lequin D, Vermeulen J, Couturier J, Peuchmaur M, Valent A, Plantaz D, Rubie H, Valteau-Couanet D, et al. Overall genomic pattern is a predictor of outcome in neuroblastoma. Journal of clinical oncology. 2009; 27:1026-1033.
72. Attiyeh EF, London WB, Mosse YP, Wang Q, Winter C, Khazi D, McGrady PW, Seeger RC, Look AT, Shimada H, Brodeur GM, Cohn SL, Matthay KK and Maris JM. Chromosome 1p and 11q deletions and outcome in neuroblastoma. The New England journal of medicine. 2005; 353:2243-2253.
73. Caron H, van Sluis P, van Hoeve M, de Kraker J, Bras J, Slater R, Mannens M, Voute PA, Westerveld A and Versteeg R. Allelic loss of chromosome 1p36 in neuroblastoma is of preferential maternal origin and correlates with N-myc amplification. Nature genetics. 1993; 4:187-190.
74. Maris JM, Weiss MJ, Guo C, Gerbing RB, Stram DO, White PS, Hogarty MD, Sulman EP, Thompson PM, Lukens JN, Matthay KK, Seeger RC and Brodeur GM. Loss of heterozygosity at 1p36 independently predicts for disease progression but not decreased overall survival probability in neuroblastoma patients: a Children’s Cancer Group study. Journal of clinical oncology. 2000; 18:1888-1899.
75. White PS, Thompson PM, Seifried BA, Sulman EP, Jensen SJ, Guo C, Maris JM, Hogarty MD, Allen C, Biegel JA, Matise TC, Gregory SG, Reynolds CP, et al. Detailed molecular analysis of 1p36 in neuroblastoma. Medical and pediatric oncology. 2001; 36:37-41.
76. Caren H, Erichsen J, Olsson L, Enerback C, Sjoberg RM, Abrahamsson J, Kogner P and Martinsson T. High-resolution array copy number analyses for detection of deletion, gain, amplification and copy-neutral LOH in primary neuroblastoma tumors: four cases of homozygous deletions of the CDKN2A gene. BMC genomics. 2008; 9:353.
77. Henrich KO, Bauer T, Schulte J, Ehemann V, Deubzer H, Gogolin S, Muth D, Fischer M, Benner A, Konig R, Schwab M and Westermann F. CAMTA1, a 1p36 tumor suppressor candidate, inhibits growth and activates differentiation programs in neuroblastoma cells. Cancer research. 2011; 71:3142-3151.
78. Caren H, Fransson S, Ejeskar K, Kogner P and Martinsson T. Genetic and epigenetic changes in the common 1p36 deletion in neuroblastoma tumours. British journal of cancer. 2007; 97:1416-1424.
79. Brodeur GM, Iyer R, Croucher JL, Zhuang T, Higashi M and Kolla V. Therapeutic targets for neuroblastomas. Expert opinion on therapeutic targets. 2014; 18:277-292.
80. Thompson PM, Gotoh T, Kok M, White PS and Brodeur GM. CHD5, a new member of the chromodomain gene family, is preferentially expressed in the nervous system. Oncogene. 2003; 22:1002-1011.
81. Fujita T, Igarashi J, Okawa ER, Gotoh T, Manne J, Kolla V, Kim J, Zhao H, Pawel BR, London WB, Maris JM, White PS and Brodeur GM. CHD5, a tumor suppressor gene deleted from 1p36.31 in neuroblastomas. Journal of the National Cancer Institute. 2008; 100:940-949.
82. Koyama H, Zhuang T, Light JE, Kolla V, Higashi M, McGrady PW, London WB and Brodeur GM. Mechanisms of CHD5 Inactivation in neuroblastomas. Clin Cancer Res. 2012; 18:1588-1597.
83. Mandriota SJ, Valentijn LJ, Lesne L, Betts DR, Marino D, Boudal-Khoshbeen M, London WB, Rougemont AL, Attiyeh EF, Maris JM, Hogarty MD, Koster J, Molenaar JJ, et al. Ataxia-telangiectasia mutated (ATM) silencing promotes neuroblastoma progression through a MYCN independent mechanism. Oncotarget. 2015; 6:18558-18576. doi: 10.18632/oncotarget.4061.
84. Michels E, Hoebeeck J, De Preter K, Schramm A, Brichard B, De Paepe A, Eggert A, Laureys G, Vandesompele J and Speleman F. CADM1 is a strong neuroblastoma candidate gene that maps within a 3.72 Mb critical region of loss on 11q23. BMC cancer. 2008; 8:173.
85. Henrich KO, Fischer M, Mertens D, Benner A, Wiedemeyer R, Brors B, Oberthuer A, Berthold F, Wei JS, Khan J, Schwab M and Westermann F. Reduced expression of CAMTA1 correlates with adverse outcome in neuroblastoma patients. Clin Cancer Res. 2006; 12:131-138.
86. Bown N, Lastowska M, Cotterill S, O’Neill S, Ellershaw C, Roberts P, Lewis I and Pearson AD. 17q gain in neuroblastoma predicts adverse clinical outcome. U.K. Cancer Cytogenetics Group and the U.K. Children’s Cancer Study Group. Medical and pediatric oncology. 2001; 36:14-19.
87. Bown N, Cotterill S, Lastowska M, O’Neill S, Pearson AD, Plantaz D, Meddeb M, Danglot G, Brinkschmidt C, Christiansen H, Laureys G, Speleman F, Nicholson J, et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. The New England journal of medicine. 1999; 340:1954-1961.
88. Islam A, Kageyama H, Takada N, Kawamoto T, Takayasu H, Isogai E, Ohira M, Hashizume K, Kobayashi H, Kaneko Y and Nakagawara A. High expression of Survivin, mapped to 17q25, is significantly associated with poor prognostic factors and promotes cell survival in human neuroblastoma. Oncogene. 2000; 19:617-623.
89. Lamers F, van der Ploeg I, Schild L, Ebus ME, Koster J, Hansen BR, Koch T, Versteeg R, Caron HN and Molenaar JJ. Knockdown of survivin (BIRC5) causes apoptosis in neuroblastoma via mitotic catastrophe. Endocrine-related cancer. 2011; 18:657-668.
90. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr. and Kinzler KW. Cancer genome landscapes. Science (New York, NY). 2013; 339:1546-1558.
91. Lasorsa VA, Formicola D, Pignataro P, Cimmino F, Calabrese FM, Mora J, Esposito MR, Pantile M, Zanon C, De Mariano M, Longo L, Hogarty MD, de Torres C, Tonini GP, et al. Exome and deep sequencing of clinically aggressive neuroblastoma reveal somatic mutations that affect key pathways involved in cancer progression. Oncotarget. 2016; 7:21840-21852. doi: 10.18632/oncotarget.8187.
92. Lovejoy CA, Li W, Reisenweber S, Thongthip S, Bruno J, de Lange T, De S, Petrini JH, Sung PA, Jasin M, Rosenbluh J, Zwang Y, Weir BA, et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS genetics. 2012; 8:e1002772.
93. Ohali A, Avigad S, Ash S, Goshen Y, Luria D, Feinmesser M, Zaizov R and Yaniv I. Telomere length is a prognostic factor in neuroblastoma. Cancer. 2006; 107:1391-1399.
94. Vasileiou G, Ekici AB, Uebe S, Zweier C, Hoyer J, Engels H, Behrens J, Reis A and Hadjihannas MV. Chromatin-Remodeling-Factor ARID1B Represses Wnt/beta-Catenin Signaling. American journal of human genetics. 2015; 97:445-456.
95. Chan RJ and Feng GS. PTPN11 is the first identified proto-oncogene that encodes a tyrosine phosphatase. Blood. 2007; 109:862-867.
96. Matozaki T, Murata Y, Saito Y, Okazawa H and Ohnishi H. Protein tyrosine phosphatase SHP-2: a proto-oncogene product that promotes Ras activation. Cancer science. 2009; 100:1786-1793.
97. Prahallad A, Heynen GJ, Germano G, Willems SM, Evers B, Vecchione L, Gambino V, Lieftink C, Beijersbergen RL, Di Nicolantonio F, Bardelli A and Bernards R. PTPN11 Is a Central Node in Intrinsic and Acquired Resistance to Targeted Cancer Drugs. Cell reports. 2015; 12:1978-1985.
98. Nakagawara A, Arima M, Azar CG, Scavarda NJ and Brodeur GM. Inverse relationship between trk expression and N-myc amplification in human neuroblastomas. Cancer research. 1992; 52:1364-1368.
99. Kogner P, Barbany G, Dominici C, Castello MA, Raschella G and Persson H. Coexpression of messenger RNA for TRK protooncogene and low affinity nerve growth factor receptor in neuroblastoma with favorable prognosis. Cancer research. 1993; 53:2044-2050.
100. Lipska BS, Drozynska E, Scaruffi P, Tonini GP, Izycka-Swieszewska E, Zietkiewicz S, Balcerska A, Perek D, Chybicka A, Biernat W and Limon J. c.1810C>T polymorphism of NTRK1 gene is associated with reduced survival in neuroblastoma patients. BMC cancer. 2009; 9:436.
101. Kang JH, Rychahou PG, Ishola TA, Qiao J, Evers BM and Chung DH. MYCN silencing induces differentiation and apoptosis in human neuroblastoma cells. Biochemical and biophysical research communications. 2006; 351:192-197.
102. Ponthan F, Lindskog M, Karnehed N, Castro J and Kogner P. Evaluation of anti-tumour effects of oral fenretinide (4-HPR) in rats with human neuroblastoma xenografts. Oncology reports. 2003; 10:1587-1592.
103. Radhakrishnan SK, Halasi M, Bhat UG, Kurmasheva RT, Houghton PJ and Gartel AL. Proapoptotic compound ARC targets Akt and N-myc in neuroblastoma cells. Oncogene. 2008; 27:694-699.
104. Liu J, Guo L, Yin F, Zheng X, Chen G and Wang Y. Characterization and antitumor activity of triethylene tetramine, a novel telomerase inhibitor. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2008; 62:480-485.
105. Gamble LD, Kees UR, Tweddle DA and Lunec J. MYCN sensitizes neuroblastoma to the MDM2-p53 antagonists Nutlin-3 and MI-63. Oncogene. 2012; 31:752-763.
106. Shalaby T and Grotzer MA. MYC as Therapeutic Target for Embryonal Tumors: Potential and Challenges. Current cancer drug targets. 2016; 16:2-21.
107. Smith JR, Moreno L, Heaton SP, Chesler L, Pearson AD and Garrett MD. Novel pharmacodynamic biomarkers for MYCN protein and PI3K/AKT/mTOR pathway signaling in children with neuroblastoma. Molecular oncology. 2016; 10:538-552.
108. Caren H, Abel F, Kogner P and Martinsson T. High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumours. The Biochemical journal. 2008; 416:153-159.
109. Mosse YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, Rolland D, Balis FM, Maris JM, Weigel BJ, Ingle AM, Ahern C, Adamson PC, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. The Lancet Oncology. 2013; 14:472-480.
110. Chen K, Lv F, Xu G, Zhang M, Wu Y and Wu Z. Phosphoproteomics reveals ALK promote cell progress via RAS/ JNK pathway in neuroblastoma. Oncotarget. 2016; 7:75968-75980. doi: 10.18632/oncotarget.12513.
111. Guan S, Zhao Y, Lu J, Yu Y, Sun W, Mao X, Chen Z, Xu X, Pan J, Sun S and Yang J. Second-generation proteasome inhibitor carfilzomib sensitizes neuroblastoma cells to doxorubicin-induced apoptosis. Oncotarget. 2016; 7:75914-75925. doi: 10.18632/oncotarget.12427.
112. Mosse YP. Anaplastic Lymphoma Kinase as a Cancer Target in Pediatric Malignancies. Clin Cancer Res. 2016; 22:546-552.
113. Lemmon MA and Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010; 141:1117-1134.
114. Kiessling MK and Rogler G. Targeting the RAS pathway by mitogen-activated protein kinase inhibitors. Swiss medical weekly. 2015; 145:w14207.
115. Ordonez R, Gallo-Oller G, Martinez-Soto S, Legarra S, Pata-Merci N, Guegan J, Danglot G, Bernheim A, Melendez B, Rey JA and Castresana JS. Genome-wide microarray expression and genomic alterations by array-CGH analysis in neuroblastoma stem-like cells. PloS one. 2014; 9:e113105.
116. Zage PE, Nolo R, Fang W, Stewart J, Garcia-Manero G and Zweidler-McKay PA. Notch pathway activation induces neuroblastoma tumor cell growth arrest. Pediatric blood & cancer. 2012; 58:682-689.
117. Gorantla B, Bhoopathi P, Chetty C, Gogineni VR, Sailaja GS, Gondi CS and Rao JS. Notch signaling regulates tumor-induced angiogenesis in SPARC-overexpressed neuroblastoma. Angiogenesis. 2013; 16:85-100.
118. Ferrari-Toninelli G, Bonini SA, Uberti D, Buizza L, Bettinsoli P, Poliani PL, Facchetti F and Memo M. Targeting Notch pathway induces growth inhibition and differentiation of neuroblastoma cells. Neuro-oncology. 2010; 12:1231-1243.
119. Ku CS, Loy EY, Pawitan Y and Chia KS. The pursuit of genome-wide association studies: where are we now? Journal of human genetics. 2010; 55:195-206.
120. Mamanova L, Coffey AJ, Scott CE, Kozarewa I, Turner EH, Kumar A, Howard E, Shendure J and Turner DJ. Target-enrichment strategies for next-generation sequencing. Nature methods. 2010; 7:111-118.